Publisher’s version / Version de l'éditeur:
Recent patents on DNA and gene sequences, 4, 1, 2010-12-01
READ THESE TERMS AND CONDITIONS CAREFULLY BEFORE USING THIS WEBSITE. https://nrc-publications.canada.ca/eng/copyright
Vous avez des questions? Nous pouvons vous aider. Pour communiquer directement avec un auteur, consultez la première page de la revue dans laquelle son article a été publié afin de trouver ses coordonnées. Si vous n’arrivez pas à les repérer, communiquez avec nous à [email protected].
Questions? Contact the NRC Publications Archive team at
[email protected]. If you wish to email the authors directly, please see the first page of the publication for their contact information.
NRC Publications Archive
Archives des publications du CNRC
This publication could be one of several versions: author’s original, accepted manuscript or the publisher’s version. / La version de cette publication peut être l’une des suivantes : la version prépublication de l’auteur, la version acceptée du manuscrit ou la version de l’éditeur.
Access and use of this website and the material on it are subject to the Terms and Conditions set forth at
Recent patents and challenges on DNA microarray probe design
Tulpan, Dan
https://publications-cnrc.canada.ca/fra/droits
L’accès à ce site Web et l’utilisation de son contenu sont assujettis aux conditions présentées dans le site LISEZ CES CONDITIONS ATTENTIVEMENT AVANT D’UTILISER CE SITE WEB.
NRC Publications Record / Notice d'Archives des publications de CNRC:
https://nrc-publications.canada.ca/eng/view/object/?id=21fae3da-54e1-4f5b-8d80-04071d3880c4 https://publications-cnrc.canada.ca/fra/voir/objet/?id=21fae3da-54e1-4f5b-8d80-04071d3880c41872-2156/10 $100.00+.00 © 2010 Bentham Science Publishers Ltd.
Recent Patents and Challenges on DNA Microarray Probe Design
Technologies
Dan Tulpan*
Institute for Information Technology, National Research Council, 100 des Aboiteaux St., Moncton, NB E1A 7R1, Canada
Received: August 23, 2010; Accepted: November 7, 2010; Revised: November 15, 2010
Abstract: The invention of microarray technology has empowered scientists to quickly transition from single gene studies
to massively parallel experiments investigating thousands of genes. The use of DNA microarrays relies on accurate design for probes that are immobilized on a surface and bind specifically to complementary targets in a complex solution. The quality of a set of DNA probes heavily relies on DNA hybridization – the process of joining two single-strands of DNA to form a double-stranded molecule, and is traditionally ensured by using specific design criteria. The design of DNA probes for microarrays requires very stringent criteria, due to the necessity of choosing unique sequences that perfectly comple-ment specific regions from large genomic data sets, while avoiding hybridization with every other region of the same ge-nome. Patents and research publications presenting various probe design methods are reviewed in this manuscript and future potential extensions of current technologies are suggested.
Keywords: Biosensor, DNA, hybridization, microarray, patent, probe design. INTRODUCTION
Since their introduction in 1995 [1], DNA microarrays have become a standard method for the genome-wide meas-urement of mRNA expression levels. While being widely used to study the effects of certain treatments, diseases, and developmental stages on gene expression, the accuracy and reproducibility of the obtained results is hampered by many factors like: quality of the biological samples, accuracy of analytical methods and quality of microarray probes. Some of these factors can be addressed with standard mathematics, chemistry and physics techniques. Others require a deeper understanding and further development of models that better simulate the reality.
One such aspect where in silico modeling and simulation play an important role is the selection of probe sequences – a process also known as probe design. The choice of appropri-ate design strappropri-ategies for probes used in microarray applica-tions requires special attention, due to its direct impact on the accuracy and interpretation of the results. Poor design strategies may lead to low quality sets of probes that cross hybridize with undesired targets. Basic questions must be addressed [2], for example: (i) How does one select the most informative probes for comprising a signature in light of amplification and cross-hybridization effects? (ii) What lev-els of fluorescent signal and signature probe involvement constitute a detected entity (e.g. pathogen, SNP or expressed gene)? (iii) What is the accuracy and sensitivity of an opti-mized detection algorithm?
*Address correspondence to this author at the Institute for Information Technology, National Research Council, 100 des Aboiteaux St., Moncton, NB E1A 7R1, Canada; Tel: +1 (506) 861-0958; Fax: +1 (506) 851-3630; E-mail: [email protected]
To design high quality microarray probes, one must take into account three fundamental properties, namely: specific-ity, sensitivity and uniformity [3]. The probe specificity property requires that each probe must be specific to its cor-responding target and its complementary sequence must not occur in any other target sequence. The probe sensitivity property demands that each probe must hybridize with its perfect match subsequence of a target, regardless of the amount of other target sequences presented to the microar-ray. The uniformity property requires that all probe sequences must perform similarly well under uniform experimental conditions.
The present article offers an overview of the develop-ment of probe design methodologies for the manufacturing of high quality microarrays used in gene expression studies, species and samples identification by looking at relevant patents issued over the past 10 years, which are openly accessible at: freepatentsonline.com, at the European and US Patent Offices, Patent Scope, or at Google Patents. The re-view is done in the light of how probe design techniques can be further improved to produce more accurate microarray chips and how the development of such chips and other simi-lar DNA-based technologies could impact the development of novel diagnostics, drugs, therapeutics and bio-sensors for a multitude of applications.
BASIC PROTOCOLS FOR MICROARRAY EXPERI-MENTS
The key aspects of microarray probe design techniques can be better identified and improved if knowledge of the usage of the microarray is acquired. First it is important to understand the three basic types of microarrays [4] classified according to the task and implicitly the sample type used to construct them, namely: gene expression (GEM)
microar-2 Recent Patents on DNA & Gene Sequences microar-2010, Vol. 4, No. 1 Dan Tulpan
rays, microarray Comparative Genomic Hybridization (MCGH), and Single Nucleotide Polymorphism microarrays (SNPM).
The basic protocols (Fig. 1) involved in such microarray experiments can be generically presented as follows:
Fig. (1). The basic steps of a microarray experiment.
1. Isolation of DNA/RNA
In this step, either the mRNA (GEM) or pieces of genomic DNA (MCGH, SNPM) is extracted from corresponding samples.
2. Labelling of DNA/RNA
This process consists of reverse transcription reactions and DNA nucleotide dye incorporation and PCR amplifica-tion may be required if sample size is insufficient to produce visible hybridization.
3. Hybridization
This is a technique that uses fluorescently labelled nucleic acid strands as targets in solution to identify com-plementary strands, sequences that are able to form stable base-pair bonding with one another. Each single-stranded DNA fragment (probe or target) is composed of combina-tions of four different nucleotides, namely adenine (A), thymine (T), guanine (G), and cytosine (C), that are all linked together and pair-wise complementary (A-T, G-C). As an example, the complementary sequence to 5’-GCATGCAT-3’ will be 3’-CGTACGTA-5’. When two complementary sequences find each other, such as the
im-mobilized DNA probe and the target DNA, cDNA, or mRNA, they will bind together and produce a visible signal. 4. Microarray Washing
Once the hybridization process ends, the solution containing the targets mixture is removed and the chip is washed with various chemicals following perfected wet-lab protocols that include various recipes for washing buffers, centrifugation, slide coasting for dye preservation, etc.
5. Microarray Scanning
This process implies the usage of commercial optical scanning devices that use filters to pass all photons emitted in a specific wavelength range to a single point detector and capture the corresponding image in a digital format. The purpose of the scanning process is to detect and/or quantify the amount of targets bound to the probes on the microarray chip.
6. Data Analysis
This process consists of analyzing images by finding hybridization spots corresponding to known genes, SNPs or chromosome segments and comparing those using different mathematical and statistical techniques that often involve data normalization, multi-chip algorithms for estimation, model-based quality control, multi-dimensional scaling, and clustering. For example, in the case of gene expression mi-croarrays, the primary focus is to apply different methods for determining which genes are differentially expressed be-tween groups of samples using covering t-tests, multiple-testing corrections, false discovery rate estimations, empiri-cal Bayes methods, principal component analysis (PCA), and analysis of variance (ANOVA, MANOVA).
THE MICROARRAY PROBE DESIGN PROBLEM Following the definition provided by [3], a generalization of the microarray probe design problem is given next. Given N DNA strands s1,…,sN, representing target sequences in
solution, a set of constraints C, and probe length n, find a set of probes S (one or more probe per target), such that all sat-isfy the constraints in C.
Lockhart et al. [5] were among the first to study in depth
the selection of probes for microarrays. They proposed a set of empirically derived criteria that are still widely used and adapted to various applications [6-8].
Some of the widely used criteria for probe design are: probe length, cross-hybridization free energies, melting tem-peratures, position of the probe within target sequence, se-quence complexity, alphabet-related scoring, base composi-tion, self-hybridization free energies, perfect match free en-ergies, existence and absence of structural patterns in the secondary structure, homology among target sequences, and information content of probe sequences.
As microarray technologies advance, so are the probe design techniques. The following sections emphasize ad-vances in probe design methodologies with direct application in gene expression, species identification, genotyping and copy number variation analysis.
RECENT PATENTS AND RESEARCH ON MI-CROARRAY PROBE DESIGN METHODS FOR GENE EXPRESSION STUDIES
Recent advances in population studies and an increased interest in the genetics of cell populations triggered the need of comparing their corresponding relative quantities of expressed mRNA sequences. This task is achieved by quan-tifying gene transcription for tens of thousands of genes at a time using spotted cDNA microarrays [9]. These microarrays rely on the selection of samples of complementary DNA clones with known sequence content (known as probes), that are spotted and immobilized on the hard surface of the microarray. Various methods with different complexity lev-els were proposed for the selection of probe sequences, some being published in international journals, while others were described in patent applications.
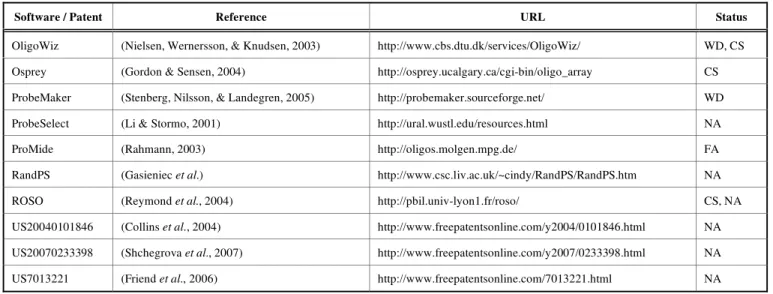
Most of these methods (see Table 1) use similar design algorithms and probe selection criteria, with only little varia-tion [6, 10-14]. While some methods trade combinatorial criteria for speed gain (ProbeSelect [6], ProMide [11], RandPS [15]), others use thermodynamically based criteria to achieve higher quality sets of probes (Osprey [16], ProbeMaker [17], OligoWiz [10], ROSO [12]).
Li and Stormo [6] introduce the ProbeSelect algorithm, which consists of seven steps. First, the program builds a suffix array [18] for the input sequences, to quickly identify the high similarity regions in the input sequences. Then, the algorithm builds a landscape for each input sequence. A landscape represents the frequency in the suffix array of all subsequences in the query input sequence. The third step consists in selecting a list with 10 to 20 candidate probes for each input sequence that minimizes the sum of the frequen-cies of their subsequences in the suffix array. The fourth step consists in searching for potential cross-hybridizations be-tween each probe and the set of input sequences. Step 5 marks the cross-hybridizations within the input sequences. The next step consists of melting temperature and
self-hybridization free energy computations for each remaining probe. The last step consists in selecting the probes, which have the most stable perfect-match free energy and hybridize the least with all the other potential targets.
Rahman [11] proposes the ProMide method, which is a fast probe selection algorithm that approximates oligo speci-ficity by longest common factor statistics. Their method uses a suffix array with the longest common substring instead of the edit distance, to speed-up the calculations, while losing accuracy.
Gasieniec et al. [15] present a probe selection method
called RandPS, which efficiently selects small sets of probes based on a randomized approach. The probe design criteria used in this work combine the criteria introduced by Lock-hart et al. [5] for Affymetrix chips, with thermodynamic criteria for homogeneity and sensitivity, and Hamming dis-tance criteria for enhanced specificity. They make the obser-vation that randomly generated sequences are expected to have properties of unique probes if evaluated through Ham-ming distance, which is defined as the number of positions where two equal length sequences differ [19]. For example, the Hamming distance between two randomly chosen se-quences of length m over a 4 letter alphabet is about 3m/4, which is a highly desired property of a system of probes. What they fail to notice is that, while calculations based on the Hamming distance measure are fast, the measure itself assumes sequence alignment symmetry and does not take into account gaps, thus denying the possibility of having asymmetrical mismatches between two aligned sequences, which reportedly happens during cross-hybridizations. Nielsen et al. [10] introduce OligoWiz, an application
which selects probes based on a set of five criteria: specific-ity, melting temperature, position within transcripts, se-quence complexity and base composition. A score is as-signed for each criterion per probe and a weighted score is computed. The probes with the best scores are further se-lected and visualized in a graphical interface.
Table 1. A Selection of Popular Probe Design Software Packages and Patents used in Gene Expression Studies. The Acronyms Used in this Table have the Following Meaning: WD = Web Download, NA = not Available (Web Site Download), FA = from Authors, CS = Client-Server
Software / Patent Reference URL Status
OligoWiz (Nielsen, Wernersson, & Knudsen, 2003) http://www.cbs.dtu.dk/services/OligoWiz/ WD, CS Osprey (Gordon & Sensen, 2004) http://osprey.ucalgary.ca/cgi-bin/oligo_array CS
ProbeMaker (Stenberg, Nilsson, & Landegren, 2005) http://probemaker.sourceforge.net/ WD
ProbeSelect (Li & Stormo, 2001) http://ural.wustl.edu/resources.html NA
ProMide (Rahmann, 2003) http://oligos.molgen.mpg.de/ FA
RandPS (Gasieniec et al.) http://www.csc.liv.ac.uk/~cindy/RandPS/RandPS.htm NA
ROSO (Reymond et al., 2004) http://pbil.univ-lyon1.fr/roso/ CS, NA
US20040101846 (Collins et al., 2004) http://www.freepatentsonline.com/y2004/0101846.html NA US20070233398 (Shchegrova et al., 2007) http://www.freepatentsonline.com/y2007/0233398.html NA
4 Recent Patents on DNA & Gene Sequences 2010, Vol. 4, No. 1 Dan Tulpan
ROSO [12] consists of a suite of 5 probe selection steps. First, it filters out all input sequences that are identical and masks also the repeated bases within them. The second step consists of a similarity search using BLAST [20]. Next step consists of eliminating probes that self-hybridize. The fourth step eliminates all probes that have undesired melting tem-peratures. The last step selects a final set of probes based on four additional criteria: GC content, first and last bases, base repetitions and self-hybridization free energies.
A number of patents describe similar methods used in the design of microarray probe sequences for gene expression pattern finding.
In patent US20040101846,Collins et al. [21] present an invention involving the use of a combination of probe selec-tion criteria and the Cluster Affinity Search Technique (CAST) based on expression scores of expression vectors. The probes are pre-screened according to a set of criteria including probe length (60nt), base composition, BLAST homology, and then an iterative clustering process is applied on experimental data obtained with the probes, such that in the end one optimum candidate probe sequence is selected from the best cluster based on a set of empirically measured performance metrics. Nevertheless, the CAST technique works only if two major assumptions are fulfilled, namely (i) expression patterns for candidate probes are representative of the expression patterns for all possible probes for a given target, and (ii) candidate probes showing gene expression patterns that differ from the pattern shown by the majority of the candidate probes are outliers.
In patent US7013221 Friend et al. [22] present methods to detect and report polynucleotide sequences relevant for gene expression studies and signature chips using sets of probes with optimum sensitivity and specificity. Their probe selection criteria include: probe length between 22nt and 35nt, binding free energies for perfect match duplex forma-tion and cross-hybridizaforma-tions, base composiforma-tion, the posiforma-tion of a probe’s complementary sequence on a target, probe se-quence complexity, limited homology (BLAST [20] or Pow-erBLAST [23]), self dimer binding free energy, the structural content of target sequences, and the information content of probe sequences. Their invention provides evidence and supports the idea that the number of probes required to relia-bly and accurately identify a particular target sequence, such as the sequence of a particular transcript, may be reduced to as few as one probe by carefully selecting probes according to a set of stringent criteria. The evidence consists of screen-ing chips with approximately 6,000 probes 60nt long used to identify changes in mRNA transcripts in inactivated and ac-tivated human lymphocytes.
Shchegrova et al. [24] describe in patent US20070233398 methods for selecting and predicting probe performance for tiling microarrays using response variable and predictors and performing multiple additive regression trees (MART) analysis to determine the functional depend-ence of observed signals on probe parameters. Their valida-tion experimental data comprises of 8 probe designs imple-mented on 16 microarrays. Each probe design uses ten or more overlapping 60-mers to identify a gene, with a total of 310,000 tiled probes. The probes overlap in 5nt increments. No a priori knowledge of the relationships between variables
and predictors is needed. Their assumptions claim is that probes, which consistently produce the same signal response will perform substantially similarly under a plurality of dif-ferent experimental conditions. Nevertheless, previous stud-ies show that DNA sequence behaviour changes when ther-modynamic conditions vary, thus influencing the ability of probes to bind the right targets [25].
RECENT PATENTS AND RESEARCH ON MI-CROARRAY PROBE DESIGN METHODS FOR SPE-CIES IDENTIFICATION
Identification of viral, prokaryotic and eukaryotic species is a problem of great interest worldwide and requires further perfection and speed-up. For example, the gold standard for detection of bacteria from patients with various diseases is blood culture, which typically takes 1–3 days to become positive. A further 1–2 days might be required for exact identification of bacteria and their differential antibiotic re-sistance. New diagnostic tests that could identify bacterial species rapidly and accurately are needed. Novel DNA-based microarray platforms now allow rapid detection and species identification of several microbial pathogens [25-27]. The same applies for the detection of viral strains, where current approaches in the laboratory, such as virus isolation and se-rological diagnosis, usually share the shortcomings of insen-sitivity, time-consuming, or cross-reaction, and are not effi-cient enough for rapid and accurate diagnosis [28-30]. In recent years, the polymerase chain reaction (PCR) has been used for detection of human viruses in clinical practice. Mul-tiplex PCR, in which two or more target sequences are am-plified simultaneously in one single reaction, is highly cost-effective and efficient. However, further species identifica-tion of PCR products remains a tedious problem involving Southern Blot or restriction fragment length polymorphism (RFLP) [31-33]. DNA microarrays, due to their capacity to accommodate different specific probes, offer an ideal way for species identification of PCR-amplified products [34, 35]. A summary of such methods is presented next (see also Table 2).
In 2002, Volokhov et al. [26] developed a rapid microar-ray-based assay for the detection and discrimination of six species of the Listeria genus. The approach used in this study involves one-tube multiplex PCR amplification of six target bacterial virulence factor genes, synthesis of fluorescently labelled single-stranded DNA, and hybridization to the mul-tiple individual oligonucleotide probes specific for each Lis-teria species and immobilized on a glass surface. They de-signed 10 individual probes per gene using only alignment and homology. A preliminary BLAST homology search was used to identify the homologous sequences of each of the six genes followed by an alignment with the ClustalX [36] soft-ware to identify sequences of regions highly conserved among all alleles of each gene.
In patent US7657381B2, Kwon et al. [37] present an invention describing a method to design DNA probes for microarrays applied in Mycobacterium species identification. They used 16sRNA targets derived from seven species of Mycobacterium and designed 5nt long probes. While the specifics of the design process have been omitted, a general overview suggests the following criteria have been used:
fixed probe length, melting temperature, and a threshold value for sequence homology.
A more advanced probe design and validation mecha-nism is reported in patent US20090053708. Here, Wong et
al. [2] present an invention consisting of a two stage process.
First step consists of identifying regions on target sequences that need to be amplified and are guaranteed to have an effi-ciency of amplification above a given threshold. Second step consists of designing probe sequences complementary to regions on the targets identified in step 1. The selection of target regions to be amplified can be done either by calculat-ing an Amplification Efficiency Score (AES), which is the probability that a forward primer ri can bind to a position i
and a reverse primer rj can bind at a position j of the target
nucleic acid, or using randomly generated forward and re-verse primer sequences. The selection of probes is done ac-cording to a combination of criteria including a GC-content from 40% to 60%, a high free energy computed based on the NN thermodynamic model, a high Hamming distance be-tween pairs of probes, and a low homology bebe-tween a probe and all potential target sequences.
Similarly, in patent EP2192182, Morishita et al. [38] present an invention describing a primer set and probes for detection of the Human Papillomavirus (HPV) types. They use a combination of criteria for designing the primer and probe sequences. For example, one of their reported primer sets has the following characteristics: 80-95% homology between each primer and the corresponding target to be am-plified by PCR, melting temperature in the range 60-70 oC, probe length 69-79nt, 50-60% GC-content, reduced homol-ogy within different HPV types, no self-hybridization, no cross-hybridization among primers.
RECENT PATENTS AND RESEARCH ON MI-CROARRAY PROBE DESIGN METHODS FOR SNP IDENTIFICATION
A single nucleotide polymorphism (SNP) is a one base variation at a specific location in the genome that is found in a significant percentage of the population. In the Human genome, more than 9 million SNPs have been reported in public databases [39, 40] with an average frequency of one in 1000nt [41, 42]. In studies of disease genetics where SNPs are used, the aim is to identify SNPs that cause changes in cellular biological processes inducing diseased states [43-46]. In pharmacogenomics studies SNPs are utilized as markers to elucidate effects of genetic polymorphisms on drug responses [47-50]. In both disease genetics and
phar-macogenomics studies, a large number of SNPs need to be genotyped in large populations, thus high throughput be-comes a critical factor that can be achieved via multiplexing. In genotyping technologies, PCR amplification of samples plays a central role and allows improved allelic discrimina-tion. One of the most efficient and accurate methods for al-lele discrimination is using hybridization. Such approaches were implemented via microarray technologies (see Table 3) and use differences in thermal stability of DNA duplexes to differentiate perfect matches versus mismatches in probe-target interactions.
An example of a microarray used in SNP identification is described in patent KR10-2004-0030447. Here, Oh et al. [51] describe an invention consisting of microarray probe selection based on limiting the melting temperature of poten-tial bindings of each probe with 5’ and 3’ ends of specific target sequences. Unfortunately, their patent application in-cludes a limited description of the design process and specif-ics of the probe sequence characteristspecif-ics. The criteria used to design such probes include probe length between 18nt and 28nt, GC-content from 20% to 80%, melting temperatures in the range 65-80 oC, less than 50% homology with other tar-gets, and a melting temperature difference between perfect matches and imperfect matches of a probe and target regions greater than 3 oC.
RECENT PATENTS AND RESEARCH ON MI-CROARRAY PROBE DESIGN METHODS FOR COPY NUMBER VARIATION ANALYSIS
The variability of genetic characteristics is what makes each human being unique and influences our susceptibility to diseases such as various types of cancer. One of the methods of choice nowadays for studying the genetics of such dis-eases is the array comparative genomic hybridization (ar-rayCGH) – a method that reshapes our understanding of the role that genetic variants play in disease development. Table 4 provides a brief summary of microarray probe design tech-niques and analysis relevant to this field.
Huang et al. [52] propose the Copy Number Analysis
with Regression and Tree (CARAT) method that uses probe intensity information to infer copy number in an allele-specific manner from high density DNA oligonucleotide arrays designed to genotype over 100,000 SNPs. The PCR primers used in this study were obtained with Primer Ex-press 1.5 System (Applied Biosystems). The CARAT method was trained on 128 samples and evaluated on test samples that included 90 normal individuals, DNA samples Table 2. A Selection of Popular Probe Design Software Packages and Patents Used in Species Identification Studies. The
Acro-nyms Used in this Table have the Following Meaning: NA = Not Available (Web Site Download)
Software / Patent Reference URL Status
NA (Volokhov et al., 2002) http://www.ncbi.nlm.nih.gov/pubmed/12454178 NA
US7657381B2 (Kwon et al., 2010) http://www.freepatentsonline.com/7657381.html NA
US20090053708 (Wong et al., 2009) http://www.freepatentsonline.com/y2009/0053708.html NA
6 Recent Patents on DNA & Gene Sequences 2010, Vol. 4, No. 1 Dan Tulpan
with varying numbers of X chromosomes (1X to 5X), and several human breast cancer cell lines that harbour both low level and high level copy number alterations. The same data set was used as a benchmark for testing a newly proposed method named PICR described in patent US2009137417 by Fu et al. [53].
In their patent, Fu et al. [53] present methodologies for accurate and efficient estimation of gene copy number using oligonucleotide microarrays. Their method incorporates both perfect and imperfect match probe-target hybridization data into a Generalised Positional-Dependent Nearest Neighbour (GPDNN) Statistical Model, which extends the already exist-ing PDNN [54, 55] to estimate gene copy numbers. They rely on data coming from Affymetrix microarrays, where the probe sets are established. Three examples are presented where their methodology is successfully used to determine: (i) copy numbers in SNP arrays samples of a HapMap [56] dataset, (ii) DNA copy number alterations of chromosome X, and (iii) RNA gene expression.
Lipson et al. [57] proposed the ProbeSpec utility for
mapping the specificity of all candidate probes for a given sequence (e.g. transcript) against a background containing a large set of sequences (e.g. a transcriptome). ProbeSpec uses an indexed search to limit the number of comparisons be-tween the target and background sequences. ProbeSpec is reportedly mentioned in patent US20060110744, as an alter-native for BLAST when a homology search is necessary. In the same patent, Sampas et al. [58] follow the traditional design approach and describe systems and techniques that rely solely on a set of successive filters aimed at designing sets of probes for CGH-based arrays. Their systems include the following selection criteria: probe length, homology search based on masking non-selectable areas on a target, duplex melting temperatures, hairpin free energies, and posi-tion on the genome. Also menposi-tioned in the same patent is the possibility of using melting temperature estimations based on the methodology developed by Breslauer et al. [59].
CURRENT & FUTURE DEVELOPMENTS
During the last 15 years, DNA microarrays played a ma-jor role in understanding the inner mechanisms of human genetics and in the development of bio sensors able to iden-tify and quaniden-tify an increased number of bacterial strains and viruses in one experiment. A major contribution to the ad-vances in DNA microarray technologies was brought by con-tinuous improvements in probe design techniques. As ac-knowledged by Friend et al. [22], the number of probe se-quences necessary to identify a specific target can be reduced to one if they are carefully selected so that sensitivity, speci-ficity and uniformity are maximized while cross-hybridi-zation and self-hybridicross-hybridi-zation are minimized.
Current probe selection methods for DNA microarrays that require high number of probes (tens or hundreds of thousands) are generally based on computationally efficient filtering mechanisms, where mathematically simpler rather than thermodynamically accurate models are employed. For example, a vast majority of probe design methods presented in patents and research publications use homology searches that employ non-optimal local alignment tools like BLAST and BLAT to screen out target sequence regions with repeats and non-unique subsequences. Only a handful of approaches employ more advanced techniques that select probes based on minimizing their cross-hybridization estimated via ther-modynamic modeling of hybridization for potential probe-target pair interactions [60-62].
While several software solutions are available to help solve the microarray probe design problem and are summa-rized in a few surveys and reviews [63, 64], thorough com-parisons and in-vitro validations of these methods still need to be performed. Thus one of the current challenges is to setup an in-vitro experimental framework where a large number of chips with probes designed with a comprehensive set of methods for the same set of targets could be compared under similar experimental conditions. Such a large scale experiment will help identify the strengths and weaknesses Table 3. A Selection of Popular Probe Design Software Packages and and Patents Used in SNP Identification Studies. The
Acro-nyms Used in this Table have the Following Meaning: NA = not Available (Web Site Download)
Software / Patent Reference URL Status
KR10-2004-0030447 (Oh et al., 2004) http://www.freepatentsonline.com/y2005/0244871.html NA
Table 4. A Selection of Popular Probe Design Software Packages and and Patents Used in Copy Number Variation Studies. The Acronyms Used in this Table have the Following Meaning: NA = Not Available (Web Site Download), FA = from Authors
Software / Patent Reference URL Status
CARAT (Huang et al., 2006) http://www.ncbi.nlm.nih.gov/pubmed/16504045 NA
ProbeSpec (Lipson et al., 2002) http://bioinfo.cs.technion.ac.il/probespec/ FA
WO2009051803 (Fu et al., 2009) http://www.freepatentsonline.com/y2010/0222225.html NA
of the proposed software solutions, thus identifying a best practices approach for the microarray probe design problem.
With the advances in sequence hybridization modeling and continuous improvements in computational power, more advanced in-silico probe design methods are expected to appear, thus leading to more powerful DNA microarray chips with higher impact on medicine, pharmacology, envi-ronment and agriculture.
ABBREVIATIONS
16sRNA = 16S ribosomal RiboNucleic Acid
A = Adenine
AES = Amplification Efficiency Score ANOVA = ANalysis Of Variance
arrayCGH = Array Comparative Genomic Hybridiza-tion
C = Cytosine
CARAT = Copy Number Analysis with Regression And Tree
CAST = Cluster Affinity Search Technique cDNA = complementary DeoxyriboNucleic Acid CGH = Comparative Genomic Hybridization DNA = DeoxyriboNucleic Acid
G = Guanine
GEM = Gene Expression Microarrays
GPDNN = Generalised Positional-Dependent Nearest Neighbour
MANOVA = Multivariate ANalysis Of VAriance MART = Multiple Additive Regression Trees MCGH = Microarray Comparative Genomic
Hybridization
mRNA = Messenger RiboNucleic Acid nt = Nucleotide
PCA = Principal Component Analysis PCR = Polymerase Chain Reaction PDNN = Positional-Dependent Nearest Neighbour RNA = RiboNucleic Acid
RFLP = Restriction Fragment Length Polymor-phism
SNP = Single Nucleotide Polymorphism
SNPM = Single Nucleotide Polymorphism Microar-ray
T = Thymine
ACKNOWLEDGEMENTS
The author would like to thank his colleagues from the Knowledge Discovery Group for valuable feedback. I am grateful for the help provided by the Bentham Sciences
Editorial Team with expanding the patent search and the anonymous reviewers for valuable comments. This work was supported by the National Research Council of Canada - Atlantic Initiative.
CONFLICT OF INTEREST
The author declares no conflict of interest in the publica-tion of this manuscript.
REFERENCES
[1] Schena M, Shalon D, Davis RW, Brown PO. Quantitative monitor-ing of gene expression patterns with a complementary DNA mi-croarray. Science 1995; 270: 467-70.
[2] Wong CWC, Sung W, Lee C, Miller LD. Method and/or apparatus of oligonucleotide design and/or nucleic acid detection. US20090053708A1, 2009.
[3] Tulpan D. Effective Heuristic Methods for DNA Strand Design. PhD Thesis, University of British Columbia, Vancouver, Canada, October 2006.
[4] NCBI – A science primer. Microarrays – chipping away at the mysteries of science and medicine. Available at: http://www.ncbi.nlm.nih.gov/About/primer/microarrays.html (Accessed on: August 16, 2010).
[5] Lockhart DJ, Dong HL, Byrne MC, Follettie MT, Gallo MV, Chee MS, et al. Expression monitoring by hybridization to high-density oligonucleotide arrays. Nat Biotechnol 1996; 14(13): 1675-80. [6] Li F, Stormo G. Selection of optimal DNA oligos for gene
expres-sion arrays. Bioinformatics 2001; 17: 1067-76.
[7] Relogio A, Schwager C, Richter A, Ansorge W, Valcarcel J. Opti-mization of oligonucleotide-based DNA microarrays. Nucleic Acids Res 2002; 30(11): e51.
[8] Bozdech Z, Zhu J, Joachimiak MP, Cohen FE, Pulliam B, DeRisi JL. Expression profling of the schizont and trophozoite stages of plasmodium falciparum with a long-oligonucleotide microarray. Genome Biol 2003; 4: R9.
[9] Brown PO, Botstein D. Exploring the new world of the genome with DNA microarrays. Nat Genet 1999; 21: 33-7.
[10] Nielsen H, Wernersson R, Knudsen S. Design of oligonucleotides for microarrays and perspectives for design of multi-transcriptome arrays. Nucleic Acids Res 2003; 31: 3491-6.
[11] Rahmann S. Fast large scale oligonucleotide selection using the longest common factor approach. J Bioinform Comput Biol 2003; 1(2): 343-61.
[12] Reymond N, Charles H, Duret L, Calevro F, Beslon G, Fayard JM. Roso: optimizing oligonucleotide probes for microarrays. Bioin-formatics 2004; 20(2): 271-3.
[13] Rimour S, Hill D, Militon C, Peyret P. GoArrays: highly dynamic and efficient microarray probe design. Bioinformatics 2005; 21(7): 1094-103.
[14] Rouillard JM, Herbert CJ, Zuker M. Oligoarray: genome-scale oligonucleotide design for microarrays. Bioinformatics 2002; 18(3): 486-7.
[15] Gasieniec L, Li CY, Sant P, Wong PWH. Efficient Probe Selection in Microarray Design. IEEE Symposium on Computational
Intelli-gence and Bioinformatics and Computational Biology,
Toronto, Ontario, Canada 2006.
[16] Gordon PM, Sensen CW. Osprey: a comprehensive tool employing novel methods for the design of oligonucleotides for DNA sequencing and microarrays. Nucleic Acids Res 2004; 32: 17. [17] Stenberg J, Nilsson M, Landegren U. ProbeMaker: an extensible
framework for design of sets of oligonucleotide probes. BMC Bioinformatics 2005; 6: 229.
[18] Manber U, Myers G. Suffix arrays: a new method for on-line string searches. Proceedings of the first annual ACM-SIAM symposium on Discrete algorithms, Society for Industrial and Applied Mathe-matics, Philadelphia, PA, USA 1990.
[19] Hamming RW. Error detecting and error correcting codes. Bell Syst Tech J 1950; 29(2): 147-60.
[20] Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol 1990; 215(3): 403-10. [21] Collins PJ, Tsalenko AM, Yakhini ZH, Webb PG, Shannon KW,
8 Recent Patents on DNA & Gene Sequences 2010, Vol. 4, No. 1 Dan Tulpan
probe sequences for use in nucleic acid arrays. US20040101846, 2004.
[22] Friend SH, Stoughton R, Linsley PS, Burchard J. Iterative probe design and detailed expression profiling with flexible in-situ syn-thesis arrays. US7013221B1, 2006.
[23] Zhang J, Madden TL. PowerBLAST: a new network BLAST ap-plication for interactive or automated sequence analysis and anno-tation. Genome Res 1997; 7: 649-56.
[24] Shchegrova SV, Webb PG, Roberts DN, Giles BS. Oligonucleotide microarray probe design via statistical regression analysis of ex-perimental data. US20070233398A1, 2007.
[25] Gao Y, Wolf LK, Georgiadis RM. Secondary structure effects on DNA hybridization kinetics: a solution versus surface comparison. Nucleic Acids Res 2006; 34: 3370-7.
[26] Volokhov D, Rasooly A, Chumakov K, Chizhikov V. Identification of Listeria species by microarray-based assay. J Clin Microbiol 2002; 40: 4720-8.
[27] Su HP, Tung SK, Tseng LR, Tsai WC, Chung TC, Chang T. Chain identification of legionella species by use of an oligonucleotide ar-ray. J Clin Microbiol 2009; 47: 1386-92.
[28] Tissari P, Zumla A, Tarkka E, et al. Accurate and rapid identifica-tion of bacterial species from positive blood cultures with a DNA-based microarray platform: an observational study. Lancet 2010; 375(9710): 224-30.
[29] Sauerbrei A, Wutzler P. Laboratory diagnosis of central nervous system infections caused by herpesviruses. J Clin Virol 2002; 25: S45-S51.
[30] Wald A, Huang ML, Carrell D, Selke S, Corey L. Polymerase chain reaction for detection of herpes simplex virus (HSV) DNA on mucosal surfaces: Comparison with HSV isolation in cell culture. J Infect Dis 2003; 188: 1345-51.
[31] Ramaswamy M, McDonald C, Smith M, et al. Diagnosis of genital herpes by real time PCR in routine clinical practice. Sex Transm Infect 2004; 80: 406-10.
[32] Hudnall SD, Chen T, Tyring SK. Species identification of all eight human herpesviruses with a single nested PCR assay. J Virol Methods 2004; 116: 19-26.
[33] Dong G, Shang S, Liang L, Yu X. Determination of the six major human herpesviruses in cerebrospinal fluid and blood specimens of children. Acta Paediatr 2005; 94: 38-43.
[34] Tafreshi NK, Sadeghizadeh M, Amini-Bavil-Olyaee S, Ahadi AM, Jahanzad I, Roostaee MH. Development of a multiplex nested con-sensus PCR for detection and identification of major human herpesviruses in CNS infections. J Clin Virol 2005; 32: 318-24. [35] Shang S, Chen G, Wu Y, Du L, Zhao Z. Rapid diagnosis of
bacte-rial sepsis with PCR amplification and microarray hybridization in 16S rRNA gene. Pediatr Res 2005; 58: 143-8.
[36] Larkin MA, Blackshields G, Brown NP, et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007; 23: 2947-8.
[37] Kwon TJ. Method of designing probe set, microarray having sub-strate on which probe designed by the method is immobilized, and computer readable medium on which program for executing the method is recorded. US7657381B2, 2010.
[38] Morishita A, Miyagawa I, Kogoh K, Kawakami A, Ogawa N. Primer set and probe for detection of human Papillomavirus. EP2192182A1, 2008.
[39] Altshuler D, Brooks L, Chakravarti A, Collins F, Daly M, Donnelly P. A haplotype map of the human genome. Nature 2005; 437: 1299-320.
[40] Rocha D, Gut I, Jeffreys AJ, Kwok PY, Brookes AJ, Chanock SJ. Seventh international meeting on single nucleotide polymorphism and complex genome analysis: “ever bigger scans and an increas-ingly variable genome.” Hum Genet 2006; 119: 451-6.
[41] Sachidanandam R, Weissman D, Schmidt SC, et al. A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature 2001; 409: 928-33.
[42] Wang DG, Fan JB, Siao CJ, et al. Large-scale identification, map-ping, and genotyping of single-nucleotide polymorphisms in the human genome. Science 1998; 280: 1077-82.
[43] Schork NJ, Fallin D, Lanchbury JS. Single nucleotide polymor-phisms and the future of genetic epidemiology. Clin Genet 2000; 58: 250-64.
[44] Emahazion T, Feuk L, Jobs M, et al. SNP association studies in Alzheimer’s disease highlight problems for complex disease analy-sis. Trends Genet 2001; 17: 407-13.
[45] Tost J, Gut IG. Genotyping single nucleotide polymorphisms by MALDI mass spectrometry in clinical applications. Clin Biochem 2005; 38: 335-50.
[46] Kwok PY, Gu Z. Single nucleotide polymorphism libraries: why and how are we building them? Mol Med Today 1999; 5: 538-43. [47] Guzey C, Spigset O. Genotyping as a tool to predict adverse drug
reactions. Curr Top Med Chem 2004; 4: 1411-21.
[48] Pirmohamed M, Park BK. Genetic susceptibility to adverse drug reactions. Trends Pharmacol Sci 2001; 22: 298-305.
[49] Marsh S, Kwok P, McLeod HL. SNP databases and pharmacoge-netics: great start, but a long way to go. Hum Mutat 2002; 20: 174-9.
[50] McCarthy JJ, Hilfiker R. The use of single-nucleotide polymor-phism maps in pharmacogenomics. Nat Biotechnol 2000; 18: 505-8.
[51] Oh J, Huh N. Polynucleotide probe having enhanced binding speci-ficity, microarray having the probe immobilized thereon, and method of designing the probe. KR10-2004-0030447, 2004. [52] Huang J, Wei W, Chen J, et al. CARAT: a novel method for allelic
detection of DNA copy number changes using high density oli-gonucleotide arrays. BMC Bioinformatics 2006; 7: 83.
[53] Fu W, Qian M, Wan L, Sun K. Microarray-based gene copy num-ber analyses. WO2009051803, 2009.
[54] Zhang L, Miles MF, Aldape KD. A model of molecular interac-tions on short oligonucleotide microarrays. Nat Biotechnol 2003; 7: 818-21.
[55] Zhang L, Wu C, Carta R, Zhao H. Free energy of DNA duplex formation on short oligonucleotide microarrays. Nucleic Acids Res 2007; 35(3): e18.
[56] Thorisson GA, Smith AV, Krishnan L, Stein LD. The International HapMap Project Web site. Genome Res 2005; 15: 1591-3. [57] Lipson D, Web P, Yakhini Z. Designing Specific Oligonucleotide
Probes for the Entire S. cerevisiae Transcriptome, WABI, Rome, Italy 2002.
[58] Sampas NM, Curry B, Tsang P, Lipson D, Yakhini ZH. Probe design methods and microarrays for comparative genomic hybridi-zation and location analysis. US 20060110744A1, 2006.
[59] Breslauer KJ, Frank R, Blöcker H, Marky LA. Predicting DNA duplex stability from the base sequence. Proc Natl Acad Sci USA 1986; 83(11): 3746-50.
[60] Bhanot G, Louzoun Y, Zhu J, Delisi C. The importance of thermo-dynamic equilibrium for high throughput gene expression arrays. Biophys J 2003; 84: 124-35.
[61] Leparc GG, Tüchler T, Striedner G, et al. Model-based probe set optimization for high-performance microarrays. Nucleic Acids Res 2009; 37(3): e18.
[62] Mueckstein U, Leparc G, Posekany A, Hofacker I, Kreil DP. Hy-bridization thermodynamics of NimbleGen Microarrays. BMC Bio-informatics 2010; 11: 35.
[63] Kreil DP, Russell RR, Russell S. Microarray oligonucleotide probes. Methods Enzymol 2006; 410: 73-98.
[64] Lemoine S, Combes F, Le Crom S. An evaluation of custom mi-croarray applications: the oligonucleotide design challenge. Nucleic Acids Res 2009; 37(6): 1726-39.