HAL Id: tel-01693271
https://tel.archives-ouvertes.fr/tel-01693271
Submitted on 26 Jan 2018HAL is a multi-disciplinary open access archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
implications dans le couplage neurovasculaire et dans
l’AVC ischémique
Antoine Anfray
To cite this version:
Antoine Anfray. Aspect vasculaire de l’interaction tPA / R-NMDA : implications dans le couplage neurovasculaire et dans l’AVC ischémique. Médecine humaine et pathologie. Normandie Université, 2017. Français. �NNT : 2017NORMC416�. �tel-01693271�
THESE
Pour obtenir le diplôme de doctorat
Spécialité Aspects cellulaires et moléculaires de la biologie Préparée au sein de l’Université de Caen Normandie
Aspects vasculaires de l’interaction tPA / R-NMDA : implications dans le
couplage neurovasculaire et dans l’AVC ischémique.
Présentée et soutenue par
Antoine ANFRAY
Thèse dirigée par Cyrille ORSET, laboratoire INSERM U1237, PhIND Thèse soutenue publiquement le 12 / 12 / 2017
devant le jury composé de
Dr Serge CHARPAK Directeur de Recherche, Université
Paris Descartes Rapporteur Pr Emmanuelle CANET-SOULAS Professeur des universités, Université
Claude Bernard Lyon 1 Rapporteur Dr Anne JOUTEL Directeur de Recherche, Université
Paris Diderot Examinateur Pr Omar TOUZANI Professeur des université, Université de
Caen Normandie Examinateur Pr Denis VIVIEN Professeur des université, Université de
Caen Normandie Examinateur Dr Cyrille ORSET Maître de conférences HDR, Université
R
EMERCIEMENTSJe tiens tout d’abord à remercier Denis Vivien pour m’avoir accueilli dans son laboratoire dès mon M1, pour nous permettre d’avoir les moyens de réaliser nos projets, et pour avoir suivi avec attention l’évolution et la réalisation de ces travaux de thèse.
Je remercie également Cyrille Orset pour l’encadrement de cette thèse, pour m’avoir transmis les savoirs et techniques indispensables à la physio, et pour m’avoir rapidement fait confiance pour mener mes travaux de thèse.
Je souhaite sincèrement remercier les rapporteurs de cette thèse, le Pr. Emmanuelle Canet-Soulas et le Dr. Serge Charpak pour avoir accepté et pris le temps d’évaluer mon travail. Je voudrais également remercier le Dr. Anne Joutel et le Pr. Omar Touzani d’avoir accepté de faire partie de mon jury de thèse.
Je voudrais également remercier le Dr. Tae-Hee Cho, qui fut mon premier encadrant à mon arrivée en stage de Master 1, et qui m’a très rapidement permis de développer mes connaissances en physiologie et chirurgie animale.
Merci à mes camarades de promo Sophie et Antoine F pour tout le chemin parcouru depuis la licence, au labo comme en dehors. Et merci aux deux autres permanents du bureau, Antoine D et Vincent pour la bonne humeur générale et communicative.
Merci à toutes les personnes avec qui j’ai apprécié travailler au cours de ces années de thèse : Antho, Arnaud, Anu, Camille B, Camille L, Marie, Matthieu, Max, Mickael, Sara D, Stefan et Vincent H. Merci à Carine et Sara M pour tous les conseils et l’aide précieuse apportée au cours de ces cinq années.
Merci à tous les membres de l’équipe PhIND (et anciennement SP2U) avec qui j’ai toujours pu échanger et trouver de l’aide en cas de besoin, et merci également aux membres de l’équipe Cervoxy. Merci à Yannick, Lolo et Karim pour les débriefs de Malherbe et de sport, Rhéda pour les pauses foot du weekend, et Ben pour les sorties VTT.
Je voudrais également remercier le personnel du CURB, et particulièrement Claudine, ainsi qu’Agnès et Jean-Michel, et plus généralement toutes les personnes de Cyceron ayant permis de près ou de loin la réalisation de ma thèse.
Je remercie tous mes amis, avec qui j’ai pu échanger et tenté d’expliquer ces travaux. Merci à Jonathan de m’avoir accueilli le soir après le labo avec le repas servi.
Un très grand merci à Mathilde pour ton aide immense et tout ton soutien.
Enfin je voudrais remercier ma famille. Clément, soutien sans faille et accompagnateur de café à 9h, cinq jours par semaine, été comme hiver, qui m’a aidé tout au long de cette thèse. Thibault, qui a toujours été là quand il a fallu décompresser, et enfin mes parents pour tout, pour leur soutien, et pour tout ce qu’ils m’ont transmis et qui m’a permis d’en arriver là.
A
VANT-
PROPOSJ’ai réalisé ma thèse dans l’unité INSERM U1237 « Physiopathology and Imaging of
Neurological Disorders » (PhIND) dirigée par le Professeur Denis Vivien. Historiquement, le
thème de recherche de cette unité concerne les sérines protéases, et plus particulièrement l’activateur tissulaire du plasminogène ou tPA. Cette protéase initialement découverte dans le sang pour son rôle dans la cascade fibrinolytique est aujourd’hui le seul traitement pharmacologique autorisé et utilisé en clinique pour lyser les caillots lors d’une ischémie cérébrale. Cependant, les effets du tPA ne se limitent pas à la fibrinolyse. Ainsi, le tPA est
également présent dans le parenchyme cérébral, où il peut avoir des rôles variés, comme dans
la mémoire ou le couplage neurovasculaire. Au-delà de ses effets physiologiques, le tPA a également des effets néfastes en conditions pathologiques. Ainsi, lors de l’ischémie cérébrale, le tPA amplifie l’excitotoxicité, responsable de la mort de nombreux neurones. Certains des mécanismes d’action du tPA, qu’ils soient pathologiques ou physiologiques, ont au moins un point commun : ils impliquent l’interaction du tPA avec les récepteurs N-Methyl-D-Aspartate.
Le laboratoire cherche donc à améliorer la compréhension des mécanismes d’actions du tPA au sein du système nerveux central et de l’unité neurovasculaire, en conditions physiologique et pathologiques, afin d’améliorer son utilisation en sauvegardant ses effets bénéfiques tout en supprimant ses effets délétères.
Mes travaux de thèse concernent l’étude de l’interaction du tPA avec les récepteurs NMDA et ses conséquences dans deux contextes différents : l’un physiologique, lors du
couplage neurovasculaire (étude 1), et l’autre pathologique, lors de la thrombolyse consécutive
à une ischémie cérébrale (étude 2).
Anisi, pour présenter ces travaux de thèse, je commencerai l’introduction par la présentation de l’acteur majeur de cette thèse : le tPA. Je continuerai par la présentation de son principal interlocuteur dans ces travaux, le récepteur NMDA. Enfin je présenterai les deux
contextes dans lequel leur interaction a été étudiée : le couplage neurovasculaire puis l’ischémie
L’activateur tissulaire du plasminogène (tPA) est une sérine protéase initialement découverte dans le sang pour sa capacité à convertir le plasminogène en plasmine, une enzyme capable de dégrader les chaînes de fibrine des caillots sanguins. Pour cette fonction pro-fibrinolytique, le tPA est le seul traitement pharmacologique aujourd’hui utilisé dans la phase aiguë de l’accident vasculaire cérébral (AVC) de type ischémique, même s’il présente plusieurs limites. Outre son rôle dans la fibrinolyse, le tPA est aussi capable de moduler différents phénomènes physiologiques et pathologiques au sein du système nerveux central et de l’unité neurovasculaire, tels que la mémoire, l’excitotoxicité ou encore le couplage neurovasculaire comme décrit plus récemment. Plusieurs fonctions du tPA impliquent son interaction avec les récepteurs N-Methyl-D-Aspartate (NMDA), qui permet de potentialiser leur signalisation. Sur le plan structurel, deux formes du tPA ont été identifiées : une forme simple chaîne (sc-tPA) et une forme double chaîne (tc-tPA). Ces deux formes, dont les proportions peuvent varier dans la solution administrée aux patients pour la thrombolyse post-AVC ischémique, partagent certaines fonctions communes mais peuvent aussi avoir des actions différentes. Le premier objectif de nos travaux visait à mieux comprendre l’implication du tPA dans le couplage neurovasculaire, un phénomène essentiel au fonctionnement cérébral permettant aux régions en activité de bénéficier d’un apport accru en sang afin de subvenir à la demande énergétique des neurones. Dans une seconde partie, nous nous sommes intéressés aux effets des formes sc-tPA et tc-tPA utilisées lors de la thrombolyse dans un modèle murin d’AVC ischémique thromboembolique.
Premièrement, nos résultats mettent en évidence la capacité du tPA vasculaire à augmenter l’hyperhémie fonctionnelle dans le cadre du couplage neurovasculaire. En effet, nous montrons chez la souris que le tPA vasculaire peut interagir avec les récepteurs NMDA présents à la surface des cellules endothéliales des artères et artérioles, et augmenter leur dilatation lors d’une activité neuronale. D’autre part, dans le cadre de l’ischémie cérébrale, nos résultats indiquent que lorsqu’ils sont utilisés pour la thrombolyse précoce, le sc-tPA et le tc-tPA ont des effets différents et parfois opposés. Le sc-tPA permet de réduire les volumes de lésion et d’améliorer la récupération fonctionnelle, alors que le tc-tPA est moins efficace pour réduire la lésion et ne diminue pas les déficits fonctionnels. De fait, nos données montrent que le tc-tPA aggrave l’altération de l’intégrité de la barrière hématoencéphalique par rapport au sc-tPA. Dans l’ensemble, ces données permettent d’améliorer les connaissances sur les mécanismes d’actions du tPA dans des phénomènes physiologiques et pathologiques importants. Nos travaux soulignent également la nécessité de prendre en compte les différences entre les formes de tPA dans l’amélioration du traitement actuel des AVC et dans l’élaboration de futures stratégies thérapeutiques impliquant cette molécule.
Mots clés : tPA, récepteurs NMDA, couplage neurovasculaire, AVC, fibrinolyse
Summary
The tissue-type plasminogen activator (tPA) is a serine protease initially discovered in the blood for its ability to convert plasminogen into plasmin, an enzyme capable of degrading fibrin chains of blood clots. tPA is the only pharmacological treatment currently used for the acute phase of ischemic stroke, although it has several limitations. Besides its role in fibrinolysis, tPA also modulates various physiological and pathological phenomena within the central nervous system and neurovascular unit, such as memory, excitotoxicity and neurovascular coupling, which has been described recently. Several functions of tPA involve its interaction with N-Methyl-D-Aspartate (NMDA) receptors, which leads to an increase in NMDA signaling. Structurally, two forms of tPA have been identified: a single chain form (sc-tPA) and a double chain form (tc-tPA). These two forms, whose proportions vary in the solution administered for thrombolysis during ischemic stroke, share some common functions but may also differ in their therapeutic action. The first objective of our work was to better understand the implication of tPA in neurovascular coupling, which is an essential phenomenon for cerebral functioning that allows active brain regions to benefit from increased blood supply in order to meet local energy demands. In the second part of our work, we investigated the effects of sc-tPA and tc-tPA in a murine model of ischemic thromboembolic stroke.
Our results establish a role for vascular tPA in increasing functional hyperemia in neurovascular coupling. We show that vascular tPA interacts with NMDA receptors present at the surface of endothelial cells of arteries and arterioles to increase their dilation during neuronal activity. In the context of cerebral ischemia, our results indicate that when administered during early thrombolysis, sc-tPA and tc-tPA have different and sometimes opposite effects. tc-tPA is less effective than sc-tPA in reducing lesion volume and protecting against functional impairment. In fact, our data show that tc-tPA worsens the integrity of the blood-brain barrier compared to sc-tPA. Overall, these data improve our knowledge of the mechanisms of action of tPA in important physiological and pathological phenomena. Our work underlines the need to take into account differences between sc-tPA and tc-tPA when trying to improve the current treatment for stroke and in the development of future therapeutic strategies involving this molecule.
Key words: tPA, NMDA receptors, neurovascular coupling, fibrinolysis
Resumé
Resumé
S
OMMAIREAvant-propos ... 1
Sommaire ... 5
Index des figures et tableaux ... 9
Liste des abréviations ... 12
Introduction ... 15
1. L’activateur tissulaire du plasminogène ... 17
1.1. Historique ... 17
1.2. Structure du tPA ... 19
1.3. Expression du tPA ... 21
1.3.1. Expression du tPA dans la circulation ... 21
1.3.1.1. Synthèse et libération par les cellules endothéliales ... 21
1.3.1.2. Clairance du tPA circulant ... 23
1.3.2. Expression périphérique du tPA ... 23
1.3.3. Expression du tPA dans le système nerveux central ... 23
1.4. Inhibition du tPA : les serpines ... 25
1.4.1. PAI-1 ... 26
1.4.2. Neuroserpine ... 27
1.5. Fonctions du tPA ... 27
1.5.1. Le tPA dans la circulation ... 27
1.5.1.1. Fibrinolyse ... 27
1.5.1.2. tPA et barrière hémato-encéphalique ... 29
1.5.2. Le tPA dans le système nerveux central ... 31
1.5.2.1. Le tPA comme neuromodulateur ... 31
1.5.2.2. Développement et migration cellulaire ... 31
1.5.2.3. Plasticité synaptique ... 32
1.5.2.4. tPA et comportement... 33
1.5.2.5. tPA et apoptose ... 34
1.5.2.6. tPA et substance blanche ... 34
1.5.2.7. Potentialisation de l’excitotoxicité ... 35
1.5.2.8. tPA et inflammation ... 37
2. Les récepteurs NMDA ... 39
2.1. Historique - le système glutamatergique ... 39
2.2. Structure des récepteurs NMDA ... 41
2.2.1. Les différentes sous-unités ... 41
2.2.2. Composition en sous-unités ... 43
2.3. Caractéristiques des récepteurs NMDA ... 44
2.3.1. Propriétés pharmacologiques ... 44
2.3.1.2. Réponse aux molécules exogènes ... 45
2.3.2. Localisation cellulaire des récepteurs NMDA ... 45
2.4. Rôles des récepteurs NMDA ... 47
2.4.1. Maturation synaptique ... 47
2.4.2. Plasticité synaptique : LTP et LTD ... 47
2.4.3. Apoptose ... 48
2.4.4. Excitotoxicité ... 48
2.4.5. Récepteurs NMDA et tPA ... 49
2.4.6. Les récepteurs NMDA des cellules endothéliales ... 51
3. Le couplage neurovasculaire ... 53
3.1. Historique ... 53
3.2. La perfusion cérébrale est primordiale pour le fonctionnement normal du SNC ... 54
3.2.1. Le SNC exerce un contrôle actif sur le DSC ... 54
3.2.2. L’unité neurovasculaire ... 55
3.3. L’activité neuronale augmente le DSC localement ... 57
3.3.1. Voies de signalisation ... 57
3.3.1.1. Voie de signalisation neuronale ... 58
3.3.1.2. Voies de signalisation astrocytaires ... 59
3.3.1.3. Cellules endothéliales et tonus vasculaire ... 61
3.3.2. Agents vasodilatateurs : NO, adénosine, lactate et O2, ... 63
3.4. Conséquences vasculaires du couplage neurovasculaire ... 64
3.4.1. Capillaire vs artériole ... 64
3.4.2. Relaxation des cellules musculaires lisses ... 65
3.4.3. Relaxation des péricytes ... 66
3.5. Facteurs influençant le couplage neurovasculaire ... 67
3.5.1. tPA et tonus vasculaire ... 67
3.5.2. Autres facteurs influençant le couplage neurovasculaire ... 69
3.6. Imagerie BOLD ... 70
3.7. Couplage neurovasculaire et pathologie ... 71
3.7.1. Couplage neurovasculaire et dépression corticale ... 71
3.7.2. Couplage neurovasculaire et maladie d’Alzheimer ... 71
3.7.3. Couplage neurovasculaire et hypertension ... 72
3.7.4. Couplage neurovasculaire et ischémie cérébrale ... 72
4. L’accident vasculaire cérébral ... 75
4.1. Généralités sur l’AVC ... 76
4.1.1. Epidémiologie ... 76
4.1.2. Différents type d’AVC ischémiques ... 76
4.1.3. Différents types d’AVC hémorragiques ... 76
4.1.4. Facteurs de risque de l’AVC ... 76
4.1.4.1. Facteurs de risques non modifiables ... 77
4.1.4.2. Facteurs de risque modifiables ... 77
4.2.1. Formation du caillot ... 77
4.2.1.1. Voie dépendante du collagène... 78
4.2.1.2. Voie dépendante du facteur tissulaire ... 78
4.2.2. Expansion du thrombus et fibrinolyse ... 79
4.3. Conséquences de l’occlusion artérielle ... 79
4.3.1. Formation et expansion de la lésion ... 79
4.3.2. Les mécanismes cellulaires ... 81
4.3.3. L’apoptose ... 82
4.3.4. L’inflammation ... 83
4.4. LE tPA comme traitement de l’AVC ... 84
4.4.1. La thrombolyse pharmacologique ... 84
4.4.2. tPA simple et double chaîne ... 85
4.4.3. L’étude OPHELIE ... 86
Objectifs ... 89
Objectifs ... 91
Résultats ... 93
1. Les récepteurs NMDA endothéliaux participent au couplage neurovasculaire : un mécanisme médié par le glutamate et le tPA circulant. ... 95
1.1. Présentation de l’étude ... 96
1.2. Matériels et méthodes ... 98
1.3. Résultats ... 105
2. La thrombolyse par le tPA simple chaîne ou le tPA double chaîne a des conséquences différentes sur les déficits après une ischémie cérébrale chez la souris. ... 125
2.1. Présentation de l’étude ... 126
2.2. Matériels et Méthodes ... 127
2.3. Résultats ... 132
Discussion ... 139
1. Le tPA vasculaire est impliqué dans le couplage neurovasculaire ... 141
1.1. Quelle est l’origine du tPA impliqué dans le couplage neurovasculaire ? ... 141
1.1.1. Les cellules endothéliales peuvent libérer du tPA en réponse à un stimulus extérieur 141 1.1.2. Libération du tPA en réponse à l’acétylcholine ... 142
1.1.3. Libération du tPA en réponse à l’élévation intracellulaire de Ca2+... 143
1.1.4. Modification du shear stress ... 144
1.1.5. Le tPA : un rôle de maintien de la relaxation vasculaire ? ... 144
1.1.6. Explorer le mécanisme de libération du tPA en réponse à l’activité neuronale ... 145
1.1.7. Sc-tPA et tc-tPA : différents effets sur le couplage neurovasculaire ? ... 146
1.2. Le tPA libéré dans la circulation augmente la dilatation des vaisseaux ... 146
1.2.1. Modèle expérimental ... 146
1.2.2. L’absence de tPA induit un déficit de réponse vasculaire : validation du modèle d’étude ... 147
1.2.4. Le tPA vasculaire agit sur les récepteurs NMDA présents sur les cellules endothéliales.
... 148
1.3. Le tPA et la vasodilatation dans l’endothélium ... 150
1.3.1. Le tPA active la vasodilatation ... 150
1.3.2. Activation des récepteurs NMDA par le tPA : quelles conséquences ? ... 151
1.3.3. Explorer l’effet de l’interaction du tPA avec les récepteurs NMDA endothéliaux .... 152
1.4. Quel rôle pour le tPA vasculaire dans le couplage neurovasculaire ? ... 154
1.5. Implication de la voie du tPA vasculaire dans le couplage neurovasculaire ... 154
1.5.1. tPA et vieillissement ... 154
1.5.2. tPA et maladie d’Alzheimer ... 155
1.5.3. tPA et ischémie cérébrale ... 155
2. Effets différentiels du sc-tPA et du tc-tPA lors de la thrombolyse. ... 157
2.1. Le tPA et la thrombolyse dans l’ischémie cérébrale ... 157
2.2. Parallèles avec l’étude OPHELIE ... 157
2.2.1. L’étude OPHELIE ... 157
2.2.2. Le choix du modèle d’étude ... 158
2.2.3. Les formes de tPA utilisées pour la thrombolyse ... 159
2.3. Les différentes formes de tPA sont a l’origine d’effets distincts dans l’ischémie cérébrale .. ... 159
2.3.1. Effets des différentes formes du tPA sur la reperfusion ... 159
2.3.2. Le sc-tPA est efficace pour lyser le caillot et réduire les déficits ... 159
2.3.3. Le tc-tPA n’améliore pas la récupération fonctionnelle ... 161
2.3.4. Evaluation des déficits moteurs après la thrombolyse ... 163
2.4. Stratégies développées pour améliorer le traitement des AVC ischémiques ... 164
2.4.1. L’échec de la génération de nouvelles stratégies fibrinolytiques ... 164
2.4.2. Comment améliorer la prise en charge aigue des AVC ischémiques ?... 164
Conclusion générale ... 167
Annexe 1 ... 1679
Autres travaux ... 175
1.1. Distant space processing is controlled by tPA-dependent NMDA Receptor signaling in the entorhinal cortex. ... 178
1.2. Hyperfibrinolysis increases blood–brain barrier permeability by a plasmin- and bradykinin-dependent mechanism. ... 192
1.3. Recombinant ADAMTS-4 enhances tPA-mediated fibrinolysis in ischemic stroke. .... ... 232
I
NDEX DES FIGURES ET TABLEAUXFigure 1 ӏ Buste d’Hippocrate ... 17
Figure 2 I Distribution de grains d’argent en autoradiographie correspondant à l’hybridation de sondes d’ADNc du tPA sur le chromosome 8 ... 19
Figure 3 I Structure et fonctions du tPA, sous forme simple et double chaîne. ... 21
Figure 4 I Effet d’un « shear stress » laminaire de 25 dynes/cm2 sur la sécrétion de tPA par des cellules endothéliales ... 22
Figure 5 I Clairance hépatique du tPA. ... 23
Figure 6 I Localisation du tPA au sein du système nerveux central chez la souris ... 24
Figure 7 I Expression du tPA au sein de différentes cellules du SNC chez la souris adulte ... 25
Figure 8 I Structure et mode d’action des serpines. ... 26
Figure 9 I Mécanisme de la fibrinolyse endogène ... 28
Figure 10 I Barrière hématoencéphalique... 29
Figure 11 I Effet du tPA sur la barrière hématoencéphalique ... 30
Figure 12 I Effet de l’absence de tPA sur la migration neuronale ... 32
Figure 13 I Elévation de la quantité d’ARN messager du tPA suite à une activation neuronale chez le rat. ... 33
Figure 14 I Les souris tPA KO sont résistantes à la morte neuronale excitotoxique ... 35
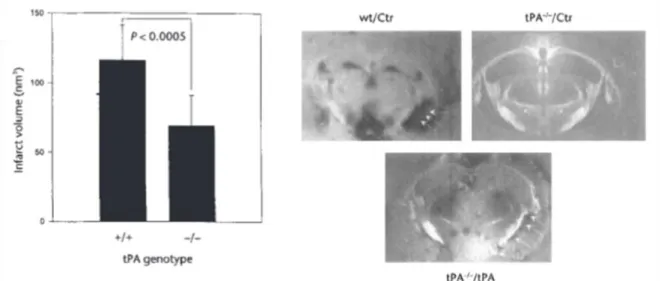
Figure 15 I Des souris déficientes en tPA présentent un volume de lésion réduit après une ischémie cérébrale ... 37
Figure 16 I Classification des récepteurs au glutamate ... 41
Figure 17 I Expression (ARNm) des sous unités GluN1 et GluN2 des récepteurs NMDA dans le cerveau de rat au cours du développement. ... 42
Figure 18 I Structure d’un récepteur NMDA ... 43
Figure 19 I La synapse glutamatergique ... 46
Figure 20 I La sous-unité GluN1 des récepteurs NMDA est exprimée dans les cellules endothéliales cérébrales de souris. ... 47
Figure 21 I L’interaction du tPA avec la sous-unité GluN1 des récepteurs NMDA potentialise la signalisation et l’entrée de Ca2+ ... 50
Figure 22 I L’inhibition par le Glunomab® de l’interaction du tPA avec les récepteurs NMDA stoppe la progression de l’EAE chez la souris ... 51
Figure 23 I Etude du volume du cerveau et des pieds chez Mr. Cane en réponse à un stimulus émotionnel ... 53
Figure 24 I Modélisation de la vascularisation cérébrale à la surface du cortex somatosensoriel des vibrisses. ... 55
Figure 25 I L’unité neurovasculaire. ... 56
Figure 27 I Figure : Dessin de C. Golgi représentant l’organisation des astrocytes autours des vaisseaux
... 59
Figure 28 I L’activité neuronale induit une augmentation du Ca2+ intracellulaire dans les astrocytes in vivo, via une activation des mGluR. ... 60
Figure 29 I L’effet vasodilatateur du glutamate et de la D-sérine sur des artères isolées de souris requiert un endothélium intact et la présence de eNOS ... 62
Figure 30 I Figure : Rôle de l’endothélium dans le couplage neurovasculaire ... 63
Figure 31 I Une stimulation électrique induit une contraction locale dépendante du Ca2+ de péricytes de la rétine de rat. ... 67
Figure 32 I Effet du tPA sur le couplage neurovasculaire ... 69
Figure 33 I Effet de l’anesthésie sur le couplage neurovasculaire ... 70
Figure 34 I Représentation schématique des deux types d’AVC ... 75
Figure 35 I Initiation de la formation d’un thrombus ... 77
Figure 36 I L’agrégation plaquettaire au niveau d’une lésion de l’endothélium ... 79
Figure 37 I Atteinte cérébrale lors d’une ischémie ... 80
Figure 38 I Evolution spatio-temporelle de la lésion lors d’une ischémie cérébrale ... 81
Figure 39 I Cascade hypothétique des événements délétères pour les neurones lors de l’ischémie cérébrale ... 81
Figure 40 I Vue simplifiée des mécanismes physiopathologiques de l’ischémie cérébrale ... 82
Figure 41 I Cellules immunitaires et inflammation lors de l’ischémie cérébrale ... 84
Figure 42 I Effets du sc-tPA et du tc-tPA sur l’excitotoxicité ... 86
Figure 43 I Modèle expérimental et paramètres physiologiques mesurés ... 99
Figure 44 I Séquence de stimulation lors de la mesure du DSC ... 100
Figure 45 I Traitement des images obtenues par laser Doppler speckle ... 100
Figure 46 I Figure : Construction du plasmide pLIVE et transfection hydrodynamique ... 102
Figure 47 I Le signal mesuré en laser Doppler speckle est plus intense lorsque le crâne est affiné chez la souris. ... 106
Figure 48 I L’hyperhémie fonctionnelle est réduite chez les souris tPA [-/-] ... 107
Figure 49 I Le tPA vasculaire augmente l’hyperhémie fonctionnelle chez les souris tPA [-/-]... 109
Figure 50 I L’effet du tPA vasculaire exogène s’ajoute à celui du tPA endogène chez les souris tPA [+/+] ... 111
Figure 51 I L’expression chronique de tPA restreinte à la circulation sanguine favorise l’hyperhémie fonctionnelle chez les souris tPA [-/-] ... 112
Figure 52 I L’inhibition de l’interaction du tPA vasculaire avec les récepteurs NMDA empêche la potentialisation de l’hyperhémie fonctionnelle par le tPA ... 115
Figure 53 I L’inhibition des récepteurs NMDA bloque la potentialisation de l’hyperhémie fonctionnelle par le tPA. ... 116
Figure 54 I Le tPA vasculaire n’a pas d’effet sur l’hyperhémie fonctionnelle en absence de glutamate vasculaire (en cours) ... 118
Figure 55 I Le tPA vasculaire augmente la dilatation des vaisseaux de plus gros diamètre ... 119
Figure 56 I Les récepteurs NMDA sont préférentiellement exprimés par les cellules endothéliales des artères et artérioles (En cours) ... 122
Figure 57 I L’interaction du tPA avec les récepteurs NMDA de cellules endothéliales en culture active la voie RhoA ... 123
Figure 58 I Description du modèle d’ischémie thromboembolique. ... 129
Figure 59 I Caractérisation de la production de sc-tPA et de tc-tPA. ... 133
Figure 60 I Protocole expérimental ... 134
Figure 61 I La thrombolyse au tPA augmente la reperfusion et tend à réduire les volumes de lésion à 24h ... 135
Figure 62 I Le sc-tPA est plus efficace que le tc-tPA pour promouvoir la reperfusion et diminuer le volume de lésion 24 heures après l’ischemie. ... 136
Figure 63 I La thrombolyse par le sc-tPA diminue les déficits fonctionnels après l’ischémie ... 137
Figure 64 I Le tc-tPA est responsable d’une ouverture exacerbée de la BHE ... 138
Figure 65 I Régulation de la réactivité vasculaire par les cellules localisées dans les régions sous-corticales et sous-corticales. ... 142
Figure 66 I Evolution spatio-temporelle de la réponse hémodynamique lors d’une stimulation sensorielle (stimulation électrique de la patte arrière chez le rat) ... 144
Figure 67 I Les récepteurs NMDA modulent la vasodilatation de l’ACM de souris en réponse au glutamate et à la D-sérine ... 149
Figure 68 I Diamètre de la lumière d’ACM de rats isolées, en condition normale ou après ischémie, en réponse à une augmentation de la pression artérielle ... 151
Figure 69 I Voies de signalisation envisagées pour la libération du tPA et son action lors du couplage neurovasculaire ... 153
Figure 70 I Le stress oxydatif réduit la quantité de NO disponible et inhibe la synthèse de prostacycline ... 156
Figure 71 I Principaux modèles d’occlusion de l’ACM chez le rongeur. ... 158
Figure 72 I La thrombolyse par le tPA est bénéfique lorsqu’elle est réalisée 20 minutes après l’ischémie, mais devient délétère lorsqu’elle est réalisée 4 heures après l’ischémie ... 160
Figure 73 I Mécanismes envisagés pour les différents effets du sc-tPA et du tc-tPA lors de la thrombolyse ... 162
Figure 74 I L’hyperfibrinolyse induit l’ouverture de la BHE par un mécanisme dépendant de la bradykinine………. 163
Figure 75 I Le sc*-tPA est un pro-fibrinolytique efficace qui ne potentialise pas l’excitotoxicité ……165
Tableau 1 I Score modifié de Rankin (modified Rankin Score ; mRS) ... 87
L
ISTE DES ABREVIATIONS20-HETE 20-hydroxyeicosatetraenoic acid
AA acide arachidonique
ACM artère cérébrale moyenne
ADAMTS a disintegrin and metalloproteinase with thrombospondin motifs – désintégrine et
métalloprotéinase à motif thrombospondines
AMPA α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid AMPc adénosine monophosphate cyclique
AP5 acide 2-amino-5-phosphonovalérique
ARNm acide ribonucléique messager
ATD domaine amino-terminal
ATP adénosine triphosphate
AVC accident vasculaire cérébral
BHE barrière hémato-encéphalique
BOLD blood-oxygen-level dependent
CAMK protéine kinase Ca2+/calmoduline-dépendante
COX cyclooxygénase
CREB C-AMP Response Element Binding protein CSPGs chondroitine sulphate protéoglycanes
DAG Diacylglycérol
DSC débit sanguin cérébral
EAE encéphalomyélite autoimmune expériementale
EET acide époxyeicosatriénoïque
EGF epidermal growth factor
eNOS endothelial nitric oxyde synthase ERK extracellular signal-related kinase Fas-L fibroblast associated ligand
FADD fas-associated protein with death domain fUS functional ultrasound
GABA γ-aminobutyric acid – acide γ-aminobutyrique GMPc guanosine monophosphate cyclique
GOT glutamate-oxaloacétate transaminase
GP glycoprotéine
HEPES 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid IP3 inositol triphosphate
IRM imagerie par résonance magnétique
IV intraveineuse
JNK c-jun N-terminal kinase
KO knockout
LBS lysine binding site
LRP lipoprotein receptor-related protein – récepteur aux lipoprotéines de faible densité LTP long term potentiation – potentialisation à long terme
LTD long term depression – dépression à long terme MAPK mitogen-activated protein kinase
mGluR récepteur métabotropique au glutamate
MLC myosin light chain – chaine légère de myosine
MMP maxtrix metalloproteinase – métalloprotéinase matricielle NMDA N-methyl-D-aspartate
nNOS neuronal nitric oxyde synthase
NO nitric oxide – oxyde nitrique
NOS neuronal nitric oxide synthase – oxyde nitrique synthase neuronale
NS neuroserpine
OPHELIE
Outcome of Patients Treated by IV Rt-PA for Cerebral Ischaemia According to the Ratio Sc-tPA/Tc-tPA
PAI plasminogen activator inhibitor – inhibiteur de l’activateur du plasminogène PDGF-CC platelet derived growth factor-CC
PDGFRβ Beta-type platelet-derived growth factor receptor
PG prostaglandine
PL phospholipase
PLAT plasminogen activator tissue type
PKG protéine kinase dépendante du guanosine monophosphate cyclique
RCL reactive center loop – boucle du centre réactif RhoA Ras homolog gene family, member A
rtPA recombinant tissue plasminogen activator sc-tPA single-chain tPA – tPA simple chaine SEM standard error of the mean – erreur standard SMCs smooth muscle cells – cellules musculaires lisses SNC système nerveux central
tc-tPA two-chain tPA – tPA double chaine TEP tomographie par émission de positons
tPA tissue-type plasminogen activator – activateur tissulaire du plasminogène uPA urokinase plasminogen activator – activateur du plasminogène de type urokinase VWF von Willebrand factor – facteur de von Willebrand
WT wild-type
1. L’
ACTIVATEUR TISSULAIRE DU PLASMINOGENEL’activateur tissulaire du plasminogène (tissue-type plasminogen activator, tPA) est une sérine protéase présente dans la circulation sanguine, produite par les cellules endothéliales, et principalement connue pour son rôle dans la fibrinolyse. Il est en effet capable d’activer le plasminogène en plasmine, qui est elle-même capable de dégrader la fibrine, un des constituants majeurs des caillots sanguins. En plus de sa présence dans le compartiment vasculaire, le tPA est retrouvé dans le compartiment cérébral. Il est exprimé par plusieurs types cellulaires cérébraux (neurones, oligodendrocytes, microglie…), et joue différents rôles notamment au niveau synaptique et au cours du développement, en conditions physiologiques ou pathologiques.
1.1. H
ISTORIQUEL’histoire du tPA est intimement liée à celle de la fibrinolyse. Elle trouve son commencement dès la période de la Grèce antique avec les travaux d’Hippocrate (Figure 1), qui observait déjà que le sang de personnes décédées ne coagulait pas. Ces observations furent ensuite réitérées par Morgani en 1761 (Morgani, 1761). Ces premières constatations ne trouveront écho qu’au milieu du XIXème siècle avec les travaux de Gabriel Andral portant sur l’étude de la coagulation sanguine en relation avec la présence de fibrine (Andral, 1843). G. Andral reporta le fait que le sang coagulé pouvait redevenir liquide, apportant ainsi les premières données sur un phénomène qui sera nommé plus tard « fibrinolyse » suite aux travaux de Denys, Marbaix et Dastre. En 1893, ils déterminèrent le caractère protéolytique de ce mécanisme (Dastre, 1893; Denys & De Marbaix, 1889). Vingt ans plus tard, Conradi démontra qu’il était possible de dégrader des caillots sanguins par l’ajout d’extrait de différents organes (Conradi, 1902), et de la même manière en 1904, Hedin démontra une activité protéolytique dans la fraction globuline du sérum (Hedin, 1903). Cette fraction sera identifiée plus tard comme étant celle contenant le plasminogène, précurseur de la principale enzyme de dégradation de la fibrine, la plasmine (Christensen & MacLeod, 1945; Feissly, 1942). Le système fibrinolytique commence alors à se dévoiler, mais il restait encore à découvrir comment s’active le plasminogène, inactif en l’état.
Figure 1 ӏ Buste
d’Hippocrate. Né sur l’île de Cos en -460 et mort vers -370 à Larissa, considéré comme le « père de la médecine ».
Des premières observations confirmées et complétées en 1915 par Fleisher et Loeb, et par Astrup et Permin en 1947, montrèrent que le plasminogène pouvait être activé par des extraits de bactéries. Ces extraits furent identifiés par Tillet et Garner comme étant la streptokinase et la staphylokinase. Ces travaux ouvrirent la voie au concept de thrombolyse, c’est-à-dire la dégradation de caillots déjà formés (Astrup & Permin, 1947; Fleisher & Loeb, 1915; Tillett & Garner, 1933). En 1952, Astrup et Stage parvinrent à isoler une molécule capable d’activer le plasminogène à partir de tissus provenant de cœurs de porc, qu’ils nommèrent fibrikinase (Astrup & Stage, 1952).
Ce n’est qu’en 1979 que cet activateur fut purifié et caractérisé, d’abord dans la circulation sanguine, avec les travaux de Binder (Binder et al., 1979), puis dans l’utérus, grâce aux travaux de Rijken (Rijken et al., 1979). Rebaptisé « activateur tissulaire du plasminogène », le tPA fut purifié à partir de surnageant de cultures de cellules de mélanome humain grâce aux travaux conjoints de Collen et Rijken (Rijken & Collen, 1981), permettant ainsi sa caractérisation.
La première utilisation du tPA chez l’humain remonte à 1981, quand W. Weimar traita deux patients victimes de thrombose de la veine rénale avec du tPA injecté par voie intraveineuse (IV). Deux ans plus tard, en 1983, Diane Pennica parvenait à cloner le gène du tPA, ouvrant la voie à la production de tPA recombinant par des cellules eucaryotes (Pennica
et al., 1983). Cette même année fut menée la première étude clinique sur des patients souffrant
d’infarctus du myocarde. L’injection IV de tPA permit une recanalisation complète des artères coronaires (Werf et al., 1986). Cette étude clinique fut suivie, 11 ans plus tard, d’une autre étude évaluant la possibilité d’utilisation du tPA comme agent thrombolytique dans le cadre de l’ischémie cérébrale (NINDS, 1995). Devant le succès de cette étude, le tPA est aujourd’hui le seul agent pharmacologique autorisé dans le traitement aigu des ischémies cérébrales, associé ou non à la thrombectomie.
1.2. S
TRUCTURE DU TPA
Figure 2 I Distribution de grains d’argent en autoradiographie correspondant à l’hybridation de sondes d’ADNc du tPA sur le chromosome 8. La majorité des sondes sont fixées entre les bandes 8p12 et 8q11.2, localisant ainsi le gène du tPA (Yang-Feng et al., 1986).
Le tPA provient du gène PLasminogen Activator Tissue type (PLAT), situé sur le chromosome 8 (Yang-Feng et al., 1986 ; Figure 2). C’est une sérine protéase de 527 acides aminés ayant une masse moléculaire d’environ 69 kDa (Pennica et al., 1983). Sa structure tridimensionnelle, maintenue par 17 ponts disulfures, permet de différencier cinq domaines, répartis sur deux chaînes A (lourde) et B (légère ; Figure 3) :
Chaîne A :
- Finger (ou fibronectin) : permet la liaison du tPA à la fibrine, formant un complexe tertiaire avec le plasminogène (Kagitani et al., 1985). Ce domaine permet également la liaison à plusieurs récepteurs membranaires, et notamment au récepteur aux lipoprotéines de faible densité (Low Density Lipoprotein Receptor-related Protein, LRP), ainsi qu’à l’annexine II (Bu et al., 1992; Hajjar et al., 1994).
- EGF-like : permet au tPA d’activer les récepteurs à l’epidermal growth factor (EGF ; Correa et al., 2011; Hurtado et al., 2007).
- Kringle 1 : domaine caractérisé par la présence d’un site actif ayant une forte affinité pour la lysine (lysine binding site, LBS), composé de deux acides aminés aromatiques (Trp242 et Trp253) formant une poche dans la structure tertiaire du tPA. Le site LBS du domaine Kringle 1 n’étant pas fonctionnel, son rôle est mal connu. Il pourrait être impliqué dans la clairance du tPA par les cellules endothéliales du foie via une glycosylation sur son Asp117 (Kuiper et al., 1988).
- Kringle 2 : ce domaine contient un site LBS actif. Il permet au tPA de se lier et d’activer différents substrats et récepteurs comme le plasminogène, le récepteur au Platelet
Derived Growth Factor-CC (PDGF-CC ; Fredriksson et al., 2004), ainsi que la sous-unité GluN1 des récepteurs au N-Methyl-D-Aspartate (NMDA; Lopez-Atalaya et al., 2008).
Chaîne B :
- Domaine catalytique : la chaîne légère du tPA n’est composée que d’un seul domaine, le site catalytique, responsable de l’activité protéolytique du tPA. Les acides aminés His322, Asp371 et Ser478 forment la triade catalytique, permettant la conversion du plasminogène en plasmine (Pennica et al., 1983).
Comme toutes les sérines protéases, le tPA existe sous deux formes : une forme simple chaîne (single-chain, sc-tPA) et une forme double chaîne (two-chain, tc-tPA) (Rijken & Collen, 1981). Initialement synthétisé sous forme simple chaîne, le tPA se transforme en tc-tPA sous l’action de protéases comme la plasmine ou les kallikréines, qui clivent le tPA au niveau de la liaison entre l’Arg275 et l’Ile276 (Rajapakse et al., 2005; Wallén et al., 1982). Sous forme
double chaîne, les deux chaînes restent reliées par un pont disulfure entre les Cys299 et Cys430, ainsi que par la création d’un pont salin entre l’Arg302 et le Glu445 (Lamba et al., 1996).
Contrairement à la majorité des sérines protéases qui sont inactives sous forme simple chaîne, le sc-tPA présente une activité protéolytique (Hoylaerts et al., 1982). La régulation de son activité est assurée par la présence d’un régulateur allostérique comme la fibrine (Rijken et al., 1982). En présence de fibrine, les deux formes ont la même activité protéolytique. En revanche, en l’absence de cette dernière, le tc-tPA présente une activité cinq fois supérieure (Petersen et
Figure 3 I Structure et fonctions du tPA, sous forme simple et double chaîne. Schéma représentant le tPA sous forme simple (sc-tPA) et double chaîne (tc-tPA) mettant en évidence les cinq domaines (Finger, EGF, Kringle 1 et 2, sérine protéase), les 17 ponts disulfures, ainsi que le site de clivage par les protéases (adapté de Chevilley et al., 2015; Hébert et al., 2016).
1.3. E
XPRESSION DU TPA
1.3.1. Expression du tPA dans la circulation
1.3.1.1. Synthèse et libération par les cellules endothéliales
Comme énoncé précédemment, le tPA a d’abord été localisé dans le compartiment vasculaire, où il est produit et libéré par les cellules endothéliales. Il est ainsi retrouvé dans le sang à une concentration d’environ 70 pM (Rijken & Lijnen, 2009). Dans l’endothélium vasculaire cérébral, le tPA est surtout retrouvé dans les microvaisseaux (< 100 µm de diamètre ; Levin et al., 1997).
Sa localisation précise au sein de la cellule n’est en revanche pas parfaitement établie. Plusieurs études le situent au sein des corps de Weibel-Palade, principalement connus pour contenir le facteur de von Willebrand (von Willebrand factor, VWF ; Sadler, 1998), mais d’autres études plus récentes laissent penser qu’il serait stocké dans un autre type de granules plus petits (Emeis et al., 1997; Huber et al., 2002; Knipe et al., 2010) qui seraient ensuite responsables de son exocytose. La libération du tPA par les cellules endothéliales se fait de
deux façons différentes : il est synthétisé en permanence et libéré de façon continue ( Angles-Cano et al., 1985; Binder et al., 1979; Levin & Loskutoff, 1982), mais les cellules endothéliales peuvent également le stocker et le libérer en réponse à certains stimuli.
- Exocytose continue :
Quand il est sécrété de manière continue, le tPA reste en contact avec la membrane des cellules endothéliales. Ce mécanisme faciliterait la fibrinolyse (détaillée ci-dessous dans la partie « 1.5.1.1 Fibrinolyse ») au niveau de la surface des cellules endothéliales, à l’endroit même où se forment préférentiellement les complexes de fibrine. L’inhibiteur de l’activateur du plasminogène de type 1 (plasminogen activator inhibitor, PAI-1), présent dans la circulation, peut ensuite agir pour faciliter le décrochage du tPA de la membrane et induire son inhibition lorsque la fibrinolyse n’est pas nécessaire (Suzuki et al., 2009).
- Exocytose contôlée :
En complément de cette exocytose constitutive, le tPA peut également être sécrété en réponse à certains stimuli. Bien que le mécanisme exact ne soit pas complètement connu, il semble que l’activation de protéines-G ainsi qu’une augmentation de la concentration du calcium intracellulaire soient importantes dans ce phénomène (Eijnden-Schrauwen et al., 1997; Knop & Gerke, 2002). Ainsi, plusieurs facteurs peuvent déclencher une sécrétion de tPA : certains facteurs de la cascade de coagulation (Emeis et al., 1997), l’hypoxie, une occlusion des vaisseaux, la thrombine, la bradykinine, la desmopressine, certains agonistes adrénergiques, l’adénosine triphosphate (ATP ; Oliver et al., 2005), ou bien encore l’augmentation de la pression sur les parois des vaisseaux (ou shear stress ; Diamond et al., 1990; Kawai et al., 1996).Dans ces études, les auteurs montrent qu’en réponse au shear stress, des cultures de cellules endothéliales libèrent du tPA dans le milieu extérieur (Figure 4).
Figure 4 I Effet d’un « shear stress » laminaire de 25 dynes/cm2 sur la sécrétion de tPA par des
cellules endothéliales. Six cultures primaires confluentes de cellules endothéliales de veine ombilicale humaine (HUVEC) ont été réalisées. Trois cultures ont été maintenues en conditions de culture normales (), alors que trois autres ont été soumises à un « shear stress » laminaire de 25 dynes/cm2 (). Les cultures de cellules ont été congelées à intervalles réguliers, et la quantité de tPA présente a été évaluée par test ELISA (Diamond et al., 1990).
1.3.1.2. Clairance du tPA circulant
La demi-vie du tPA dans la circulation est d’environ six minutes (Verstraete et al., 1985). Sa clairance s’effectue grâce à des récepteurs membranaires situés sur les hépatocytes. Plusieurs récepteurs sont impliqués : les récepteurs au mannose, sur les cellules endothéliales hépatiques (Kuiper et al., 1988), les récepteurs LRP (Yepes et al., 2003), ainsi qu’une voie dépendante des galectines (Nagaoka et al., 2002) au niveau des hépatocytes (Figure 5).
1.3.2. Expression périphérique du tPA
L’expression du tPA a d’abord été mise en évidence dans le sang, mais il a depuis été montré que le tPA est présent dans de nombreux organes tels que les poumons, les muscles, le cœur ou l’utérus (Rouf et al., 1996). Son expression est souvent associée à un remodelage des tissus et de leur vascularisation, comme lors de la mise en place de l’endomètre par exemple (Lockwood & Schatz, 1996).
1.3.3. Expression du tPA dans le système nerveux central
La présence du tPA au sein du système nerveux central (SNC) a été mise en évidence pour la première fois en 1981 par Soreq et Miskin. Ces derniers l’ont détecté dans les cellules endothéliales cérébrales et dans les neurones à différents temps au cours du développement, lui prédisant ainsi d’autres rôles que son implication dans la fibrinolyse (Soreq & Miskin, 1981). En 1993, Sappino et collaborateurs ont pour la première fois caractérisé la présence du tPA au sein des différentes structures cérébrales. Sa présence a été détectée dans la plupart des régions
Figure 5 I Clairance hépatique du tPA. Trois voies d’endocytose ont été décrites, assurant la recapture du tPA vasculaire : une voie dépendante des récepteurs au mannose à la surface des cellules endothéliales hépatiques, une voie dépendante des récepteurs LRP, et une voie dépendante des galectines, ces deux dernières se trouvant au niveau des hépatocytes (adapté de Nagaoka et
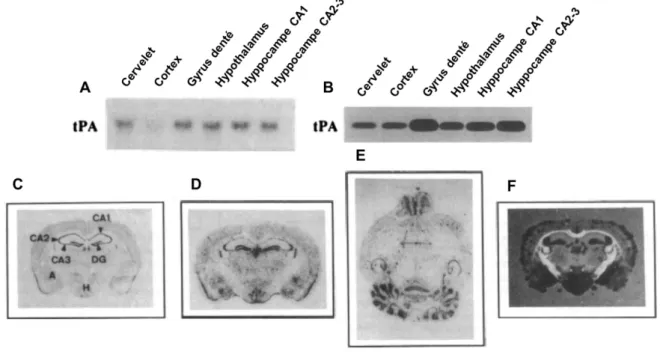
cérébrales, exprimé principalement par les neurones, alors que son activité protéolytique n’a été détectée que dans l’hippocampe et l’hypothalamus (Sappino et al., 1993 ; Figure 6).
Figure 6 I Localisation du tPA au sein du SNC chez la souris. A) Northern-blot mettant en évidence les acides ribonucléiques messagers (ARNm) du tPA selon différentes régions cérébrales. B) Activité protéolytique du tPA révélée par zymographie in-situ au sein de différentes régions cérébrales. C) Coupe coronale de cerveau de souris colorée au cresyl-violet : A = amygdale, H = hypothalamus, DG = Gyrus Denté, CA1, 2 et 3 = corne d’Ammon. D) et E) ARNm du tPA mis en évidence par hybridation in-situ sur une coupe coronale (D) et horizontale (E) de cerveau de souris. F) Activité protéolytique du tPA révélée par zymographie in-situ sur une coupe coronale de cerveau de souris (adapté de Sappino et al., 1993).
A l’échelle cellulaire, le tPA est exprimé par la majorité des types cellulaires du SNC : les neurones (Sallés & Strickland, 2002), les astrocytes (Buisson et al., 1998), les oligodendrocytes (Correa et al., 2011) et la microglie (Siao et al., 2003 ; Figure 7).
- Dans les neurones : le tPA peut être observé dans les dendrites et au niveau des synapses, où il est stocké dans des vésicules présynaptiques (Parmer et al., 1997; Shin
et al., 2004). Il peut alors être libéré au sein de la synapse lors de l’activité neuronale
(Lochner et al., 2006). Cependant, il semble que tous les neurones n’expriment pas le tPA, et que son expression soit réduite aux neurones glutamatergiques excitateurs pyramidaux (Louessard et al., 2016).
- Dans les astrocytes : une fois libéré dans la synapse, le tPA peut être recapté par les astrocytes via les récepteurs LRP et son domaine Kringle 1. Les astrocytes seraient ensuite capables de libérer ce tPA endocytosé par un mécanisme impliquant la
protéinase kinase C suite à l’activation des récepteurs kaïnates membranaires. Cet effet pourrait également être à l’origine de la clairance du tPA au sein du SNC (Cassé et al., 2012).
- Dans les oligodendrocytes : la présence de tPA dans les oligodendrocytes est confirmée par plusieurs études (Correa et al., 2011; Leonetti et al., 2017; Louessard et al., 2016), principalement dans les oligodendrocytes matures où son expression diminue avec l’âge.
- Dans la microglie : la microglie est également capable de synthétiser du tPA. Ce tPA peut ensuite avoir un rôle dans son activation (Rogove et al., 1999; Tsirka et al., 1995).
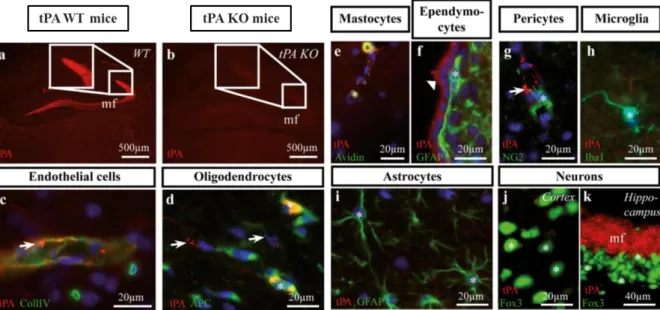
La présence du tPA a également été mise en évidence au sein des mastocytes et des ependymocytes dans le SNC (Louessard et al., 2016).
Figure 7 I Expression du tPA au sein de différentes cellules du SNC chez la souris adulte. Le tPA (ici en rouge) est particulièrement présent au niveau des fibres moussues de l’hippocampe chez des souris wild type (WT ; a ; vs absent chez les souris tPA KO, b). Cette figure démontre également la présence du tPA au niveau de différents types cellulaires du SNC (e-k ; Louessard et al., 2016).
1.4. I
NHIBITION DU TPA :
LES SERPINESDès 1961, Brakman & Astrup postulaient l’existence d’un inhibiteur sanguin des activateurs du plasminogène après avoir observé la présence d’un inhibiteur de la fibrinolyse qui ne ciblait pas directement la plasmine (Brakman & Astrup, 1963). Ces molécules seront identifiées plus tard comme étant les serpines. Les serpines sont des protéines monomériques glycosylées comprenant généralement entre 350 et 400 acides aminés pour un poids moyen de 40 à 60 kDa. La caractéristique principale des serpines est de posséder une conformation secondaire formant un domaine (le core domain) constitué de trois feuillets β et de huit ou neuf
hélices α. Les serpines possèdent également une boucle, appelée boucle du centre réactif (reactive center loop, RCL), qui permet leur interaction avec les protéases. La serpine se fixe sur la protéase par l’intermédiaire de la RCL, puis la protéase clive cette boucle, formant une liaison covalente entre la serpine et la protéase. Le complexe formé par la serpine et la protéase entre alors dans une conformation stable, inhibant cette dernière. Les serpines peuvent aussi exister sous une forme latente, pour laquelle l’interaction avec la protéase n’est pas possible (Silverman et al., 2001 ; Figure 8).
A l’heure actuelle, quatre serpines sont connues pour inhiber le tPA : PAI-1 et PAI-2, la neuroserpine (NS) et la protéase nexine-1 (Osterwalder et al., 1996; Sprengers & Kluft, 1987). Dans le parenchyme cérébral, PAI-1 et la NS sont les principaux inhibiteurs du tPA. La NS y est exprimée de manière physiologique et en grande quantité par les neurones et les astrocytes, alors que PAI-1 est exprimé seulement dans certaines conditions de stress par les astrocytes réactifs (Docagne et al., 1999; Hino et al., 2001). Dans la circulation sanguine, PAI-1 est le principal inhibiteur du tPA, alors que la NS est très peu exprimée (Miranda & Lomas, 2006).
1.4.1. PAI-1
PAI-1 est une protéine de 50 kDa composée de 379 acides aminés. Au niveau périphérique, il est synthétisé par de nombreux tissus comme le foie, le tissu adipeux, les poumons, les reins ou le cœur. PAI-1 est principalement retrouvé dans le plasma, où il est synthétisé par les cellules endothéliales, les cellules musculaires lisses (smooth muscle cells,
Figure 8 I Structure et mode d’action des serpines. A) Structure native. B) Structure latente : le repliement de la boucle du RCL au sein du feuillet βA empêche l’interaction avec la protéase. C) Forme clivée par la protéase. D) Interaction entre la serpine et la protéase au niveau de la RCL. La boucle clivée va s’insérer dans le feuillet βA. La protéase est entraînée au pôle opposé. E) La protéase est au pôle opposé. Une liaison covalente se forme entre la serpine et la protéase, ce qui inhibe cette dernière (Silverman et al., 2001).
A-C sheet : feuillet β A à C ; hA-I : hélice α A à I de la serpine ; P1 : site de clivage par la protéase.
SMCs) et les plaquettes (Brogren et al., 2004; Sprengers & Kluft, 1987). Dans la circulation, PAI-1 est présent en excès par rapport au tPA. La majorité du tPA est donc complexé à PAI-1, ce qui inhibe son activité. De plus, PAI-1 est capable de se fixer aussi bien au sc-tPA qu’au tc-tPA (Rijken & Lijnen, 2009). Les complexes formés entre le tPA et PAI-1 sont très stables, ce qui rend l’inhibition du tPA presque définitive (Zhou et al., 2001).
L’association de PAI-1 avec le tPA ne gêne pas son élimination. En effet, la clairance du tPA par les hépatocytes peut se faire aussi bien sous forme libre que sous forme complexée (Nagaoka et al., 2002).
Enfin, PAI-1 est également présent dans le SNC, principalement au niveau astrocytaire et, bien que ceci soit plus débattu, au niveau neuronal (Hino et al., 2001).
1.4.2. Neuroserpine
La NS est une molécule composée de 410 acides aminés, pour un poids de 54-60 kDa. Elle est retrouvée presque exclusivement dans le SNC ; toutefois une faible quantité est aussi retrouvée dans les reins , le cœur, le pancréas et les testicules (Schrimpf et al., 1997). Dans le SNC, la NS est synthétisée principalement par les neurones, mais aussi par les astrocytes. Elle n’est en revanche pas exprimée par les cellules endothéliales. La NS est ainsi très peu présente dans le compartiment sanguin, et n’a donc aucune influence sur le tPA vasculaire (Docagne et
al., 1999; Hastings et al., 1997; Osterwalder et al., 1996). Contrairement à PAI-1, l’inhibition
du tPA par la plasmine est peu stable. Il est probable que le clivage de la RCL par le tPA forme une boucle trop longue, empêchant le complexe tPA/NS de se stabiliser. Le tPA peut alors s’en libérer et retrouver son activité (Barker-Carlson et al., 2002). De ce fait, la durée de vie moyenne des complexes tPA-NS n’excède pas une dizaine de minutes (Miranda & Lomas, 2006).
1.5. F
ONCTIONS DU TPA
1.5.1. Le tPA dans la circulation
1.5.1.1. Fibrinolyse
Il existe dans le sang un mécanisme enzymatique capable de dégrader les caillots de fibrine qui peuvent se former dans la circulation : le système fibrinolytique, ou fibrinolyse. Il s’articule autour d’une enzyme principale, la plasmine, qui est capable de dégrader les chaînes de fibrine. La plasmine est sécrétée par le foie sous la forme d’une pro-enzyme, le
plasminogène. Ce plasminogène en lui-même est inactif : il a besoin d’être activé (Collen & Lijnen, 1991).
Il existe deux activateurs du plasminogène chez l’homme : le tPA et l’activateur du plasminogène de type urokinase (urokinase plasminogen activator, uPA). Bien qu’ayant des caractéristiques différentes, leurs actions aboutissent toutes les deux à l’activation du plasminogène en plasmine. Comme décrit précédemment (voir la partie « 1.3.1.1 Synthèse et
libération par les cellules endothéliales »), le tPA est synthétisé par les cellules endothéliales
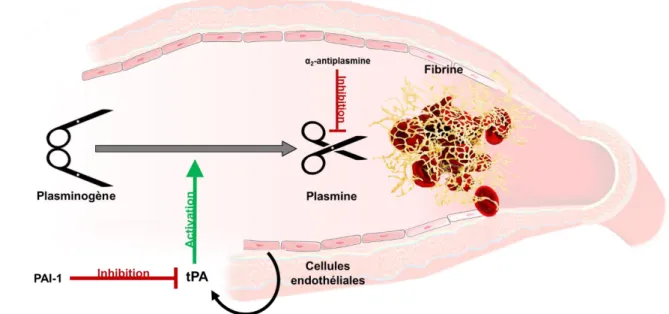
et libéré de manière constitutive ou bien en réponse à certains stimuli. En conditions normales, le tPA n’est pas un très bon activateur du plasminogène. En revanche, en présence de fibrine, sa capacité à activer le plasminogène est multipliée par 100 (Hoylaerts et al., 1982). En effet, la fibrine contient un site de liaison avec le tPA proche d’un autre site de liaison avec le plasminogène, facilitant ainsi leur interaction. Le tPA va ainsi se fixer à la fibrine et au plasminogène, pour convertir ce dernier en plasmine. Par ce mécanisme, le tPA active le plasminogène principalement au niveau du thrombus (Medved & Nieuwenhuizen, 2003). De plus ce mécanisme permet de placer la plasmine à l’abri de ses inhibiteurs, améliorant ainsi l’efficacité de la fibrinolyse.
Figure 9 I Mécanisme de la fibrinolyse endogène. Le tPA est sécrété par les cellules endothéliales dans la circulation sanguine. Il peut alors activer le plasminogène circulant en plasmine à proximité d’un caillot de fibrine. La plasmine est alors capable de dégrader la fibrine, et ainsi de lyser le caillot. La régulation de la fibrinolyse peut se faire par inhibition de la plasmine (α2-antiplasmine) ou du tPA (PAI-1).
Il existe deux niveaux de régulation de la fibrinolyse, principalement afin d’éviter une fibrinolyse exacerbée, ou hyperfibrinolyse, qui en plus d’augmenter le risque d’hémorragie, provoque une altération de la barrière hémato-encéphalique (BHE ; Marcos-Contreras et al., 2016). Le premier échelon d’inhibition se situe au niveau de la plasmine directement, qui peut être notamment inhibée par l’α2-antiplasmine (Holmes et al., 1987). La fibrinolyse peut également être inhibée par la diminution de la quantité de plasmine circulante, et donc par l’inactivation du tPA par le PAI-1 (Figure 9).
1.5.1.2. tPA et barrière hémato-encéphalique
La BHE est une barrière physiologique qui assure une protection entre le parenchyme cérébral et la circulation sanguine. Son rôle est d’empêcher le passage des molécules du sang vers le parenchyme cérébral, à l’exception de certaines molécules nécessaires au cerveau. Elle est formée par les cellules endothéliales des vaisseaux reliées entre elles par des jonctions serrées, par une lame basale composée de collagène IV, de laminine et de fibronectine, par des péricytes, et par les pieds astrocytaires qui viennent former un manchon autour du vaisseau
(Figure 10 ; Abbott et al., 2006).
Figure 10 I Barrière hématoencéphalique. La BHE est formée par les cellules endothéliales reliées par des jonctions serrées, une lame basale, les péricytes, et les pieds astrocytaires (adapté de Abbott et
En conditions pathologiques, le tPA est capable d’altérer la BHE, via une interaction avec les récepteurs LRP situés à la surface des cellules endothéliales. En plus d’augmenter le risque hémorragique, ce mécanisme mène au passage du tPA vasculaire vers le parenchyme cérébral (Yepes et al., 2003). L’interaction du tPA avec les récepteurs LRP mène également au décrochage des pieds astrocytaires de la lame basale (Cheng et al., 2006). Le tPA pourrait également dégrader la BHE par l’activation de métalloprotéinases matricielles (matrix
metalloproteinase, MMP), notamment MMP-9, 2 et 3 (Lijnen, 2001). Ces MMPs activées pourraient participer à la dégradation de la matrice extracellulaire, fragiliser les jonctions serrées, et altérer la lame basale des vaisseaux cérébraux (Suzuki et al., 2009; Wang et al., 2003). De plus, le tPA est également capable de d’interagir et d’activer le PDGF-CC via son domaine Kringle 2 et son domaine catalytique. Le PDGF-CC peut ensuite stimuler les récepteurs PDGF-α des péricytes et astrocytes, dont l’activation peut altérer la perméabilité de la BHE et ainsi aggraver le risque hémorragique (Su et al., 2008; Figure 11).
Figure 11 I Effet du tPA sur la barrière hématoencéphalique. Le tPA interagit avec les récepteurs LRP des cellules endothéliales et des astrocytes, menant entre autres à la production de MMP-9 au sein de ces cellules. MMP-9 provoque alors une altération de la BHE. De plus, l’activation du PDGF-CC par le tPA contribue également au détachement des pieds astrocytaires de la lame basale, par l’activation des récepteurs PDGFR-α. L’activation du PDGF-CC est facilitée par l’interaction du tPA avec les récepteurs LRP sur les astrocytes (Mehra et al., 2016).
En conditions physiologiques, il a été montré que le tPA vasculaire pouvait aussi traverser la BHE, également par une interaction avec les récepteurs LRP (Benchenane et al., 2005; Mehra et al., 2016).
1.5.2. Le tPA dans le système nerveux central
1.5.2.1. Le tPA comme neuromodulateur
Lors de l’activité neuronale, il a été montré que la dépolarisation des neurones glutamatergiques provoquait, en plus de la libération du glutamate dans la synapse, une exocytose de tPA (Gualandris et al., 1996). Dans la synapse, le tPA peut agir comme un neuromodulateur, puisqu’il est capable de potentialiser la signalisation induite par l’interaction du glutamate avec son récepteur post-synaptique, le récepteur NMDA (Nicole et al., 2001). De plus, le tPA est impliqué dans l’activation du cycle des vésicules synaptiques et leur recrutement au niveau de la zone active, augmentant ainsi la quantité de neurotransmetteur libéré dans la synapse. Il est également impliqué dans le recyclage de ces vésicules (Wu et al., 2015). Au niveau de la synapse, le tPA a également un rôle de gliotransmetteur. En effet, le tPA peut être endocytosé par les astrocytes via le récepteur LRP, puis exocytosé de manière régulé dans la synapse (Cassé et al., 2012).
1.5.2.2. Développement et migration cellulaire
Le développement cérébral est le fruit d’un enchaînement complexe d’évènements, incluant prolifération cellulaire, différenciation, migration, et mise en place de connexions et d’interactions entre les différents types cellulaires (Gupta et al., 2002; Nadarajah et al., 2003; van den Ameele et al., 2014). Ces différentes phases nécessitent le plus souvent la dégradation de la matrice extracellulaire (MEC ; Lu et al., 2011). Pour cela, les cellules sécrètent notamment des métalloprotéases telles que les MMPs ou les désintégrines et métalloprotéinase à motif thrombospondines (a disintegrin and metalloproteinase with thrombospondin motifs, ADAMTS), ainsi que des sérines protéases telles que le tPA. Ainsi, des souris déficientes en tPA présentent une migration neuronale retardée, notamment mise en évidence par une accumulation des cellules granulaires au sein de la couche moléculaire du cervelet (Seeds et
Plus récemment, des travaux du laboratoire ont permis de montrer que le tPA est impliqué dans la migration des progéniteurs d’oligodendrocytes au cours du développement. Cet effet implique une activation des récepteurs à l’EGF présents sur les progéniteurs d’oligodendrocytes (Leonetti et al., 2017).
1.5.2.3. Plasticité synaptique
En conditions physiologiques, le tPA est fortement exprimé dans certaines régions cérébrales comme l’hippocampe ou l’amygdale (Teesalu et al., 2004). Ces deux régions jouent un rôle important dans les processus de mémoire et d’apprentissage. Le mécanisme moléculaire décrit comme support de ces deux fonctions est la potentialisation à long terme (Long Term
Potentiation, LTP). Ce mécanisme consiste en un renforcement de l’efficacité synaptique après
une stimulation excitatrice répétée (Nabavi et al., 2014). Ce phénomène a initialement été découvert en 1973 chez le lapin (Bliss & Lømo, 1973), et il a été rapidement mis en évidence que le système glutamatergique avait un rôle central dans ce mécanisme, impliquant particulièrement les récepteurs NMDA (Collingridge et al., 1983).
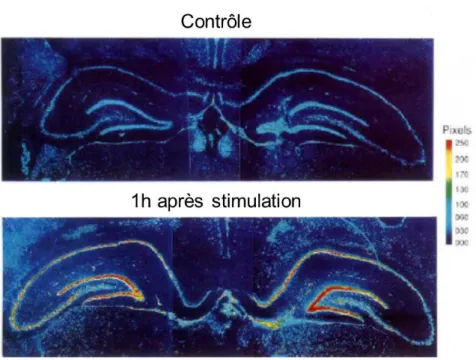
Le tPA joue également un rôle dans ce processus. En effet, en 1993, Qian et collaborateurs ont observé une augmentation rapide des ARN messagers codant pour le tPA suite à l’activation des neurones hippocampiques (Qian et al., 1993; Figure 13).
Figure 12 I Effet de l’absence de tPA sur la migration neuronale. Cellules granulaires (gn) dans la couche moléculaire du cervelet de souris (P10) WT et tPA KO. Microphotgraphies montrant l’accumulation des cellules granulaires dans la couche moléculaire en l’absence de tPA (adapté de Seeds
L’implication du tPA dans la LTP est également visible sur le comportement des souris. La surexpression du tPA par les neurones chez la souris induit une augmentation des performances d’apprentissage et de mémorisation grâce à une augmentation de la LTP (Madani
et al., 1999). A l’inverse, chez des souris déficientes en tPA (knockout, KO), l’absence de tPA
se traduit par un déficit d’apprentissage. Cet effet peut être annulé par l’infusion de tPA directement dans l’hippocampe (Pawlak et al., 2002). En outre, des travaux du laboratoire ont mis en évidence le fait que les effets du tPA sur la LTP seraient plutôt médiés par le sc-tPA plutôt que par le tc-tPA (Parcq et al., 2012).
En plus de son interaction avec les récepteurs NMDA, le tPA pourrait agir sur la LTP à travers plusieurs mécanismes : par la dégradation de la MEC, qui faciliterait la création de nouvelles synapses (Baranes et al., 1998), par sa capacité à activer le pro-brain-derived growth
factor (BDNF) en sa forme mature, un facteur de croissance neuronal nécessaire à la LTP (Pang
& Lu, 2004), ainsi que par sa capacité à cliver la reeline, une protéine importante dans la régulation des fonctions synaptiques (Trotter et al., 2014).
1.5.2.4. tPA et comportement
Nous l’avons vu dans le paragraphe précédent, le tPA est impliqué dans la plasticité synaptique. Ce rôle lui confère une influence dans plusieurs types de comportement impliquant la mémoire (Calabresi et al., 2000).
Figure 13 I Elévation de la quantité d’ARN messager du tPA suite à une activation neuronale chez
le rat. Hybridation in situ des ARNm du tPA dans l’hippocampe de rat en conditions normales et 1h après stimulation électrique du gyrus denté (adapté de Qian et al., 1993).