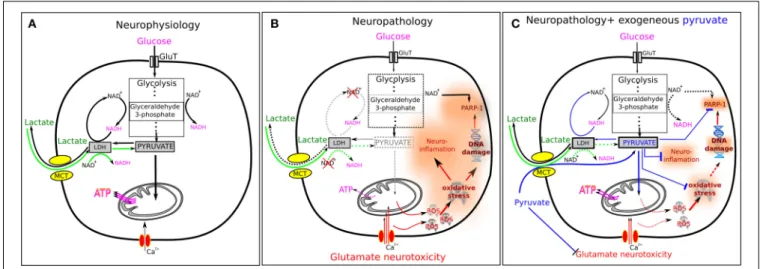
A unique array of neuroprotective effects of pyruvate in neuropathology
6
0
0
Texte intégral
Figure
Documents relatifs