T
T
H
H
È
È
S
S
E
E
En vue de l'obtention du
D
D
O
O
C
C
T
T
O
O
R
R
A
A
T
T
D
D
E
E
L
L
’
’
U
U
N
N
I
I
V
V
E
E
R
R
S
S
I
I
T
T
É
É
D
D
E
E
T
T
O
O
U
U
L
L
O
O
U
U
S
S
E
E
Délivré par l'Université Toulouse III - Paul Sabatier Discipline ou spécialité : Chimie-Biologie-Santé
JURY
P.Gonzalez-Duarte Professeur Universitat Autonoma de Barcelona G. Guillaumet Professeur d'Université (Orléans)
N. Ndifon Peter Professeur d'Université (Yaoundé) B. Martin-Vaca Professeur d'Université (Toulouse)
P.Faller Professeur d'Université (Toulouse) E. Gras Chargé de Recheche CNRS (Toulouse)
Ecole doctorale : Sciences de la matière
Unité de recherche : Laboratoire de Chimie de Coordination/SPCMIB Directeur(s) de Thèse : P.Faller Professeur d'Université, Toulouse
E. Gras Chargé de Recherche CNRS(Toulouse)
Rapporteurs : P. Gonzalez-Duarte Professeur Universitat Autonoma de Barcelona Présentée et soutenue par Rodrigue Leuma Yona
Le 27 janvier 2009
Je tiens tout d’abord à remercier l’Association France Alzheimer qui a permis que ce travail puisse se réaliser dans de bonnes conditions.
Ce travail a été effectué au Laboratoire de Chimie de Coordination et au Laboratoire de Synthèse et Physico-Chimie des Molécules d’Intérêt Biologique de Toulouse. Je remercie monsieur Brunot Chaudret et monsieur Michel Baltas de m’avoir accueilli dans leurs laboratoires respectifs.
Monsieur Robert Martino, Le Dierecteur de l’Ecole Doctorale de Sciences de la Matière, pour sa compréhension.
Je remercie très particulièrement le Professeur Gérald Guillaumet, Président de l’Université d’Orléans, Le Professeur Pilar Gonzalèz-Duarte, de l’Universitat Autonoma de Barcelona de me faire l’honneur de juger ce travail en tant que rapporteurs. Je leur en suis reconnaissant.
Je remercie le Professeur Ndifon Peter Nteke, Professeur à l’Université de Yaoundé 1, actuellement Chargé de Mission au Ministère de la Recherche Scientifique au Cameroun qui m’a initié à la recherche en D.E.A et qui a bien voulu me faire l’honneur et la plaisir de sa présence à ce jury de thèse.
Je remercie également Madame Blanca Martin-Vaca, Professeur à l’Université Paul Sabatier de Toulouse, d’avoir accepté de participer à ce jury de thèse.
Ces travaux ont été effectués sous la co-direction de Peter Faller, Professeur à l’Université Paul Sabatier et Docteur Emmanuel Gras, Chargé de Recherche CNRS. Les mots me manquent pour leur exprimer ma profonde gratitude et il est irréaliste de penser que quelques lignes suffiront :
Pour le premier, je lui suis reconnaissant de m’avoir offert l’opportunité depuis le Cameroun de rejoindre son groupe, sans lequel tout ceci n’aurait pas été possible. Je le remercie encore plus d’avoir assuré la co-direction scientifique de ce travail, sa perpétuelle disponibilité à l’écoute, aux échanges, aux différentes explications des méthodes possibles et envisageables, m’ont marqué scientifiquement de façon indélébile. Ses conseils, ses enseignements, ses encouragements et sa gentillesse ont contribué à mener à bien ce travail. Je lui suis aussi infiniment reconnaissant de m’avoir offert en 2007 l’opportunité de retourner au Cameroun présenter mes travaux à l’Université de Yaoundé 1 et voir ma famille, dont ma fille pour la première fois. Ceci a été une source de remotivation.
Au second, Emmanuel, résumer ces 3 années est impossible, tant j’ai reçu. D’abord merci d’avoir effectué toutes les procédures pour mon accueil à Toulouse, de m’avoir accueilli à l’aéroport et, d’avoir largement contribué à mon intégration à Toulouse. Au départ ayant une formation de Chimiste Inorganicien, je n’avais que très peu de connaissance en chimie organique, mais ta disponibilité à la transmission et à celui de pédagogue infaillible, tes
m’ont largement inspiré et m’inspireront encore.
Je vous suis à tous les deux pour ce que j’ai appris sur le plan scientifique et humain à vos côtés infiniment reconnaissants. Vous avez fait que ceci soit possible et j’en garde un souvenir mémorable.
Je remercie, Béatrice Mestre-Voegtle, mon parrain de thèse pour ces conseils durant ces 3 années.
Monsieur et Madame Gorrichon, merci pour vos conseils, pour votre soutien et vos éclairements parfois, et pour la location de l’appartement.
Je remercie également les membres du SPCMIB Alain, Chantal A., Corinne, Évelyne, Ghislaine, Jean-Marc, Nadine, Marie, Nathalie, Christiane, Christian, Colette. Tous les autres membres du SPCMIB du second et du premier étage.
Je remercie les gens qui ont participé à la réalisation de ce travail :
Marie Lise à IPBS pour le prêt de divers matériels, Serge Mazères pour les traitements de donnés de fluorescence, Pierre et Yannick du Service RMN pour l’initiation aux appareils, Vincent Collière pour les expériences de MET et pour sa gentillesse, Le service d’analyse de masse, Cécile Caubet à Purpan pour les Expériences de Dot Blot que nous avons réalisé ensemble, Chantal Zedde pour les purifications HPLC, Emmanuel Mothes pour la suite des expériences qu’elle effectue, Pascal du service informatique, Berkouk Soroya la sécrétaire de l’école doctorale..
Je tiens à remercier aussi tout particulièrement Christelle Hureau-Sabatierpour son aide lors de la phase de rédaction ; ton support et tes encouragements ont été très bénéfiques et je t’en suis reconnaissant.
Je remercie du cœur toutes les personnes que j’ai côtoyé durant ces années et avec qui j’ai passé de bons moments.
Au SPCMIB, je pense à Hélène Bonin, ces 3 trois années ont été remarquables avec toi comme collègue de labo (Bonne Chance pour ta soutenance !!!!), nos échanges et discussions constructives resteront des bons souvenirs, Bruno pour ta gentillesse au labo, Caroline Marette pour m’avoir aidé au début de ma thèse et pour ta gentillesse. Arnaud, Sébastien, Céline pour la bonne humeur et l’ambiance détendu. Au LCC je pense Luc, Vanessa, Christine qui m’ont accueilli dans l’équipe. Tous les membres de l’équipe K qui m’ont souvent été d’un grand secours en me prêtant des produits, ou par des discussions constructives, et aussi souvent la bonne humeur.
Je pense aussi particulière au stagiaire Christophe qui a travaillé sur les études physico-chimiques.
conseils, et Soephi pour son accueil aimable lors de la soirée barbecue. J’ai été très touché par votre générosité à toutes les deux.
A mon amie Nathalie, merci pour toute ton aide et tout ce que tu as fait pour moi.
Je tiens aussi à remercier tous mes amis qui ont fait de mon séjour à Toulouse un moment de bonheur et de convivialité : Alex, Faustin, Richard, Georges, Paul Ervé, Chills, Sylvain, Ernestine, Danielle, Alioum, Karim, Marie-Claire, Mireille, Huguette, Élodie Simone, Ramadan, Gilles, Igor, Patrick, Rostand, Harold. Pardon à tous ceux que j’ai oublié, vous avez fait que ces 3 années soit très agréables.
Je pense très particulièrement à mon frère à Bordeaux, Arnaud Maxime, tu es mon principal soutien, et tu es toujours là pour moi. Merci infiniment, courage et bonne chance pour ta soutenance de thèse à venir.
À mes frères : Arsène Élisée, Isidore, Patrick, Brice, Nicole en Allemagne, Caroline en Belgique, Merci pour vos encouragements et votre soutien permanent.
À mes Parents pour leurs soutiens et pour tout ce qu’ils ont fais pour moi. Ce travail représente peu comparé aux lourds sacrifices que vous avez consentis pour moi. Je vous en serais éternellement redevable. Votre absence à ma soutenance me manquera. Je n’oublie pas mes frères et sœurs au Cameroun Edith et son mari Victor et mes petites nièces, Gwladys Léonie, Arsène Sorian, votre affection à mon égard sont une source de motivation considérable.
Enfin, à ma fille Ashley Stecy, mon affection pour toi est inimaginable, et à Marlyse pour son support, son affection et ses critiques. Je comprends bien grâce à toi qu’on demande beaucoup à ceux qu’on affectionne tout particulièrement. Merci d’être là pour moi.
CHAPITRE I : Rappels bibliographiques ... 3
I Les démences neurodégénératives... 3
I.1 Historique : Un siècle déjà… ... 3
I.2 Manifestation Clinique... 4
I.3 Prévalence, Incidence et charge financière ... 6
I.4 Facteurs à risques de la Maladie ... 8
I.4.1 Facteurs génétiques ... 8
I.4.2 Facteurs environnementaux ... 9
I.4.3 Facteurs Vasculaires ... 10
I.4.4 Facteurs nutritionnels... 11
I.4.5 Stress Oxydant ... 11
I.5 Système Cellulaire perturbés par la Maladie d’Alzheimer ... 12
I.5.1 Les Cellules des tissus nerveux... 12
I.5.2 Dysfonctionnement des systèmes de neurotransmetteurs dans la MA ... 13
I.5.3 Le zinc « Libre » Comme Modulateur... 16
I.5.4 Toxicité généré par le dérèglement du calcium ... 16
I.5.5 Inflammation... 16
I.6 Les Lésions Caractéristiques de la Maladie D’Alzheimer... 17
I .6.1 Dégénérescences neurofibrillaires ... 17
I.6.2 Plaques séniles (plaque amyloïdes) ... 19
I.6.3 Relation entre tau et Aβ ... 19
I.6.4 Hypothèse de la cascade amyloïde ... 20
I.7 Implication des métaux ... 23
II Les plaques séniles... 23
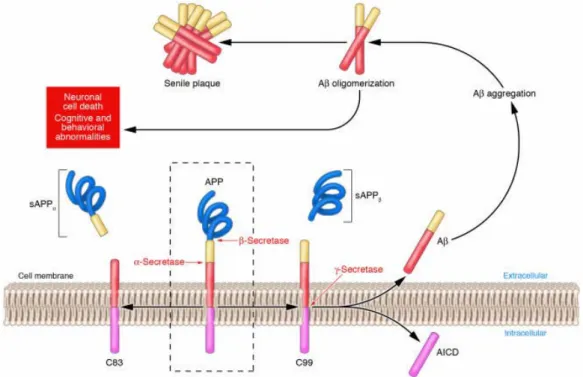
II.1 Origine de l’Aβ: la protéine APP ... 23
II.1.1 La protéine APP... 23
II.1.2 De l’APP à Aβ ... 24
II.1.3 Le peptide β-amyloïde (Aβ) ... 26
II.1.4 Agrégation des peptides Aβ... 27
II.2 Toxicité due à l’agrégation ... 29
III Interaction diverses avec le peptide Aβ ... 31
III.1 Interaction de l’Aβ avec les membranes ... 31
III.2 Interaction de l’Aβ avec les ions métalliques... 32
IV Les Diverses Voies Thérapeutiques... 32
IV.1 Généralité ... 32
IV.2 Approches symptomatiques et traitements utilisés ... 33
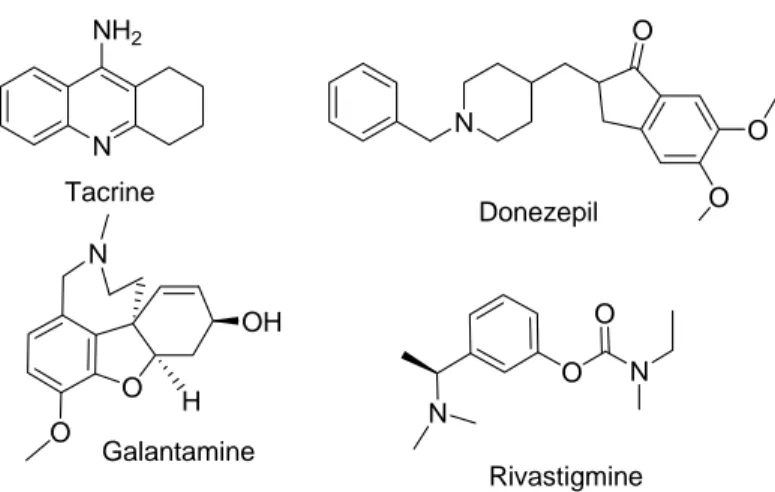
IV.2.1 Les Inhibiteurs des cholines estérases... 33
IV.2.2 Régulation du Ca2+ : Antagonistes des récepteurs du glutamate de la famille NMDA... 35
IV.2.3 Les Anti-inflammatoires non stéroïdiens ... 36
IV.2.4 Les antioxydants ... 37
IV.3 Agents thérapeutiques ciblant les causes de la maladie ... 39
IV.3.1 Les modulateurs de sécrétases ... 39
IV.3.2 Les Composés favorisant la diminution des taux de cholestérol ... 42
IV.3.3 L’approche vaccinale ... 42
IV.3.4 Inhibiteurs d’agrégation de l’Aβ : agents déstructurant les feuillets-β ... 43
IV.3.5 Inhibiteurs de la phosphorylation ou de la polymérisation de tau ... 45
V. 0 Généralité ... 47
V.1 TEP ... 48
V.1.1 Définition... 48
V.1.2 Radioisotope pour la TEP... 49
V.1.3 Protocole... 50
V.1.4 Quantification Par Imagerie Nucléaire... 51
V.1.5 Challenge... 52
V.1.6 Sites de liaisons des Aβ ... 53
V.1.7 Ligands pour le Diagnostic précoce Par TEP... 54
VI Conclusion ... 59
Références Chapitre I ... 61
CHAPITRE II : Synthèse en série Benzothiazole ... 75
I Introduction... 75
I.1 Généralités ... 75
I.2 Réactivité benzothiazoles... 76
I.2.1 Généralités ... 76
I.2.2 Basicité (au sens de Bronsted et de Lewis)... 77
I.2.3 Formation de benzothiazole métallés... 78
I.2.4 Cycloaddition ... 78
I.2.5 Ouverture du cycle ... 79
I.2.6 Réaction Radicalaire ... 79
I.2.7 Réaction de substitution électrophile (S.E.)... 80
I.3 Applications thérapeutiques... 81
II. Synthèse du noyau benzothiazole ... 82
II.1 Généralités ... 82
II.2 A partir d’aniline ... 83
II.2.1 Voie A... 83
II.2 .2 voie B : réaction de Hugerscoff ... 84
II.3 Création da la liaison Ar-S métallocatalysé ... 85
II.4 Réaction de Diels-Alder ... 86
II.5 A partir d’aminothiophenol ... 87
III. Synthèse Dérivés 2-arylbenzothiazole... 88
III.1 Généralités... 88
III.2 Couplage de Stille ... 89
III.2.1 Définition ... 89
III.2.2 Cycle Catalytique ... 89
III.2.3 Application ... 90
III.3 Couplage de Miyaura Suzuki ... 90
III.3.1 Définition ... 90
III.3.2 Le Cycle Catalytique ... 91
III.3.3 Application ... 92
III.4 A partir des anilines... 93
III.4.1 Généralités... 93
III.4.2 A partir de l’aminothiophenol ... 94
III.4. 3 Via la cyclisation de Jacobson ... 95
III.4. 4 Réaction aza-Wittig... 97
III.4. 4 Via réarrangement [3,3] sigmatropique ... 98
III.4.7 Photocyclisation ... 99
III.4.8 A partir de réactif de Grignards... 100
IV Résultats Personnels ... 100
IV.1 Introduction ... 100
IV.2 Dérivés arylbenzothiazoles non substitués sur le noyau benzothiazole... 101
IV.2.1 Généralités ... 101
IV.2.2 Voie A : Synthèse en solution catalysé par un acide de Lewis... 101
IV.2.2.1 Généralités ... 101
IV.2.2.2 Mécanisme ... 102
IV.2.2.3 Résultats ... 102
IV.2.2.4 Avantages et inconvénients de la méthode ... 105
IV.2.3 Voie B: Synthèse Sous Champ Micro-onde ... 104
IV.2. 3.1 Généralité sur les micro-ondes... 106
IV.2.3.2 formation arylbenzothiazoles sous champ microonde ... 106
IV.2.3.3 Résultats ... 107
IV.2.3.4 Avantages et inconvénients de cette méthode... 110
IV.3 Voie C : Synthèse à partir du chlorure d’acyle ... 111
IV.4 Synthèse de 2-(aminoaryl)benzothiazoles ... 111
IV.4.1Généralité ... 111
IV.4.1 Réduction des dérivés nitro... 112
IV.4.2 Formylation des amines. ... 113
IV.4.2 Réduction des amides... 115
IV.5 Réaction de benzoylation ... 116
IV.6. Synthèses d’autres dérivés N-alkylés... 117
IV.6.1 Réaction d’amino-réduction... 117
IV.6.1.1 Généralités ... 117
IV.6.1.2 Résultats ... 118
IV.6.1.3 Synthèse parallèle ... 119
V Synthèse de dérivés substitués sur le noyau benzothiazole... 122
V.1 Généralités... 122
V.2 Synthèse des précurseurs halogénés... 123
V.2.1 Synthèse des noyaux 2-aminobenzothiazole ... 123
V.2.1.1 Généralités... 123
V.2.1.2 Résultats ... 124
V.2.2 Synthèses des partenaires Bromés... 125
V.2.2.1 Généralités... 125
V.2.2.2. Résultats ... 126
V.2.2.3 Autres benzothiazoles partenaires de couplages ... 129
V.3 Synthèse des Partenaires Bores ... 130
V.3.1 Généralités... 131
V.3.2 Synthèse des Organotrifluoroborates de Potassium ... 131
V.3.2.1. Généralités... 131
V.3.2.2. Résultats ... 131
V.3.3. Synthèse de dérivés Dioxazaborocanes... 132
V.3.3.1 Généralités... 132
V.3.3.2 Choix du groupement protecteur de l’aniline... 133
V.3.3.3 Voie A : Protection avec les Benzyles ... 131
V.3.3.4 Voie B : Protection avec le Boc ... 134
V.4.1. A partir des dérivés organotrifluoroborate. ... 137
V.4.1.1 Généralités... 137
V.4.1.2 Résultats ... 137
V.4.2 A partir des dérivés dioxazaborocanes ... 138
V.4.2.1 Généralité ... 138
V.4.2.2 Résultats ... 138
V.5 Déprotection des arylbenzothiazoles... 140
V.5.1 Déprotection benzyle... 140
V.5.1.1 Généralité ... 140
V.5.1.2 Résultats ... 140
V.5.2 Déprotection du Boc... 141
V.5.2.1 Généralités... 143
V.6 Déméthylation (obtention du PIB) ... 143
V.6.1 Généralités... 143
V.6.2 Résultats ... 143
VI Conclusion ... 144
Références Chapitre II... 145
CHAPITRE III : Etude Physico-Chimique ... 154
I Préparation des échantillons... 154
I.1 Spectroscopie UV-Visible ... 154
I.1.1 Définition ... 154
I.1.2 Principe ... 154
I.1.3 Application... 155
I.2 Mesure du spectre UV-visible des composés ... 156
I.3 Préparation des Fibres amyloïdes Aβ(1-40) et Aβ(1-42)... 156
I.3.1 Solution stock de fibres... 156
I.3.2 Solution de travail ... 157
II. Caractérisation des agrégats amyloïdes ... 157
II.1 Processus de la spectroscopie de Fluorescence ... 157
II.2 Méthodes de caractérisations... 160
II.2.1 Généralités ... 160
II .2.2 Titrage avec la Thioflavine T ... 161
II.2.2.1 Généralités ... 161
II.2.2.2 Préparation des solutions de ThT ... 162
II.2.2.2.1 Solution stock ... 162
III.2.2.2.2 Solution de Travail ... 162
III.2.2.2.3 Mesure de la fluorescence ... 162
II. 2.3 Microscopie Electronique à Transmission (MET) ... 164
II.2.3.1 Préparation des grilles ... 164
II.2.3.2 Images MET ... 164
III. Détermination des constantes d’affinités... 165
III.1 Détermination de la constante de dissociation : titrage direct... 165
III.1.1 Principe du titrage ... 165
III.1.2 Calcul constante de dissociation ... 166
III.1.3 Analyse des données ... 167
III.2 Détermination de la constante d’inhibition : titrage indirect... 169
III.2.1 Titrage par compétition ... 169
IV Détermination de la constante de dissociation de la ThT... 172
IV.1 Généralité ... 172
IV.2 Préparation des solutions ... 173
IV.2.1 Solution stock de ThT ... 173
IV.2.2 Solution de travail ... 173
IV.3 Mesure de la fluorescence de la ThT ... 173
IV.4 Calcul de la constante de dissociation pour Aβ(1-40). ... 174
V. Titrages des dérivés des composés synthétisés... 175
V.1 Généralité ... 175
V.2 Détermination Kd : titrage direct du peptide et du tampon ... 176
V.2.1 Solution tampon ... 176
V.2.2 Cas du 2-(2’-fluorophenyl)-1,3- benzothiazole... 176
V.3 Détermination Ki : titrage par compétition ... 181
V.3.1 Généralité ... 181
V.3.2 Préparation des échantillons de titrage ... 181
V.3.2.1 Titrage dans la cuve... 181
V.3.2.2 Titrage sur la cuve à 96 puits... 181
V.3.3 Ki des échantillons avec Aβ (1-40) ... 182
V.3.3.1 Titrage compétition 2-(2-furyl)-1,3-benzothiazole ... 182
V.3.3.2 Titrage compétition 2-(1,3-benzothiazol-2-yl)-N-methylaniline ... 184
V.4 Valeurs des constantes d’affinités des dérivés benzothiazoles ... 185
V.5 Etude relation structure–activité (SAR) ... 190
V.5.1 Lipophilie des dérivés ... 191
V.5.2 Apport de la charge positive et de l’azote intra-cyclique ... 191
V.5.3 Isomérie de position ... 191
V.5.4 Densité électronique des noyaux aromatiques ... 192
V.5.5 Formation de liaisons hydrogènes ... 192
V.6 Titrage Dérivés BTA-ethyl... 195
V.6.1. Protocole de titrage... 195
V.6.2 Etude Structure Activité du substituant sur l’azote extra cyclique de BAT-1198 V.6.3 Affinité avec les fibres Aβ (1-42)... 199
VI Conclusion ... 199
Références Chapitre III ... 201
Conclusion Générale ... 206
°C degré celsius
µl microlitre
A absorbance
Ach acétycholine AchE achetylcholinestérase AcOt Acétate d’éthyle
ADDLs « Aβ-derived diffusible plaque » ADN acide désoxyribonucléique ApoE apoliprotéine E
APP « amyloid protein precusor »
ARN acide ribonucleique
Aβ amyloïde-beta
BACE beta site APP cleaving enzyme
Bn benzyle
Boc ter-butylcarbamate
BTA-1 4-(1,3-benzothiazol-2-yl)-N-methylaniline BuChE butylcholinestérase
C83 fragment C83 (possédant 83 acides aminés) C99 : fragment C99 (possédant 99 domaines) CCM : chromatographie sur couche mince ChaT cholinestérase
DCM dichlorométhane
DMF diméthylformamide
DMSO diméthylsufoxide
éq. équivalent
ESI-MS « electrospray ionization mass spectroscopy »
EtOH ethanol
EtP éther de pétrole
g grammes h heure
HMBC « heteronuclear Multiple Bond correlation » HPLC chromatographie liquide haut performance HSQC « Heteronuclear Single quantum Correlation »
IR Infra-Rouge
IRM Imagerie par Résonance Magnétique Kd constant de d’association
kDa kilodalton Ki constante d’inhibition
Koff vitesse cinétique de dissociation Kon vitesse cinétique d’association M molaires m millilitre M .O MicroOnde MA maladie d’Alzheimer mg microgramme min minute mM millimolaire
NaH hydrure de sodium
NMP N-methyl pyrrilidone
Pd/C palladium activé sur charbon PHFs paires hélicoïdales de filaments
PhMe toluene
PIB 2-[4-(methylaminophenyl]-1,3-benzothiazol-6-ol pM picomolaire
RAS Relation Structure activité
RMN Résonnance Magnétique Nucléaire ROS « réactive oxygen species »
s seconde S.E substitution Electrophile SiO2 silice
SPECT Tomographie par Emission Monophotonique T.A température ambiante
TEP Tomographie par Emission de Positons THF tetrahydrofurane
ThT Thioflavine T
TMS tetraméthylsilane
UV/Vis Ultra Violet/Visible
Introduction générale
Les travaux de thèse qui m’ont été confiés s’inscrivent dans le cadre général de la Maladie d’Alzheimer.
Nous nous sommes plus particulièrement intéressés à la nature des interactions entre de petites molécules organiques et les agrégats amyloïdes. Un objectif à long terme de ces travaux développés en étroite entre la chimie organique de synthèse et l’analyse physico-chimique est la mise au point de nouveaux ligands plus spécifiques des plaques séniles avec en arrière plan la possibilité d’intégration d’un adduit fluoré qui permettra d’accéder à l’imagerie TEP.
Nous avons souhaité nous diriger vers ce but en nous appuyant sur une approche rationnelle. Cette dernière est basée sur une connaissance plus approfondie des liaisons des marqueurs avec les plaques séniles (fibres amyloïdes) et permettra de s’engager dans une boucle d’optimisation schématisée ci-dessous :
Squelette connu pour son aff inité envers les agrégats
amyloïdes
Mise au point des stratégies d'accès simples et efficaces Constitution d'une banque de dérivés Détermination des affinités Evaluation
des spécificités analyses structurales (RMN HR-MAS)
Docking -design rationnel : Nouveaux squelettes
Mes travaux de thèse représentent l’amorce de cette boucle et avaient donc pour but de fournir un socle à ce travail de longue haleine.
Afin de mieux comprendre les interactions ligands–amyloïdes, nous avons entrepris : Dans un premier temps d’explorer les voies de synthèses à la fois simples et originales (permettant une éventuelle automatisation) permettant d’accéder aux dérivés comportant un motif benzothiazole de la famille de la Thioflavine T.
Dans un second temps la caractérisation et la détermination des constantes d’affinités de ces dérivés à travers une étude relation structure-activité (RAS).
Ce mémoire sera donc logiquement développé en trois parties. Le chapitre I fera le point sur l’ensemble des éléments bibliographiques relatifs à la Maladie d’Alzheimer, aux agrégats amyloïdes et à l’imagerie TEP. Les deux chapitres suivants traceront le tableau des résultats expérimentaux obtenus durant les trois années de thèse. Le chapitre II sera principalement consacré à la description des travaux de synthèse organique que nous avons mis en place pour accéder de manière simple, rapide et reproductible aux dérivés benzothiazoles. Le chapitre III présentera l’ensemble des méthodes mises en œuvre pour déterminer l’affinité des composés obtenus avec les agrégats amyloïdes, dont l’obtention in vitro sera également décrite.
Chapitre I
I Les démences neurodégénératives... 3
I.1 Historique : Un siècle déjà… ... 3
I.2 Manifestation Clinique ... 4
I.3 Prévalence, Incidence et charge financière ... 6
I.4 Facteurs à risques de la Maladie ... 8
I.4.1 Facteurs génétiques... 8
I.4.2 Facteurs environnementaux ... 9
I.4.3 Facteurs Vasculaires ... 10
I.4.4 Facteurs nutritionnels ... 11
1.4.5 Stress Oxydant ... 11
I.5 Système Cellulaire perturbés par la Maladie d’Alzheimer ... 12
I.5.1 Les Cellules des tissus nerveux ... 12
I.5.2 Dysfonctionnement des systèmes de neurotransmetteurs dans la MA... 13
I.5.3 Le zinc « Libre » Comme Modulateur ... 16
1.5.4 Toxicité généré par le dérèglement du calcium ... 16
1.5.5 Inflammation... 16
I.6 Les Lésions Caractéristiques de la Maladie D’Alzheimer ... 17
I .6.1 Dégénérescences neurofibrillaires ... 17
I.6.2 Plaques séniles (plaque amyloïdes) ... 19
I.6.3 Relation entre tau et Aβ ... 19
I.6.4 Hypothèse de la cascade amyloïde... 20
I.7 Implication des métaux ... 23
II Les plaques séniles... 23
II.1 Origine de l’Aβ: la protéine APP... 23
II.1.1 La protéine APP ... 23
II.1.2 De l’APP à Aβ... 24
II.1.3 Le peptide β-amyloïde (Aβ)... 26
II.1.4 Agrégation des peptides Aβ ... 27
II.2 Toxicité due à l’agrégation ... 29
III Interaction diverses avec le peptide Aβ ... 31
III.1 Interaction de l’Aβ avec les membranes... 31
III.2 Interaction de l’Aβ avec les ions métalliques... 32
IV. Les Diverses Voies Thérapeutiques... 32
IV.1 Généralité ... 32
IV.2 Approches symptomatiques et traitements utilisés ... 33
IV.2.1 Les Inhibiteurs des cholines estérases ... 33
IV.2.2 Régulation du Ca2+ : Antagonistes des récepteurs du glutamate de la famille NMDA ... 35
IV.2.3 Les Anti-inflammatoires non stéroïdiens... 36
IV.2.4 Les antioxydants ... 37
IV.3 Agents thérapeutiques ciblant les causes de la maladie ... 39
IV.3.1 Les modulateurs de sécrétases ... 39
IV.3.2 Les Composés favorisant la diminution des taux de cholestérol ... 42
IV.3.3 L’approche vaccinale... 42
IV.3.4 Inhibiteurs d’agrégation de Aβ : agents déstructurant les feuillets-β ... 43
IV.3.5 Inhibiteurs de la phosphorylation ou de la polymérisation de tau ... 45
IV.3.6 Chélateurs d’ions métalliques... 46
V.1.1 Définition ... 48
V.1.2 Radioisotope pour la TEP ... 49
V.1.3 Protocole ... 50
V.1.4 Quantification Par Imagerie Nucléaire ... 51
V.1.5 Challenge ... 52
V.1.6 Sites de liaisons des Aβ... 53
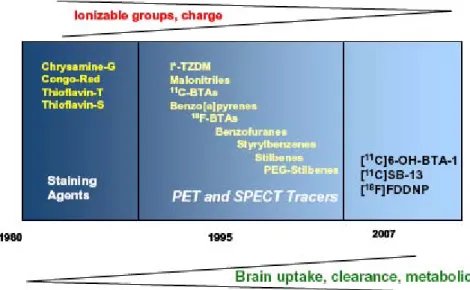
V.1.7 Ligands pour le Diagnostic précoce Par TEP ... 54
VI Conclusion ... 58
CHAPITRE I : Rappels bibliographiques
I Les démences neurodégénératives
I.1 Historique : Un siècle déjà…
L’état de démence des personnes âgées était considéré par la plupart des psychiatres à la fin du XIXe siècle comme habituel et lié à un vieillissement normal. Ce sont les travaux de l’école de Munich, autour de Kraepelin, un des deux psychiatres allemands à croire à l’intérêt de l’étude histologique du cerveau dans les maladies mentales, qui ont conduit à mieux comprendre ces maladies. Plusieurs médecins rejoignent alors le groupe dont Aloïs Alzheimer qui s’était initié à l’étude microscopique du cerveau.
Alois Alzheimer 1864-1915
C’est en 1906, lors d’une réunion de psychiatres allemands à Tübingen, qu’Alzheimer décrit l’observation d’une femme de 51 ans (Auguste D) qui a présenté un délire de jalousie suivie d’une désintégration des fonctions intellectuelles. Grâce à des techniques de coloration, l’examen au microscope (ci-dessous) du cerveau de la patiente a révélé dans le cortex cérébral, des lésions jusque-là inconnues, caractérisées par des amas anormaux de fibrilles dans les neurones, les dégénérescences neurofibrillaires.
Peu de temps après ces observations, la communauté médicale de l’époque donna à cette maladie le nom de celui qui l’avait mis en évidence, la maladie d’Alzheimer (MA).
Les connaissances sur cette maladie n’ont évolué que vers la fin du XXe siècle. Une compréhension plus étendue de la maladie a conduit à une amélioration de sa définition. C’est une démence dégénérative du cerveau par opposition aux autres causes de démences (vasculaire, toxique ou carentielle). Elle associe les troubles prédominants de la mémoire, des troubles cognitifs et / où du comportement ayant un retentissement sur la vie quotidienne des patients. Cette démence est associée à deux types de lésions : les plaques séniles et les dégénérescences neurofibrillaires. Si ces lésions peuvent se développer assez tôt dans la vie, la maladie ne s’exprime habituellement que tardivement.
I.2 Manifestation Clinique
Cette démence dont l’étiologie reste encore mal connue, est associé à des lésions histopathologiques qui la définissent : les plaques séniles ou amyloïdes qui sont extracellulaires (pathologie Aβ) et les dégénérescences neurofibrillaires (pathologie Tau) qui son des enchevêtrements intracellulaire (Figure 1-I).1 Ces lésions sont principalement accumulées dans le cortex (partie superficielle du cerveau) et dans l’hippocampe.
Figure 1-I : Plaques et enchevêtrements dans le cerveau de patient atteint de la maladie d’Alzheimer post mortem (d’après Laferla, 2005). (a) Microphotographie de plaque amyloïdes marqués par un des anticorps spécifiques anti-Aβ42. (b) Microphotographie de
À ces 2 phénomènes s’ajoute une perte de synapse et de neurones. Ce phénomène a lieu dans un premier temps dans la partie interne du lobe temporal du cerveau (cortex entorhinal et l’hippocampe) qui est la région vers laquelle convergent les informations pour êtres mise en mémoires (Figure 2-I). Les lésions se développent ensuite vers les lobes pariétaux, occipitaux et frontaux, pour envahir presque tout le cerveau, sans toutefois atteindre les centres de commandes de la motricité.
Figure 2-I : Représentation des zones du cerveau impliquées dans la maladie d’Alzheimer
d’aprèsVanderberghe et al. 2
Au final, le cerveau des malades présente une large gamme d’altérations neuropathologiques (Figure 3-I) incluant l’atrophie corticale, la réduction sélective de certains systèmes de neurotransmetteurs (comme le système cholinergique) qui est retrouvés en particulier dans les zones associés aux plaques amyloïdes et aux neurofibrilles et une diminution du métabolisme énergétique. On observe également dans le cerveau de ces malades un stress oxydant anormalement élevé et synonyme d’inflammation.3
du lobe temporal (partie basse) et des lobes frontaux (partie gauche). (b) Imagerie TEP révélant la consommation de glucose : le patient atteint de la MA montre une forte diminution du métabolisme énergétique dans le cortex frontal et les lobes temporaux.
Les manifestations de cette maladie dont les lésions se développent longtemps à bas bruit, impliquent une cascade d’évènements. L’idée émerge que les deux lésions fréquentes au cours du vieillissement ont un effet synergique et provoquent un processus dégénératif qui porte atteinte, progressivement mais inexorablement aux fonctions supérieures au sein des aires corticales associées à la mémoire, au jugement et aux fonctions intellectuelles.
Les plaques séniles qui sont observées normalement au cours du vieillissement et les dégénérescences neurofibrillaires qui sont parfois observées dans d’autres démences dégénératives ne sont pas spécifiques de la maladie d’Alzheimer. C’est leur association chez un même patient qui est caractéristique de cette maladie éminemment complexe et spécifiquement humaine pour laquelle il n’existe pas de modèle animal totalement satisfaisant qui reproduise complètement la pathologie humaine.
Elle se manifeste par des troubles de la mémoire, en particulier des évènements récents (mémoire épisodique) dans les stades initiaux de la maladie. L’aggravation des troubles conduit à une perte de cohérence dans le discours et une inintelligibilité de l’expression écrite, ainsi qu’à des détériorations dans d’autres phénomènes interférant avec l’humeur, la raison ou le jugement. Il en découle une altération de la personnalité, des troubles neurologiques (aphasie, crises épileptiques) et une démence prononcée.
Au stade final le patient devient totalement dépendant. Les délires et l’agitation viennent compliquer la prise en charge du malade par son entourage, ce qui rend la phase terminale de la maladie particulièrement difficile.
I.3 Prévalence, Incidence et charge financière
De toutes les maladies neurodégénératives comme les démences fronto-temporales, la démence à corps de Léwy, les démences vasculaires, la maladie de Creutzfeldt-Jacob, la maladie d’Alzheimer est la forme de démence la plus répandue. Cette pathologie neurodégénérative affecterait plus de 30 millions de personnes dans le monde (dont la moitié dans les pays occidentaux dans lesquels l’âge de la population est élevé).
En France on estime à 850 000, le nombre de personnes atteinte d’Alzheimer, et l'on dénombre 200 000 nouveaux cas tous les ans.4 L’affection touche plus de femmes que
d’hommes puisque au-delà de 75ans, les proportions sont de 13,2 pour les hommes et de 20,5 chez les femmes. Au-delà de 85 ans, d’après une étude PAQUID, la prévalence s’accroit de manière exponentielle avec une proportion de 25 % de sujets atteints. Les formes précoces ne sont pas rares puisqu’elles concernent 32 000 cas avant 60 ans et 1000 cas avant 50 ans d’après France Alzheimer. Il s’agit dans cette dernière situation le plus souvent de formes monogéniques avec présence de mutations autosomiques à transmission dominante et à pénétrance complète.
Dans le monde, on dénombre plus de 30 millions de personnes vivant avec des démences dont la grande majorité d’Alzheimer,5 elle constitue dans les pays développées la quatrième cause de décès précédé par les maladies cardiaques, le cancer et les accidents cérébraux.
Les perspectives pour les années à venir sont inquiétantes ; Si rien ne vient ralentir la tendance actuelle, de l’ordre de 1 200 000 cas en 2012 (soient 2 déments pour 100 habitants) et de 2 100 000 cas en 2040 (soient 3 déments pour 100 habitants) seront recensés en France.
Si rien n’est fait, la situation ne va donc qu’empirer avec le vieillissement de la population aussi bien dans les pays en voie de développement que les pays développés.
L’incidence s’élève à 200 000 nouveaux cas en France par an et l’espérance de vie après l’apparition des symptômes est en moyenne de 8,5 ans. Le niveau de la dépendance est très variable et actuellement 40 % des patients sont pris en charge par une institution privée ce qui signifie que 60 % des frais sont à la charge des familles.
La dépense moyenne de prise en charge d’un patient est 22 000 euros par an, soit une charge annuelle de 18,8 milliard d’euros (une partie aux frais de l’état et l’autre à la charge des familles) qui se repartit en 75 % de dépenses médico-sociales et 25 % de dépenses médicales. Ces dépenses s’élèvent actuellement à 0,6 % du PIB, mais passeront à 0,8 % en 2020 et à 1,8 en 2040. La prise en charge des personnes atteintes de démences représentera alors si la tendance actuelle continue, 7 % des dépenses de santé. À ceci il faut ajouter l’impact qui est difficilement quantifiable sur la famille qui entoure le malade.
Cette maladie est devenue un problème majeur de santé publique et un véritable fléau social. C’est pourquoi en 2007 elle a été déclarée en France Grande Cause Nationale comme l’avait été il y a quelques années le Cancer. Toute la société est sollicitée, et la construction
des structures d’accueil adaptées devient indispensable, et plus que jamais la communauté scientifique est mobilisée.
I.4 Facteurs à risques de la Maladie
Les dégénérescences neurofibrillaires et les plaques amyloïdes ont été considérées initialement comme les lésions cérébrales étroitement liées à la maladie. Cependant, les causes exactes de l’apparition de ces lésions restent encore inconnues. En quête de réponse, les chercheurs étudient les facteurs qui semblent de nature à agir sur la progression de la maladie. On les appelle les « facteur à risques ». Le caractère polyfactoriel de cette maladie résulte de l’interaction entre un terrain génétique et des facteurs environnementaux.
I.4.1 Facteurs génétiques
L’implication des gènes dans la maladie d’Alzheimer est double :
D’une part, il existe des formes monogéniques exceptionnelles, caractérisés par un début précoce (inférieur à 60 ans) et par l’atteinte d’un sujet sur deux à chaque génération (formes autosomiques dominantes) qui représente environ 5 % des cas. Elle est liée à un facteur héréditaire. Elle est encore appelé « maladie d’Alzheimer familiale ». À ce jour près de, 150 mutations sur trois gènes ont été observées et sont responsables de la transmission de la maladie. Les trois gènes susceptibles de porter ces mutations sont :
(i) Un gène du chromosome 21 codant pour la protéine APP (Amyloid Precusor Protein), précurseur du peptide β-amyloïde (Aβ) impliqué dans la maladie ; c’est la raison pour laquelle la plupart des personnes atteintes du syndrome de Down (trisomie 21) expriment 1,5 fois plus l’APP que les individus sains et souffrent de démence de type Alzheimer dès l’âge de 40 ans.6
(ii) deux gènes situés sur le chromosome 1 et 14 codant respectivement pour les protéines préséniles 2 et 1 (ces protéines s’associent aux γ-sécrétase pour conduire à la génération de l’Aβ à partir de l’APP.
D’autre part, dans les formes dites sporadiques de la maladie, de loin les plus courantes, sont impliquées des facteurs de risque génétique (polymorphisme de l’ADN) qui constituent simplement un terrain génétique. Ainsi, il semble que les variations du gène l’apoliprotéine (ApoE) localisé sur le chromosome 19, codant pour la production de trois protéines isoformes E2, E3, E4, qui sont des transporteurs des lipides sanguins avec les
plus fréquente ; L’ApoE4 est associée à une élévation de la concentration plasmique du LDL-cholesterol (anthérogène) et donc du risque cardio–vasculaire (Infarctus du myocarde) ; ApoE 2 est associé à l’inverse à une diminution du LDL cholestérol et du risque cardio-vasculaire.
De nombreuses études ont mis en évidence une forte association entre l’allèle ε4 et la MA. Le risque de développer cette affection est plus fréquente entre 60 ans et 80 ans et augmente avec le nombre d’allèles ε4 présents. Au contraire, ceux qui sont porteurs d’ApoE2 ont un risque plus faible comparé à l’ensemble de la population.7
Ce facteur génétique n’est qu’un facteur de risque, cela revient à dire qu’il n’est ni nécessaire ni suffisant pour développer la Maladie d’Alzheimer. Il est possible que l’ApoE soit impliqué dans la cascade amyloïde que nous décrirons plus tard. Cette protéine exercerait un rôle de molécule chaperonne et conduirait, après fixation sur le peptide Aβ, à sa modification en forme soluble ayant la propriété de s’agréger sous forme amyloïde au sein des plaques séniles.
Cet allèle a également été associé à d’autres maladies neurodégénératives comme les démences vasculaires, les démences à corps de Lévy et la maladie de Creutzfeldt Jacob.
Un gène du chromosome 12 codant pour l’α2-macroglobuline, impliqué dans la
dégradation de l’Aβ, ainsi qu’un locus sur le chromosome 10 ont été identifiés comme d’autres facteurs de risque.8
I.4.2 Facteurs environnementaux
Dans 95 % des cas, la MA est de forme sporadique, sans antécédents familiaux et à l’étiologie inconnue.9 Comme c’est le cas dans la plupart des autres maladies liées à l’âge (diabète, maladie de Parkinson, maladie cardiovasculaire…..), l’âge, l’environnement et d’autres facteurs s’y rattachant jouent un rôle non négligeable.
Age et sexe :
L’âge est un fort facteur à risque dans la MA. En effet, la prédominance de la pathologie double tous les 5 ans après 65 ans,10 par contre, la démence de type Alzheimer n’est pas comme semble le croire l’opinion populaire un processus normal de vieillissement.
Cette pathologie comme nous l’avons vu touche plus de femme que des hommes puisque au-delà de 75 ans, les proportions chez les femmes sont de 20,5% contre 13,2% chez
D’autres facteurs ont également été évoqués pour expliquer cette observation : la diminution des taux d’œstrogènes suite à la ménopause, mais également une dérégulation du système de transport des métaux plus importante chez les femmes pourraient favoriser le développement de la maladie.12
Carence Alimentaire :
Les carences nutritionnelles limitant les apports en vitamines C et D chez personnes âgées, pourraient être considérées comme un facteur aggravant. Des études épidémiologiques ont montré que l’utilisation de compléments alimentaires vitaminés pourrait réduire le risque de développer la maladie.13
Cholestérol :
Le cholestérol est un constituant essentiel des membranes et joue un rôle fondamental dans le développement, le maintien de la plasticité et le fonctionnement des neurones.14
Des études épidémiologiques ont montré qu’un niveau élevé de cholestérol pouvait être corrélé avec le risque plus élevé de développement de la maladie.15 Chez les souris transgéniques nourries selon un régime riche en cholestérols, le nombre de plaques amyloïdes est nettement plus élevé que chez les souris de contrôle.16
De plus, un traitement par les statines (inhibiteurs de la biosynthèse du cholestérol) prescrit aux personnes de taux de cholestérol trop élevé) réduit fortement les taux d’Aβ40 et Aβ42 intracellulaires et secrétés in vitro (cultures neuronales). Leur utilisation pourrait réduire considérablement le risque d’Alzheimer.17, 18
I.4.3 Facteurs Vasculaires
Des études suggèrent que les facteurs vasculaires constituent un facteur de risque vis-à-vis des démences survenant chez les personnes âgées. Preuve est que les diabétiques et les sujets victimes d’un accident vasculaire cérébral courent un rique accru (multiplié par 1,9 pour les premiers) de développer une démence.
I.4.4 Facteurs nutritionnels
Il a été montré que les acides gras polyinsaturés oméga-3 sont impliqués dans la physiologie du cerveau qui est exceptionnellement riche en acide gras. Des études épidémiologiques ont montré que de trop faibles concentrations en oméga -3 constituent un facteur de risque vis-à-vis de plusieurs types de pathologies de démences dont celle d’Alzheimer. Cause ou conséquence, la répartition des acides gras polyinsaturés au sein des phospholipides cérébraux est perturbée au cours de la maladie d’Alzheimer. Ces observations attirent l’attention sur l’importance, dans la prévention de démence d’un apport nutritionnel suffisant en acides gras oméga-3, en particulier en acide α-linoléique (ALA) et en potentiel de protection de ces lipides, présumés capable de moduler l’inflammation cérébrale.
1.4.5 Stress Oxydant
La protection de l’organisme vis-à-vis des dommages oxydants est réalisée par un grand nombre d’antioxydants, enzymatiques ou non. Par exemple, les super oxydes dismutases Mn-SOD et Cu/Zn-SOD sont responsables de la conversion de l’anion superoxyde en H2O2 et O2. L’H2O2 est quant à lui détoxifié en O2 et H2O par la catalase et les
peroxydases. Cependant, lorsque les taux de ROS dépassent la capacité antioxydante de la cellule, les phénomènes délétères de stress oxydant apparaissent au niveau des lipides, des protéines ou encore de l’ADN de celle-ci.
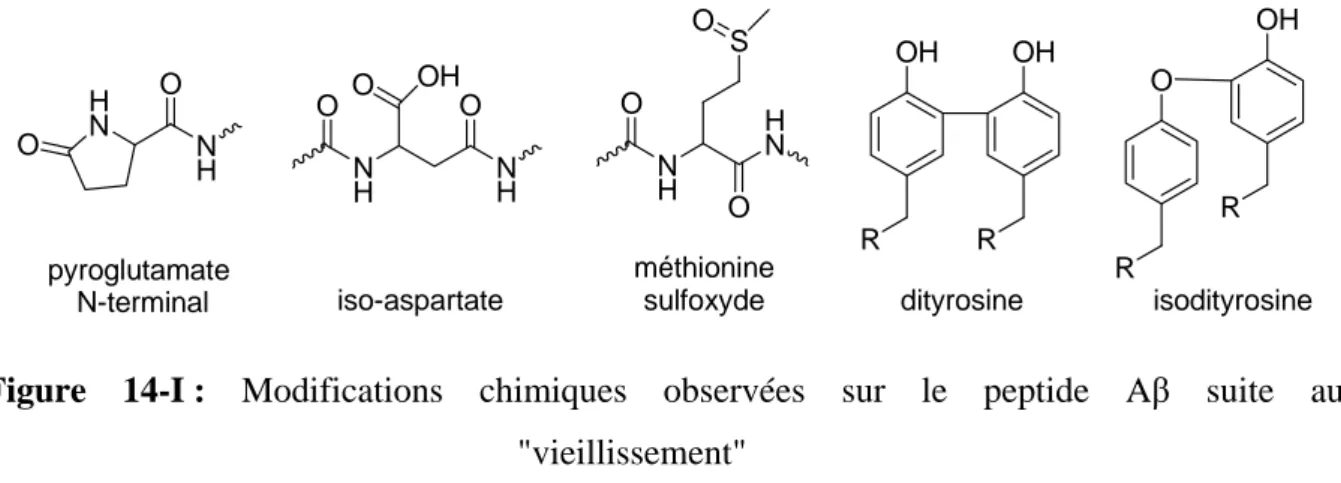
L’attaque de radicaux sur une double liaison d’acides gras insaturés (acide arachidonique, acide docosohenènoïque…) initie des réactions en chaînes sur d’autres acides gras. Les produits formés incluent le 4-hydroxy-2-nonèn-1-al (HNE), le 2-propèn-1-al (acroléine) (figure 4-I), le malondialdéhyde et les isoprostanes. Toutes ces espèces, ainsi que des formes oxydées du cholestérol (4-cholesten-3-one par exemple) ont été retrouvées à des niveaux élevés dans les cerveaux atteints d’Alzheimer, à proximité des plaques et des neurofibrilles.19, 20 OH H O H O HNE acroléine
Figure 4-I : Structures du 4-hydroxy-2-nonèn-1-al (HNE) et du 2-propèn-1-al (acroléine) La présence de dommages sur les acides nucléiques (en particulier la formation de la 8-hydroxy-2’-désoxyguanosine (8-OHdG) et de modifications des protéines (carbonylation et
nitration, adduits covalents avec l’HNE ou l’acroléine via des additions électrophiles de Michael…) est également observée dans les régions présentant des neurofibrilles ou des dépôts amyloïdes, ainsi que sur les protéines Aβ et tau elles-mêmes.21
Enfin, les marques de stress oxydants (catalase, superoxyde dismutase (SOD), glutathionnes peroxydase et glutathionne réductase) sont augmentées dans l’hippocampe et le complexe amygdalien des malades d’Alzheimer.22
Toutes ces observations sont en faveur d’un stress oxydant élevé dans la maladie d’Alzheimer. Les modifications engendrées (oxydation des lipides, oxydations des protéines et de l’ADN) conduisent à une toxicité générale dans le cerveau. Par exemple, les espèces réactives formées lors du stress oxydant peuvent déclencher des évènements conduisant à l’apoptose23. D’autre part, la perte de l’intégrité des membranes suite à l’oxydation des lipides peut conduire à des dysfonctionnements cellulaires, comme l’inhibition des ATPases et la perte de l’homéostasie du calcium, rendant les neurones vulnérables à l’excitotoxicité et à l’apoptose. La conséquence ultime de tous ces dysfonctionnements cellulaires est la mort neuronale.
Le peptide Aβ est capable de produire des espèces réactives de l’oxygène (en particulier H2O2) qui ont été détectées sur cultures cellulaires mais également in vivo. 24, 25
I.5 Système Cellulaire perturbés par la Maladie d’Alzheimer
I.5.1 Les Cellules des tissus nerveux
I.5.1.1 Les neurones et la transmission neuronale
Le neurone est un type de cellule différenciée qui compose, avec les cellules gliales, les tissus nerveux. Il se compose d’un corps cellulaire et de 2 types de prolongements : Les dendrites et l’axone. Son rôle est d’assurer la transmission du signal nerveux. Ils sont incapables de toute forme de division cellulaire. Cette incapacité a pour effet un traumatisme qui endommage le système nerveux de façon irréversible.
Lors de la transmission neuronale, les dendrites reçoivent une information d’autres neurones sous la forme d’un potentiel membranaire électrique (potentiel d’action). Celui-ci engendre un flux d’ions Na+ dans les cellules au travers des canaux ioniques suivi d’un flux de K+ à travers la membrane spécifique sur le neurone pour restaurer le potentiel de repos. Cet influx nerveux se propage de proche en proche le long de l’axone, puis est transféré au
neurone suivant dans la synapse. Le signal électrique y est alors converti en signal chimique : l’axone du neurone présynaptique contient des neurotransmetteurs chimiques qui sont libérés au niveau de la synapse sous l’influence du potentiel d’action. Le neurotransmetteur diffuse dans la fente synaptique et se lie à des récepteurs spécifiques sur le neurone postsynaptique, initiant une potentialisation de la membrane et propageant ainsi le signal.
Par ailleurs on notera que la mémoire et l’apprentissage sont associés à la transmission neuronale par un processus appelé potentialisation à long terme. Ce phénomène consiste en un renforcement de l’activité et de l’efficacité des transmissions synaptiques sur de longues durées, via des changements morphologiques durables de neurones.
I.5.1.2 Les Cellules gliales
Les cellules gliales représentent des composantes majeures du système nerveux central. Elles sont trois fois plus nombreuses que les neurones. Sans être des acteurs directs de la neurotransmission, elles jouent un rôle de soutien qui contribue à la définition des contacts et au maintien des capacités de transmission des neurones.
On trouve dans le système nerveux central plusieurs types de cellules gliales, dont les astrocytes et les cellules microgliales. Les astrocytes assurent le transport, un support mécanique aux neurones et les approvisionnements en nutriments. Ils ont un rôle de digestion et d’élimination de « déchets ». Les cellules microgliales constituent quant à elles la première ligne de défense contre les envahisseurs étrangers, avec un rôle comparable à celui des macrophages.
I.5.2 Dysfonctionnement des systèmes de neurotransmetteurs dans la MA
Certains systèmes de neurotransmetteurs (Cholinergique et glutamatergique) sont particulièrement affectés lors de la MA, en particulier dans les zones du cerveau les plus touchées par la maladie tel que l’hippocampe et le cortex.
I.5.2.1 L’acétylcholine
L’acétylcholine (ACh) est un neurotransmetteur jouant un rôle important dans l’apprentissage et l’attention. Son taux est régulé par deux enzymes. Elle est synthétisée à partir de la choline par la choline acétyltransférase(ChAT) et hydrolysée par l’acétycholine estérase (AchE) (Schéma1-I).
N O O AChE ChAT N OH + O O + H
Schéma 1-I : Libération de la choline à partir de l'acétylcholine acétycholine
choline
+H2O
Dans les parties endommagées du cerveau des malades d’Alzheimer, on observe une perte importante de neurones cholinergiques avec de faibles niveaux d’acétylcholine dans l’espace intersynaptique (Figure 5-I). Ce faible niveau d’acétylcholine ne peut être relié seulement à l’activité de l’acétylcholinestérase. En effet, l’activité de cette enzyme diminue lors de la progression de la maladie. L’existence d’une enzyme de la même famille, la butyrylcholinestérase(BChE) qui reconnait également l’acétylcholine comme substrat et dont l’activité croit au cours de la maladie, pourrait également expliquer ce déficit en acétylcholine.26
Figure 5-I : Représentation schématique du Système cholinergique (d’après Scarpini et al., 2003). ChAT: Choline acétyltransférase, ACh : Acétycholine, AChE : acétylcholinestérase, BChE : butyrylcholinestérase, récepteur de type M : muscariniques et N : nicotiniques.
Le déficit en acétylcholine se traduit par une diminution des fonctions cognitives suite à la décroissance de l’activation des récepteurs cholinergiques.
De plus, la maladie d’Alzheimer présente également une diminution du nombre de récepteurs nicotiniques (Type N) dans le cortex cérébral et l’hippocampe27, ce qui constitue un handicap supplémentaire pour la transmission du signal cholinergique.
I.5.2.2 Le glutamate : phénomène d’excitotoxicité
Le glutamate est un neurotransmetteur excitateur majeur du système nerveux central. Les neurones à glutamate sont impliqués dans le processus cognitif du cortex tel que l’apprentissage et la mémoire, dont le processus de potentialisation à long terme.
Lors de la MA, les dysfonctionnements du métabolisme énergétique et des systèmes de transport de glutamate, ainsi que le stress oxydant conduisent à l’accumulation de ce neurotransmetteur dans les synapses. Or le glutamate endogène induit un effet neurotoxique (appelé excitotoxicité) via l’activation excessive des récepteurs qui lui sont associés, en particulier les récepteurs du NMDA (N-méthyl-D-Aspartate).
Le récepteur du NMDA est couplé à un canal ionique perméable au Ca 2+(et au Na+ de même canal) ( Figure 6-I), Au potentiel de repos de celui-ci, ce canal calcique est bloqué par des ions Mg2+. Une dépolarisation de la membrane permet d’expulser le Mg2+. Dans ces conditions et si le glutamate est fixé sur le recepteur, le canal ioniqe s’ouvre, permettant un influx de Ca2+ dans la cellule. L’hyperactivation des récepteurs du NMDA par le glutamate en excès est accompagné d’une entrée massive des ions calcium dans la cellule neuronale qui est toxique pour celle-ci, comme cela sera évoqué par la suite.28
Figure 3-I : Représentation du récepteur du NMDA activé par le glutamate (d'après Lipton, 2006). "NR1" et " NR2": sous-unités 1 et 2 du récepteur du NMDA. "Glu or NMDA": site de liaison du glutamate ou du NMDA. "Gly": site de liaison activateur de la glycine. "Zn2+": site
I.5.3 Le zinc « Libre » Comme Modulateur
Parmi les espèces libérées au niveau des synapses de l’hippocampe, on trouve également des ions métalliques, en particulier le Zn(II).29
Le Zn(II) « libre » c’est-à-dire rapidement échangeable, est contenu dans les vésicules synaptiques de certains neurones situés principalement au niveau de l’hippocampe. Il est libéré de manière dépendante dans la fente synaptique lors de la transmission du signal nerveux, à des concentrations pouvant atteindre 300 µM. Le Zn(II) peut alors pénétrer dans le neurone post-synaptique par l’intermédiaire de canaux perméable au Zinc29, Il a un rôle de neuromodulateur, permettant le transport de l’information d’une cellule à l’autre. Comme le Zn(II) mais à des concentrations beaucoup plus faible, le Cu(II) échangeable est également libéré dans les fentes synaptiques. Cependant, ce rôle ne semble pas très bien établi.30
1.5.4 Toxicité généré par le dérèglement du calcium
L’ion calcium est le messager intracellulaire le plus important ; il joue un rôle fondamental dans le processus d’apprentissage et de mémoire et est impliqué dans le suivi et a mort neuronale.
L’incapacité des neurones à réguler l’homéostasie du Ca2+ est un aspect de la MA intimement responsable de la mort des neurones.
En effet, des taux élévés de calcium intracellulaire ont de nombreuses conséquences délétères. Ainsi, une concentration de calcium élevée génère un stress via la surconcentration du calcium dans les mitochondries. De plus, une dérégulation de l’homéostasie calcique à un impact sur les fonctions neuronales dépendant du calcium telle que la libération des neurotransmetteurs et la plasticité synaptique.
Enfin, l’excès de Ca2+ intracellulaire cause une activation de kinase, des caspaces et d’autres facteurs d’induction d’apoptose, conduisant à la perte des neurones.31
1.5.5 Inflammation
La mort des cellules dans la MA est accompagnée de phénomènes inflammatoires, les marqueurs de cette inflammation étant significativement plus élevés dans le cerveau des malades que chez les contrôles, (par exemple NFkB…..) et de la production d’espèces oxygénées réactives à la suite de l’activation du complément.
Les cellules microgliales et les astrocytes sont alors à l’origine de la libération de médiateurs inflammatoire (cytokine, Chémokines, facteur de croissance, facteur de transmission, tout ce phénomène issu du processus d’inflammation peut faciliter le développement de stress oxydant et / où induire l’apoptose.32
I.6 Les Lésions Caractéristiques de la Maladie D’Alzheimer
La maladie d’Alzheimer est caractérisée par deux types de lésions cérébrales : les plaques séniles extracellulaires (plaques amyloïdes) et les enchevêtrements neurofibrillaires intraneuronaux, i.e. «neurofibrillary tange en aglais » (dégénérescence neurofibrillaire) (Figure 7 -I).
Figure 7-I : Image de microscopie fluorescente sur des sections de cortex frontal (A) patient atteint de la maladie d’Alzheimer (B) patient âgé non atteint de démence (d’après Mathis et al., 33),coloration à la Thioflavine T .
La caractérisation de ces deux types de lésions avec certitude ne peut se faire à l’heure actuelle sur le cerveau des patients que post mortem. Des détections in vivo pourraient être effectuées en utilisant des techniques d’imagerie médicale comme la TEP (Tomographie par Emission de positons) la SPECT (Tomographie par Emission Monophotonique) ou l’IRM (L’Imagerie par Résonnance Magnétique). Nous aurons l’occasion de revenir sur ces techniques.
I .6.1 Dégénérescences neurofibrillaires
Les enchevêtrements de neurofibrilles sont constitués d’agrégats de protéines tau sous forme hyperphosphorylée.34 Ce type de lésions impliquant la protéine tau n’est pas spécifique à la maladie d’Alzheimer, il est également retrouvé dans d’autres maladies
neurodégénératives regroupées sous le terme "taupathies", incluant le syndrome de Down, la démence fronto-temporale avec Parkinsonisme liée au chromosome 17 (FTDP-17), la maladie de Pick ou encore la paralysie supranucléaire progressive. 35
La protéine tau est responsable de l’assemblage et de la stabilisation des microtubules, composants essentiels du cytosquelette des neurones. Dans son état natif, tau est une protéine comprenant entre 352 et 441 acides aminés (de masse 55 à 62 kDa) et existe sous six isoformes produites à partir d’un gène unique situé sur le chromosome 17.
Des mutations génétiques sur ce chromosome conduisent à la démence frontale temporale avec Parkinsonisme (FTDP-17), mais aucune mutation conduisant à la maladie d’Alzheimer n’a été répertoriée à ce jour.35
Ces protéines ont la particularité de posséder dans leur séquence trois à quatre répétitions d’un même domaine qui permet la liaison aux microtubules. Cette liaison est par ailleurs fortement régulée par la phosphorylation de la protéine au niveau de résidus sérine et thréonine.
La protéine tau pathologique est hyperphosphorylée. Suite à cette hyperphosphorylation, tau se dissocie des microtubules36, compromettant ainsi leur stabilisation et leur fonctionnement. Cela conduit à une altération des transports axonaux et dendritiques ainsi qu’à une dégénérescence des neurones. De plus, la protéine hyperphosphorylée libérée montre une solubilité nettement plus faible. Elle est aussi plus sensible aux modifications chimiques induites par le stress oxydant et subit des changements conformationnels suite à ces oxydations. Elle s’agrège alors, selon un processus de polymérisation avec une étape de nucléation 37, sous forme de paires hélicoïdales de filaments (PHFs: "paired helical filaments"). Ces dernières s’associent ensuite en structures plus grandes : les neurofibrilles. Par ailleurs, il est également suggéré que les ions AlIII et FeIII, retrouvés liés à tau dans des cerveaux de patients post mortem, soient impliqués dans la polymérisation des protéines tau hyperphosphorylées. 38, 39
Ces structures en filaments hélicoïdaux présentes dans le cytoplasme sont responsables de neurodégénérescences. Des modèles de protéines tau hyperphosphorylées sont toxiques sur cultures cellulaires.40 De plus, le nombre et la localisation des enchevêtrements de neurofibrilles dans le cerveau post-mortem ont pu être corrélés avec le
niveau de démence, alors qu’une telle relation n’existe pas dans le cas des plaques amyloïdes.
41
I.6.2 Plaques séniles (plaque amyloïdes)
Les plaques séniles sont des lésions extracellulaires, sous la forme d’agrégats sphériques denses de protéines organisées en fibrilles. Le peptide β-amyloïde (Aβ) en est le composant principal. Ces lésions extra cellulaires sont présentes principalement dans les régions limbiques du cerveau tel que l’hippocampe et le complexe amygdalien, ainsi que dans des régions corticales et subcorticales spécifiques.
Le peptide Aβ contient de 39 à 43 acides aminés. Les formes les plus abondantes sont celles avec 40 et 42 acides aminés (Aβ40 et Aβ42).
Ces deux formes jouent un rôle important dans la maladie d’Alzheimer, mais, le peptide Aβ42 est considéré comme plus neurotoxique que la forme Aβ40 par sa propriété plus insoluble et oligomérisante. 42
Il est important de noter que des agrégats de type amyloïde impliquant d’autres protéines se retrouvent également dans des maladies aussi différentes que le diabète de type 2, la maladie de Huntington ou les maladies liées aux prions.
I.6.3 Relation entre tau et Aβ
La relation entre les lésions de la maladie d’Alzheimer observées histologiquement (plaques amyloïdes et enchevêtrements de neurofibrilles) et les composants qui leur sont associés (Aβ et tau) a longtemps été controversée. De même, l'origine du processus neurodégénératif a fait l’objet d’un long débat entre chercheurs : elle pourrait être due à la production anormale d'Aβ ou à la dégénération du cytosquelette neuronal causée par tau. Récemment des études ont montré plutot une hierachisation dans le sens Aβ vers tau.
Parmi les kinases qui entrent en jeu dans la phosphorylation de tau, deux kinases sont importantes dans la pathologie d’Alzheimer : La 3γ-kinase synthase glycogène (GSK-3) et le mitogène activé protéine kinase (MAPK). Ces deux kinases sont capables de phosphoryler tau et sont colocalisées avec les neurofibrilles dans le cerveau des patients atteints. Des travaux montrent qu’une stimulation de MAPK par Aβ peut augmenter la phosphorylation de tau. L’activation de GSK-3 suite à l’agrégation d'Aβ induit aussi l’hyperphosphorylation de tau, et conduit consécutivement à la mort neuronale. Une étude sur des cultures neuronales de rats a
également décrit que le peptide Aβ induit non seulement l’hyper phosphorylation de tau, mais aussi diminue l’activité de la choline acétyl transférase. 43
Plus récemment, des études réalisées sur des souris triplement transgéniques [APP × PS1 × tau] ont égalemment montré le lien hiérarchique entre Aβ et tau36 L'emploi d'anticorps spécifiques a montré que ces souris développent les pathologies associées à Aβ et à tau, mais de façon non synchrone. En effet, la détection d'Aβ intracellulaire est possible dès trois mois et celle des dépôts amyloïdes extracellulaires vers six mois alors que les premiers changements conformationnels de tau ne sont visibles qu'au bout de dix à douze mois et l'hyperphosphorylation de tau seulement à partir de douze à quinze mois44 Toutefois, il faut noter que tau lui-même est essentiel pour la neurotoxicité associée à Aβ. Ainsi, dans un modèle cellulaire en présence d'Aβ fibrillaire, on observe une dégénérescence des neurones isolés de souris transgéniques surexprimant tau (humain ou de souris), alors que les cellules issues d’animaux délétères pour tau ne dégénèrent pas. Cette dégénérescence est rétablie suite à la réexpression de tau. 45 Le groupe de Laferla et Oddo a montré que le taux d’Aβ soluble et de tau (et pas seulement de Aβ), aggrave le déclin cognitif dans des souris transgéniques modèles ayant des plaques et des neurofibrilles.
De manière globale, le peptide Aβ et ses différentes formes agrégées sont à l'origine d'un niveau élevé de stress oxydant qui va engendrer, entre autres phénomènes complexes, la stimulation des kinases et ainsi conduire à l'hyperphosphorylation de tau.
I.6.4 Hypothèse de la cascade amyloïde
La maladie d’Alzheimer est une maladie complexe et multifactorielle, qui touche principalement les synapses de certains neurones situés dans l’hippocampe et le cortex. C’est au niveau de ces zones que les principaux acteurs de la maladie exercent leurs effets délétères (Figure 8-I).
Figure 8-I : Localisation des principaux facteurs conduisant à la maladie d'Alzheimer (d'après Mattson, 2004). ER: réticulum endoplasmique.
L’hypothèse de la cascade amyloïde (Figure 9-I) fait intervenir l’Aβ comme le déclencheur de tous les cas d’Alzheimer, plaçant les pathologies associées à tau, ainsi que d’autres modifications dégénératives, en aval de celui-ci46-48.
Figure 9-I : Hypothèse de la cascade amyloïde pour les formes familiales de la maladie d’Alzheimer (d’après Cai et al., 2007).
Selon cette hypothèse, l'origine de la dégénérescence neuronale et synaptique observée dans la maladie d'Alzheimer provient d’une protéolyse altérée de la protéine APP. Des causes
génétiques et environnementales (le taux de cholestérol par exemple) et une altération générale du fonctionnement du cerveau liée à l'âge sont responsables de l'augmentation de la production d'Aβ (et plus particulièrement de sa forme la plus toxique, Aβ42). Celui-ci est alors libéré au niveau de la membrane présynaptique des neurones, où l'APP est accumulé.
Dans le cas des neurones à acétylcholine, la présence de l'acétylcholine estérase dans cette zone favorise la fibrillation d'Aβ et la formation de plaques amyloïdes. De même, les ions métalliques ZnII et CuII libérés lors de la transmission synaptique causent l'agrégation du peptide au niveau des synapses.
Les ions CuII libérés peuvent aussi être à l'origine de toxicité, via l’induction de stress oxydant. Celui-ci conduit en particulier à l'oxydation des membranes et à la perte de leur intégrité.49
L'interaction directe du peptide Aβ hydrophobe avec ces membranes et sa pénétration entraîne également la perméation de celles-ci. Il en résulte une augmentation du flux de Ca2+ à l'intérieur de la cellule neuronale, entraînant de nombreux effets délétères. Elle génère du stress oxydant supplémentaire.
Cette augmentation active également des kinases, conduisant à l'hyperphosphorylation de tau. Il s'en suit une déstructuration des axones et une perte des synapses qui conduit à l'altération de la mémoire et des fonctions cognitives. Une concentration intracellulaire de Ca2+ excessive peut également déclencher des facteurs d'induction d'apoptose et conduire à la mort neuronale.
D’autre part, la dérégulation des synapses perturbe le fonctionnement des systèmes de neurotransmetteurs cholinergiques et favorise ainsi la production supplémentaire d'Aβ et son accumulation. Les dysfonctionnements synaptiques et le stress oxydant augmentent par ailleurs l'influx de Ca2+ dans la cellule au niveau des neurones à glutamate, via la suractivation des récepteurs au NMDA. Ce phénomène conduit à une toxicité accrue au niveau de ce type de neurones.
Enfin, l'inflammation déclenchée suite à ces évènements produit davantage de stress oxydant et accentue la toxicité générale.
I.7 Implication des métaux
Parmi tous les facteurs biologiques en relation avec la maladie d’Alzheimer, les métaux tiennent une place essentielle. Ils sont impliqués dans de nombreuses autres maladies neurodégénératives comme la maladie de Parkinson, la maladie de Huntington, la maladie de Creutzfeldt-Jacob, la maladie de Wilson, la sclérose latérale amyotrophique ou la maladie du prion. Dans le cas de la maladie d’Alzheimer, de nombreuses études ont montré que le métabolisme des ions métalliques est altéré. Les concentrations de cuivre, de zinc et de fer sont plus élevées que la normale, et plus particulièrement dans les lésions caractéristiques de la maladie (plaques amyloïdes) et à proximité de celles-ci. 50, 51
Les métaux jouent donc certainement un rôle non négligeable dans la Maladie d’Alzheimer. L’intérêt pour l’étude des ions métalliques en relation avec la pathologie est donc croissant.
II Les plaques séniles
II.1 Origine de l’Aβ: la protéine APP
II.1.1 La protéine APP
Le gène des protéines APP est situé sur le chromosome 21; son épissage alternatif conduit à la formation de plusieurs ARN messagers, puis de glycoprotéines dont 3 isoformes principales ayant une longue partie N-terminale située dans la partie extracellulaire, une partie transmembranaire et une courte région C-terminale située dans le cytoplasme. Le nombre d’acides aminés de ces protéines est variable (entre 695 et 770).
Elles sont exprimées dans tous les tissus, mais les neurones expriment plus particulièrement la protéine APP695 que l’on trouve en forte quantité dans le cerveau.52 En raison de l’évidence de la perturbation du métabolisme de l’APP, de nombreuses recherches se sont focalisées sur la compréhension des rôles physiologiques normaux de l’APP et de ses dérivés. 53
L’APP est exprimé de façon ubiquitaire dans l’organisme, mais ses fonctions physiologiques ne sont pas encore bien connues. Cependant, des travaux de Grimm et al., 54 ont récemment montré que ces peptides (Aβ) réguleraient le métabolisme du cholestérol et des sphingomyélines par un processus impliquant le clivage de l’APP.