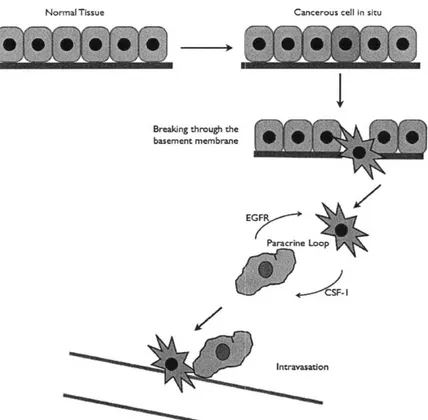
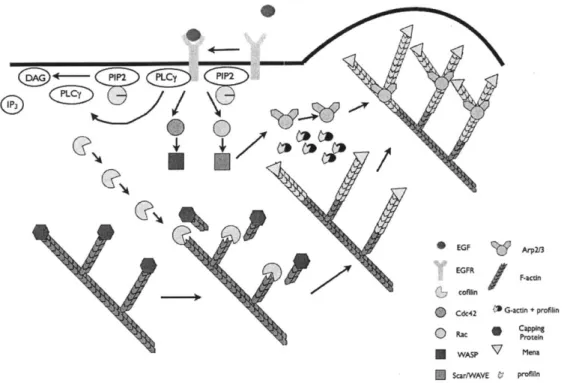
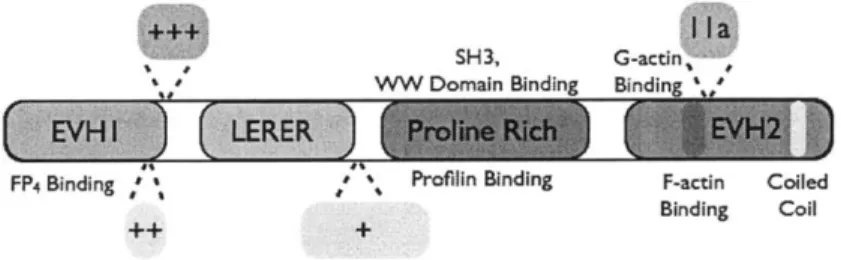
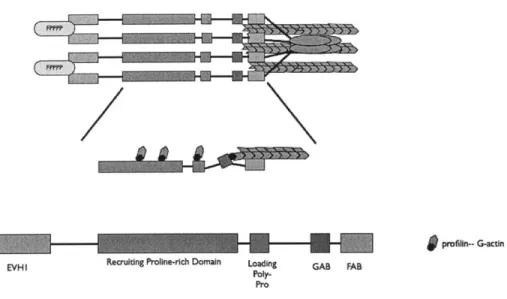
The 5' inositol phosphatase SHIP2 regulates EGF-elicited protrusion in MTLn3 cells
Texte intégral
Figure
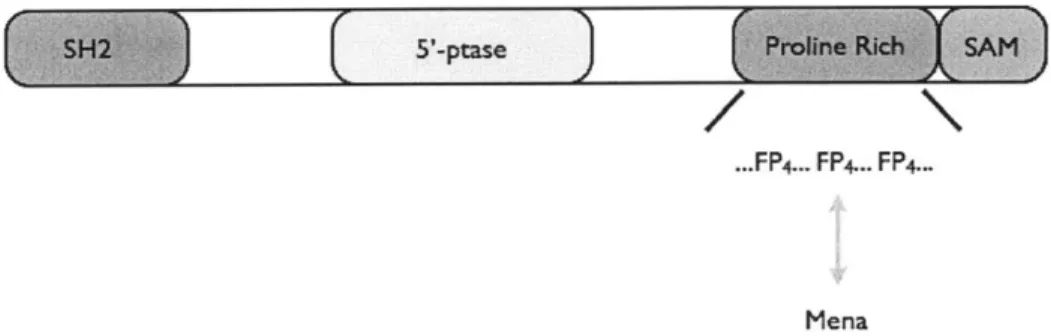
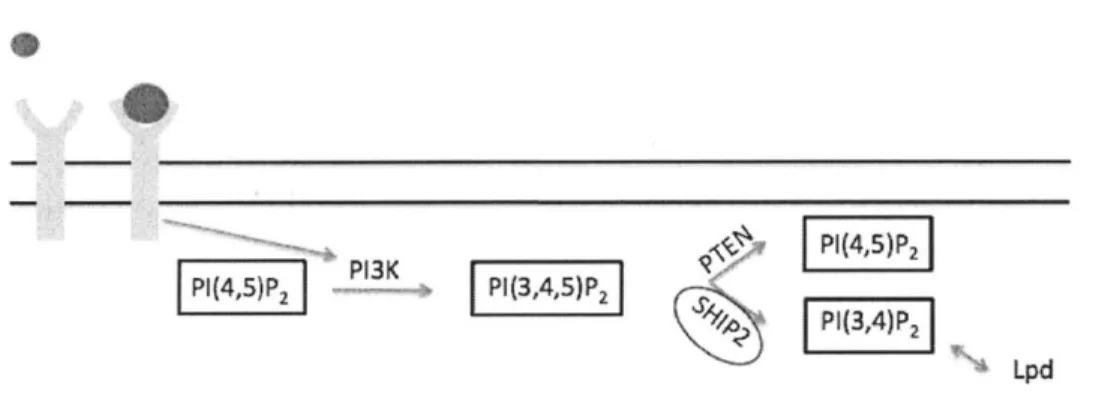
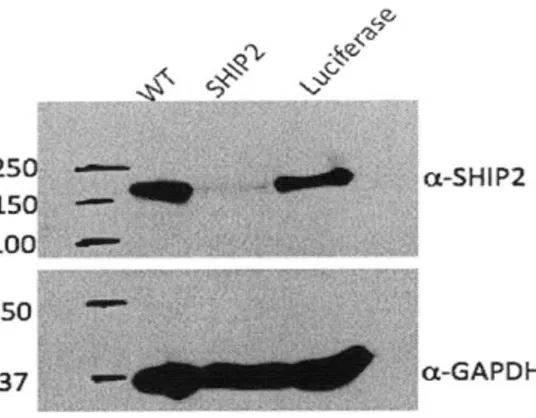
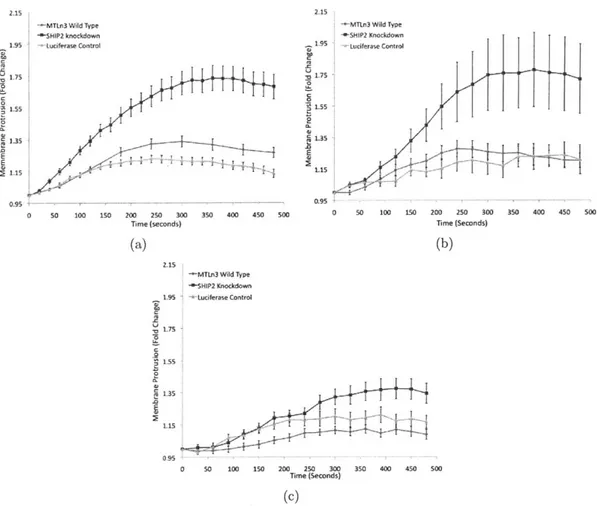
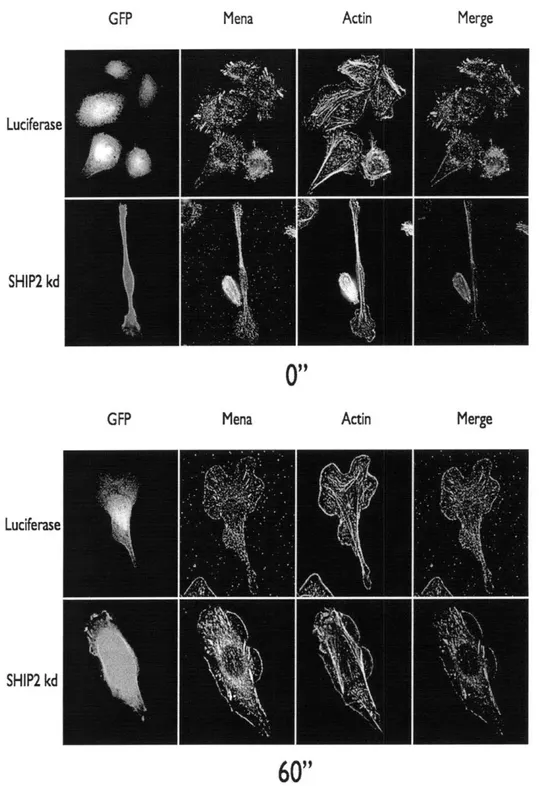
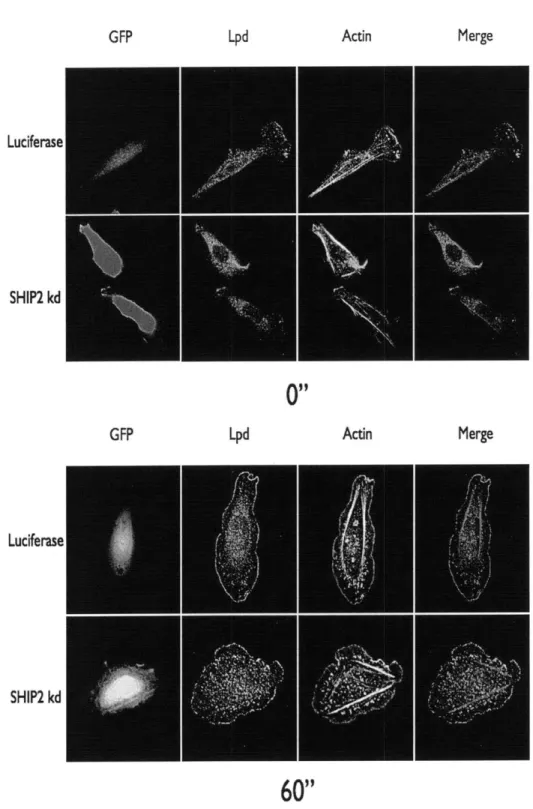
Documents relatifs
In our context each expert is a Kalman filter fed by a subset of sensors, and a gating network serves as a mediator between individual filters, basing its decision on sensor inputs
Ce réseau de correspondants devait faciliter la collecte des familles qui, en France, sont susceptibles d'être dispersées dans plusieurs régions : « Notre idée était
Les fonds Rodolphe Töpffer (1799-1846) de la Ville de Genève sont issus pour l’essentiel de trois sources, qui ont fait entrer petit à petit dans ses collections, dans la
To establish the effects of blocks of linked genetic polymorphisms on gene transcription, we repeated fat transcriptome profiling in a series of reciprocal BN.GK and GK.BN
L’analyse des items hors échelles montre que si les objectifs minimaux ont été atteints pour plus d’un répondant sur deux (« déterminer une filière d’étude qui
A 1988 study on the teaching of business ethics in Canadian business schools, carried out by Jang Singh (1988), found that of the 55% of business schools which offer courses
Rapporter dans la corbeille, le plus d’objets correspondant à la couleur de son équipe en un temps donné. • Le meneur relance les objets déposés par les élèves dans
En ce qui a trait spécifiquement à l’analyse par réconciliation, cette étude avait également pour objectifs secondaires 1) l’évaluation du respect de l’exigence d’Agrément
