Stability of the Maternal Gut Microbiota During Late Pregnancy and Early Lactation
9
0
0
Texte intégral
Figure
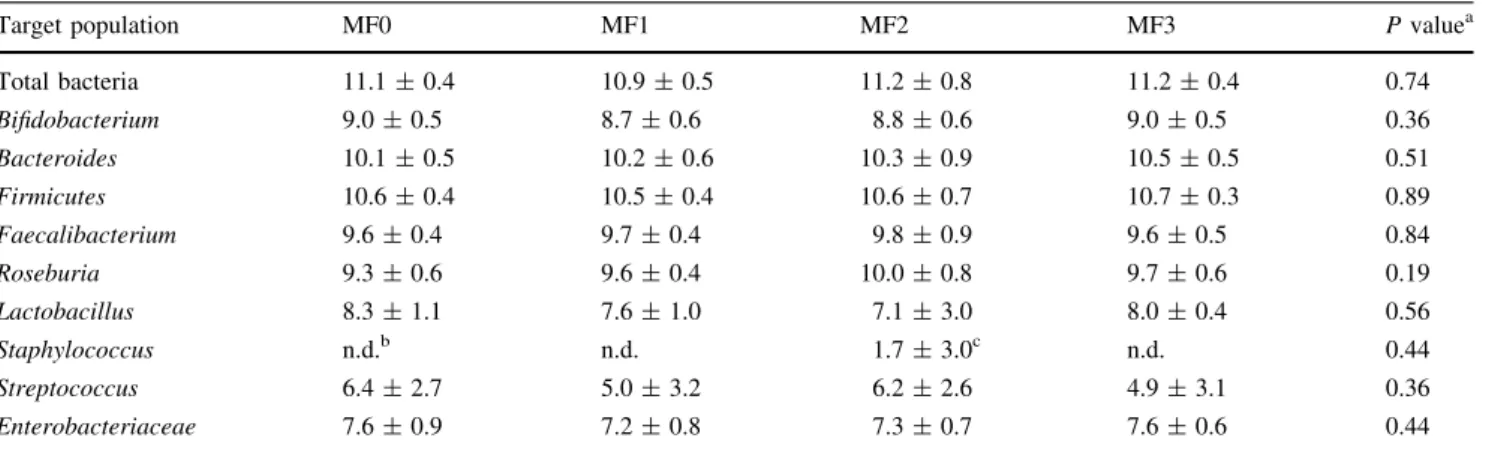
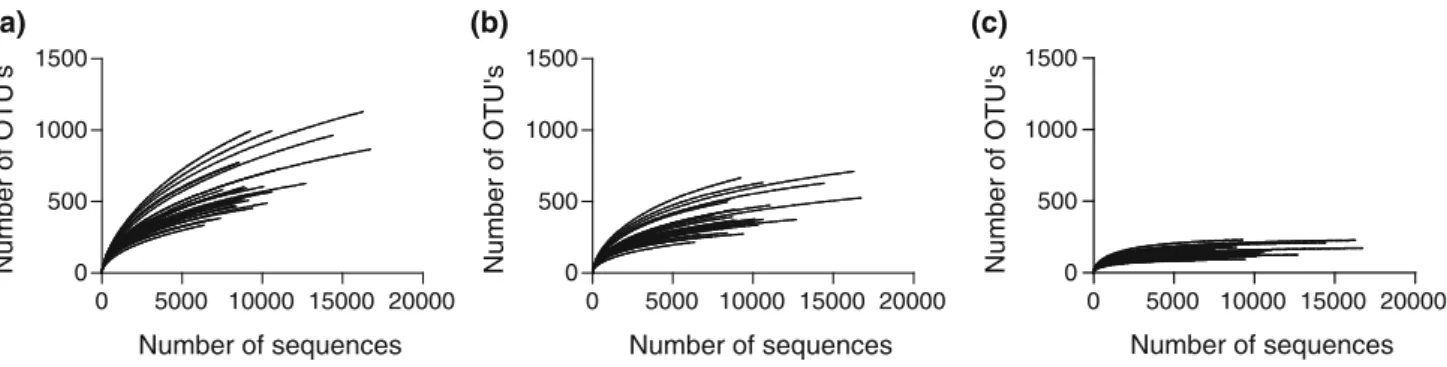
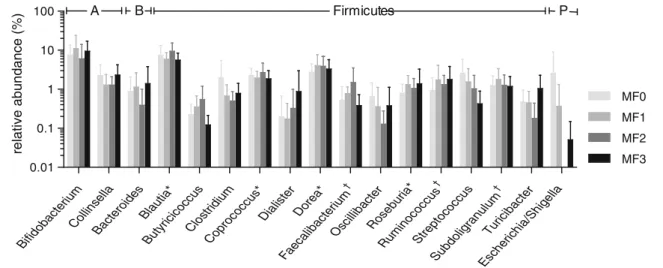
Documents relatifs