Pour l'obtention du grade de
DOCTEUR DE L'UNIVERSITÉ DE POITIERS UFR des sciences fondamentales et appliquées
Laboratoire Signalisation et transports ioniques membranaires - STIM (Poitiers) (Diplôme National - Arrêté du 7 août 2006)
École doctorale : Biologie-santé - Bio-santé (Limoges)
Secteur de recherche : Aspects moléculaire et cellulaire de la biologie Cotutelle : Université libanaise
Présentée par : Ferdos Alaa El Din
Le syndrome de Rendu-Osler-Weber :
aspects génétiques, moléculaires et épidémiologiques
Directeur(s) de Thèse : Alain Kitzis, Raghida Abou Merhi Soutenue le 12 juin 2015 devant le jury Jury :
Président Myrna El Hachem Professeur, Université libanaise
Rapporteur Alain Calender Professeur des Universités, Université de Lyon 1
Rapporteur Georges Nemer Professeur associé, Université américaine de Beyrouth, Liban Membre Alain Kitzis Professeur des Universités, Université de Poitiers
Membre Raghida Abou Merhi Professeur, Université libanaise Membre Sylvie Patri Professeur, CHU de Poitiers
Pour citer cette thèse :
Ferdos Alaa El Din. Le syndrome de Rendu-Osler-Weber : aspects génétiques, moléculaires et épidémiologiques [En ligne]. Thèse Aspects moléculaire et cellulaire de la biologie. Poitiers : Université de Poitiers, 2015. Disponible sur l'Intranet de l'Université de Poitiers <http://theses.univ-poitiers.fr>
Thèse en cotutelle
Pour obtenir le grade de Docteur Délivré par
L’Université de Poitiers
(École Doctorale Biologie et Santé) et
L’Université Libanaise
(École Doctorale des Sciences et Technologie) Spécialité : Génétique et Maladie humaine
Présentée par
Ferdos ALAA EL DIN
Le syndrome de Rendu-Osler-Weber :
Aspects génétiques, moléculaires et épidémiologiques
Soutenue publiquement le 12 Juin 2015 devant le jury composé de
M. Alain KITZIS, MD-PhD, Université de Poitiers Directeur
Mme Raghida ABOU MERHI, Professeur, Université Libanaise Co-Directeur Mme Myrna MEDLEJ-HASHIM, Professeur,Université Libanaise Examinateur
Mme Sylvie PATRI, PhD,CHU de Poitiers Examinateur
M. Alain CALENDER, MD-PhD, Université de Lyon 1 Rapporteur M. George Nemer, Professeur associé, Université américaine de Beyrouth Rapporteur
I
Aspects génétiques, moléculaires et épidémiologiques
Résumé
La télangiectasie hémorragique héréditaire (HHT) est une maladie rare (1/10.000). Son incidence est plus élevée (pouvant atteindre 1/1000) dans certaines zones géographiques dont la région Poitou-Charentes. Cette maladie autosomique dominante est causée par des mutations d’un des trois gènes identifiés ENG, ACVRL1 et SMAD4 codant pour des protéines de la voie BMP spécifiquement exprimés dans les cellules endothéliales. Le nombre croissant de mutations détectées chez les patients et l’expressivité variable de certaines mutations nous a ammené à déterminer les conséquences de mutations afin d’établir une corrélation génotype/phénotype. Cette corrélation est importante pour le conseil génétique et évidemment le diagnostic prénatal. Dans ce contexte, nous avons étudié aux niveaux cellulaire et moléculaire les effets de plusieurs mutations. L’effet délétère de ces mutations sur la protéine et/ou l’épissage de l’ARN a été évalué. Nous avons montré que sur les 23 mutations d’ACVRL1 : 1) 18 mutations faux-sens affectent la fonctionnalité de la protéine en réponse à BMP9 et 3 mutations sont de simples polymorphismes, 2) la mutation exonique c.733A>G (p.Ile245Val) affecte l’épissage de l’exon 6, 3) La mutation c.1048+5G>A de l’intron 7 en dehors du site consensus induit un épissage aberrant de l’exon 7. En ce qui concerne l’ENG, nous avons analysé 4 mutations et nous avons montré que la mutation c.1088G>A (p.Cys363Tyr) a un impact sur l’activité du récepeteur et que les mutations c.1134G>A (p.Ala378Ala) et c.1060C>T (p.Leu364Leu) altèrent l’épissage de l’exon 8. Ce travail montre l’importance de l’étude approfondie de toute nouvelle mutation par des études in silico, in vitro et in cellulo à différents niveaux cellulaires. Des études in vivo ultérieures peuvent compléter et appuyer la stratégie expérimentale que nous avons suivie.
Mots clés : Télangiectasie hémorragique héréditaire, mutations, fonctionnalité, épissage, in silico, in cellulo, in vivo.
II
Rendu-Osler-Weber syndrome:
Genetic, molecular and epidemiological aspects
Abstract
Hereditary hemorrhagic telangiectasia (HHT) is a rare disease (1/10.000). Its incidence is higher in certain geographic areas including the Poitou-Charentes region (1/1000). This autosomal dominant disease is caused by mutations in one of three identified genes ENG, ACVRL1 and SMAD4 encoding BMP pathway proteins specially expressed in endothelial cells. The increasing number of mutations detected in patients and the variable expressivity of certain mutations has taken us to determine the consequences of mutations to establish a genotype/phenotype correlation. This correlation is important for genetic counseling and obviously for prenatal diagnosis. In this context, we investigated the effects of several mutations at the cellular and molecular levels. The deleterious impact of these mutations on the protein and/or RNA splicing was evaluated. We have shown that for the 23 mutations of ACVRL1: 1) 18 missense mutations affect the functionality of the protein in response to BMP9 and 3 are polymorphisms, 2) exonic mutation c.733A>G (p. Ile245Val) affects the splicing of the exon 6, 3) mutation c.1048+5G>A in intron 7 off the consensus site induces an aberrant splicing of exon 7. Concerning the ENG, we analyzed 4 mutations and we showed that the mutation c.1088G> A (p.Cys363Tyr) has an impact on the activity of the receptor and that the mutations c.1134G> A (p.Ala378Ala) and c.1060C> T (p.Leu364Leu) inhibit the splicing of exon 8. This work shows the importance of the comprehensive study of any new mutation by in silico, in vitro and in cellulo studies at different cellular levels. The in vivo studies can further complement and support the experimental strategy that we followed.
Keywords: Hereditary hemorrhagic telangiectasia, mutations, functionality, splicing, in silico, in cellulo, in vivo.
III
Ce travail a été effectué sous la co-direction de l’université libanaise et l’université de Poitiers et a été soutenu financièrement par LASER (Lebanese Association for Scientific Research), AMRO France- HHT (Association française de la Maladie de Rendu-Osler-Weber), l’équipe de génétique des maladies rares- CHU de Poitiers et l’équipe de recherche ER031 de génomique et santé à l’université libanaise. Je suis extrêmement reconnaissante à l’effort commun de tous les collaborateurs qui a aboutit à la réalisation de ce projet.
Je tiens à remercier sincèrement, mon directeur de thèse en France Pr. Alain KITZIS. Je vous remercie, monsieur, de m'avoir guidé tout au long de ces quatre années de thèse. Vous m’avez reçu dans votre laboratoire et je crois que vous m’avez passé votre passion pour la génétique. Vous étiez toujours la principale référence scientifique pour mon travail et vous m’avez aidé sans cesse pour résoudre les différents problèmes que nous avons affrontés. Votre soutien précieux et votre encouragement m’ont motivé dans mon projet pour être toujours à la hauteur de vos attentes. Merci aussi de m'avoir donné la possibilité de développer mes connaissances et mes compétences en m'autorisant à suivre de multiples formations et réunions et assister au cours pour consolider et élargir mes idées.
Je remercie grandement mon co-directeur au Liban Pr. Raghida ABOU MERHI. J’avais l’honneur de vous connaître pendant mon cursus universitaire et j’étais toujours attirée par votre dynamisme et votre engagement professionnel. C’était un privilège de travailler avec vous. Vous étiez toujours à mes côtés à chaque fois j’avais besoin de l’aide. Vos commentaires et vos suggestions étaient bénéfiques sur les plans personnel et scientifique. J’apprécie tout ce que vous avez fait pour m’introduire dans le monde de recherche et je tiendrai à cœur tous vos conseils et vos directives.
Je remercie ardemment, Dr. Sylvie PATRI. Je suis très reconnaissante pour ton orientation et ton soutien. J’apprécie bien ta patience, ta disponibilité même dans les moments où tu étais accablée du travail. Ton expertise technique était bénéfique pour la réussite des manipulations. Ta présence m’a beaucoup rassuré au cours des années de la thèse et j’ai bien profité de tes remarques avisés et tes conseils techniques.
Un remerciement spécial pour mes collègues et mes amis Dr. Vincent THOREAU et Dr. Montserrat Rodriguez. J’avais de la chance de vous connaître au laboratoire. Vous étiez
IV
toujours disponibles et prêts à donner rapidement toute aide et tout éclaircissement indispensables aux techniques. Votre contribution était un des piliers pour la réussite du travail. J’ai une reconnaissance profonde pour votre partage d’expérience qui m’a soutenu moralement et scientifiquement. Vous m’avez accueilli tout le temps, même à court préavis ou discussion. Je suis contente de vous connaître.
Je remercie ma collègue au laboratoire Valérie Lepine. Ta vivacité d’esprit et ton habilité d’encourager les gens autour de toi m’a fortement soutenu moralement et j’ai profité plus ou moins de ton sens d’organisation.
Un grand merci pour tous les membres du laboratoire à Poitiers. Vous m’avez bien accueilli parmi vous et vous étiez une source de force pour dépasser les difficultés que j’ai eues. Vous étiez une superbe famille qui a toujours écouté mon état d’âme et m’a encouragé. J’apprécie votre soutien et votre encouragement. Nous avons partagé des beaux moments de convivialité qui on dissipé la sensation d’être une étrangère. J’ai admiré votre dévouement pour le travail et la bonne ambiance que vous veillez à la garder au laboratoire. Je n’oublierai jamais les bons souvenirs avec vous. Merci pour les étudiants qui ont fini leurs stages ou leurs thèses, je remercie Ghina, Raed, Ayman et mes amis au Pôle Biologie Santé.
Je remercie les membres du jury, Pr. Georges NEMER, Pr. Myrna MEDLEJ-HASHIM, Pr. Alain CALENDER de me faire l'honneur d'évaluer mon travail de thèse. Vos suggestions précieuses et vos commentaires ont amélioré considérablement ma thèse.
Je remercie la plateforme EDST/PRASE représenté par Pr. Bassam BADRAN pour les facilités du travail qu’il nous en fourni. Je remercie aussi le personnel de l’EDST qui fait leur meilleur pour faciliter notre travail. Je remercie les membres de l’équipe ER031, Jihane, Imane, Amani et Louna pour votre aide dans les expériences au Liban.
Finalement, toute ma reconnaissance à mon père Mohammad et ma mère Siham pour vos sacrifices et votre soutien. Je suis fière d’être votre fille, vous étiez toujours formidables, patients et à mes côtés. Je remercie mes sœurs Lara et Sara, mes frères Ali, Jamil et Walid. Vous êtes magnifiques et vous formez une très belle famille et je souhaite que vous atteigniez tous vos objectifs dans la vie. Je remercie de même ma large famille bien aimée.
V
Résumé ……….….I Remerciements………...………..III Liste des Figures ………..…..………..………....X Liste des Tableaux………..………..……….………....XII Abréviations……….………..XIII Liste des activités atteintes pendant la thèse………..……...….XVII
Chapitre 1. REVUE DE LA LITTÉRATURE………..………1
Avant propos……….2
I- LA MALADIE DE RENDU-OSLER-WEBER ………..………..2
1- Définition………2
2- Symptômes et diagnostic clinique……….…...………...…….2
3- Epidémiologie...………..………….….4
II- LA GÉNÉTIQUE DE HHT………..………..…….7
1- Le gène Endogline (ENG)………...………....8
1-1 La structure de l’ENG………..……….8
1-2 Fonction de l’ENG………..………...11
2- Le gène ACVRL1 (ALK1)………...12
2-1 La structure d’ALK1………...………....…12
2-2 Fonction d’ALK1………....14
3- Le gène MADH4 ou Smad4………....16
3-1 La structure de Smad4 …....……….………..………..16
3-2 Mutations de Smad4……….17
3-3 Fonction de Smad4………..…….18
4- Les théories de « Second Hit » et « LOH »……….……...18
VI
III- VOIE DE SIGNALISATION ENG/ALK1/Smad4………..……….…23
1- La signalisation d’ALK1……….24
1-1 Les ligands d’ALK1………..………..24
1-1-1 Le ligand BMP9………...………..………25
1-1-2 BMP10 : ligand physiologique d’ALK1 peu connu………...…………27
1-2 Récepteur de type II spécifique d’ALK1……….……28
1-3 Réponse intracellulaire………...………..29
1-4 Gènes cibles transcriptionnels impliqués dans la voie BMP9/ALK1/Smad 1,5,8…………...…....30
2- La signalisation d’ENG……….………..31
3- HHT est une maladie BMP dépendante………..……..32
IV- LA PHYSIOPATHOLOGIE DE HHT……….…………...…………...…35
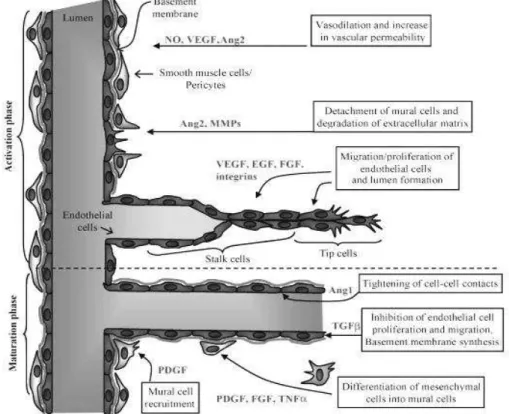
1- L’Angiogenèse………..36
1-1 La phase d’activation...……….……….…………..36
1-2 La phase de maturation……….37
2- Rôle de la voie de signalisation BMP9/ALK1/ENG/ Smad 1,5,8…………...…….38
2-1 Fonction vasculaire du récepteur ALK1………...…………38
2-2 Fonction vasculaire du BMP9 et BMP10………...40
2-3 Fonction vasculaire du récepteur d’ENG………….……….…………..………41
2-4 Invalidation du récepteur BMPR2………...42
2-5 Invalidation de Smad1/5/8/4……….………42
2-6 Invalidation des gènes Id1 et Id3………..………43
3- HHT est dû à un déséquilibre de l’angiogenèse………....……....43
4- Modèles moléculaires et cellulaires proposés pour les MAVs observées dans HHT……….45
5- Traitement de HHT………...…….48
V- OBJECTIFS DU PROJET………..………..52
Objectif principal de la thèse……….52
VII
1- Introduction……….56
1-1 Etudes des mutations d’ACVRL1 répertoriées dans la littérature………..……....56
1-2 Test de diagnostic des mutations faux-sens d’ACVRL1 dans HHT2………..…..….57
1-3 Mutations d’épissage dans ACVRL1………..…….………58
1-4 Epissage alternatif……….…………..……….………58
1-5 NMD (Non-sens Mediated mRNA Decay)………..……….…...60
2- Objectif……….………65
3- Matériels et Méthodes………..……….65
3-1 Culture cellulaire……….……….………..………..65
3-2 Les plasmides……….……….……….…..…………..………66
3-3 Extraction d’ADN des patients………...66
3-4 Séquençage et détection des mutations………...……….67
3-5 Test fonctionnel………...………...………..68
3-5-1 Mutagenèse dirigée……….……...…...…….68
3-5-2 Test fonctionnel des mutations faux-sens d’ACVRL1………..…...68
3-6 Western blot………..………70
3-7 Immunocytochimie et microscopie à fluorescence……….………….71
3-8 Biotinylation de surface……….……….…..72
3-9 Etude d’épissage……….….……….…73
3-9-1 Amplification des régions d’ADN d’intérêts……….….74
3-9-2 Purification du produit PCR……….…………...………...…75
3-9-3 Vérification sur gel……….…...………..………..76
3-9-4 Procédure de clonage dans pSpliceExpress………..………..….…………..76
3-10 Programmes informatiques (Etudes in silico)………...….80
4- Résultats et Discussion……….………..……….80
4-1 Les mutations étudiées d’ACVRL1………….……….………80
VIII
4-3 Etude de la maturation des récepteurs mutés………...………...……….…87
4-4 Etude de la localisation des récepteurs par microscopie confocale………..…….………..87
4-5 Etude de la localisation de certains mutants d’ALK1 par biotinylation de surface……….87
4-6 Etude d’épissage des mutations d’ACVRL1………...……….…81
4-6-1 Etude de la mutation intronique………..…..………81
4-6-2 Etude de la mutation c.733A>G (p.Ile245Val)………...………..91
4-6-3 Etude de la mutation c.1249A>T (p.Ile417Phe)……….……..94
4-6-4 Etude d’épissage de certaines mutations exoniques d’ACVRL1……….……….94
2- Conclusion………..………...……….98
Chapitre 3. ETUDE DES MUTATIONS D’ENG……….………...……..100
1- Introduction ………...101
1-1Etudes des mutations d’ENG répertoriées dans la littérature………....101
2- Objectif………..…….103
3- Matériels et Méthodes……..………..………...103
3-1Test de diagnostic des mutations faux-sens d’ENG dans HHT1…….………...103
3-2 Etude d’épissage……….104
4- Résultats et Discussion………..…104
4-1 Etude fonctionnelle de la mutation faux-sens c.1088G>A (p.Cys363Tyr)……….….…..104
4-2 Etude d’épissage des mutations d’ENG………...105
5- Conclusion………...109
Chapitre 4. ETUDE D’EXPRESSION DES GÈNES DANS LE SANG……….110
1- Introduction……….………..111
2- Objectif………...111
3- Matériels et Méthodes………...111
3-1 Préparation des organes………..………..…....112
3-2 Extraction des ARN totaux………..………..……112
3-3 Réverse transcription (RT)………...………..…112
IX
Chapitre 5. CONCLUSION GÉNÉRALE ET PERSPECTIVES……….……...117
1- Conclusion générale……….……….118
1-1 La mutabilité d’ACVRL1……….…..118
1-2 Démarche d’étude(s) d’une mutation………..………...…119
1-3 Une mutation pourrait avoir des conséquences sur plusieurs mécanismes cellulaires?...120
1-4 Le modèle d’haploinsuffisance……….………..…121
1-5 Fiabilité des outils informatiques………..………...……..121
2- Perspectives……….…………...122
2-1 Etudes in vivo………..……….…..122
2-2 Etudes biologiques……….123
2-3 Test diagnostique pour HTAP………123
2-4 Test diagnostique pour la polypose juvénile………...…………..…….123
2-5 Séquençage de BMP9 par NGS………...……….…..124
2-6 Etude de l’effet fondateur………..……….…124
ANNEXE………....125
X
Liste des Figures
Figure 1 : Symptômes de la maladie de Rendu-Osler-Weber ……….………...…3
Figure 2 : Epidémiologie de la maladie de Rendu-Osler-Weber……….………....5
Figure 3 : Structure de l’ADNc de l’ENG………...………...……….9
Figure 4 : Structure de l’endogline………...………...………..10
Figure 5 : Invalidation totale de l’ENG chez les souris……...………..11
Figure 6: Modèle d’activation de l’eNOS en condition normale et en cas d’absence de l’endogline...12
Figure 7 : Structure de l’ADNc de l’ACVRL1………...………13
Figure 8: Structure de la protéine d’ALK1………....14
Figure 9 : Invalidation totale d’ACVRL1chez la souris………....15
Figure 10: Structure typique des Smads………...……….17
Figure 11 : Maturation de BMP9 et BMP10 et leur activité……….……….27
Figure 12 : Modèle de signalisation d'ALK1 avec TGF-β comme ligand……….…………30
Figure 13 : Effet différentiel des deux isoformes de l'endogline sur la signalisation………....…32
Figure 14: La voie de signalisation ENG/BMP9/ALK1/Smads 1,5,8 impliqués dans HHT…….…...34
Figure 15 : Les deux phases de l'angiogenèse. ………...……...………...38
Figure 16 : Rôles des deux sous-groupes de BMP dans la balance angiogénique………....…44
Figure 17 : Dérégulation de la balance angiogénique dans la maladie de Rendu-Osler-Weber…..….45
Figure 18 : Modèle hypothétique du rôle de l’endogline dans l’apoptose des cellules endothéliales et la génération des MAVs dans HHT1………...…………...46
Figure 19 : Concept de traitement des vaisseaux anormaux d’HHT..………..………….51
Figure 20 : Réactions de trans-esterification………...………..…59
Figure 21: Schéma positionnant les principales protéines sur le pré-ARNm, ainsi que les séquences en cis les plus déterminantes pour la réaction d’épissage………...………60
Figure 22 : Assemblage du complexe de surveillance………...………63
Figure 23: Modèle des voies de dégradation des ARNm soumis au NMD………..64
Figure 24 : Principe de la méthode de la mutagenèse dirigée du kit QuikChange® II XL Site-Directed Mutagenesis………..……….…...….68
XI
Figure 27 : Représentation schématique de la localisation des 14 nouvelles mutations au niveau de la
protéine ALK1………..…….81
Figure 28 : Représentation schématique de la localisation des 8 mutations déjà décrites au niveau de la protéine ALK1 ……….…….82
Figure 29 : Réponse au BMP9 des protéines produites par les plasmides portant les 14 mutations faux-sens nouvelles…………...……….……….……84
Figure 30 : Réponse au BMP9 des protéines produites par les plasmides portant les 8 mutations faux-sens décrites dans la base de données………...……….……….84
Figure 31 : Analyse de la maturation des protéines ALK1 (WT et mutées) ………..………..85
Figure 32 : Analyse de la maturation des protéines ALK1……….………..…85
Figure 33 : Conservation des acides aminés affectés par les mutations étudiées chez plusieurs espèces...132
Figure 34 : Localisation des récepteurs produits par les mutations étudiées……..……….135
Figure 35 : Résultat de biotinylation réalisée pour certaines protéines mutées afin de préciser leur localisation cellulaire………….……….88
Figure 36 : Résultat de RT-PCR de la mutation c.1048+1G>A………90
Figure 37 : Résultat de l’étude d’épissage de la mutation c.1048+5G>A ………91
Figure 38 : Prédiction du programme PolyPhen 2 concernant l’effet sévère de la mutation p.Ile245Val sur la protéine ALK1………...……...………92
Figure 39a : Résultat de RT-PCR de la mutation c.733A>G (p.Ile245Val)……….…………...92
Figure 39b : Résultat de RT-PCR de la mutation c.733A>G (p.Ile245Val)…….………..93
Figure 40 : Résultat de RT-PCR de la mutation c.1249A>T (p.Ile245Phe). ……….……….94
Figure 41 : Résultat de RT-PCR des mutations de l’exon 8 d’ACVRL1……….………..96
Figure 42 : Résultat de RT-PCR des mutations de l’exon 3 d’ACVRL1……….………..98
Figure 43 : Résultat de RT-PCR des mutations de l’exon 8 d’ACVRL1……….…………..98
Figure 44 : Réponse au BMP9 du récepteur ENG codé par les plasmide portant la mutation faux-sens c.1088G>A (p.Cys 363Tyr)……….……….105
Figure 45 : Résultat de RT-PCR des mutations de l’exon 8 d’ENG…….……...………...……108
Figure 46 : Expression d’ALK1 chez la souris ………...………115
XII
Liste des Tableaux
Tableau 1 : Séquences des amorces utilisées dans la réaction de mutagenèse dirigée pour reproduire
les 14 nouvelles mutations………...………130
Tableau 2 : Séquences des amorces utilisées dans la réaction de mutagenèse dirigée pour reproduire
les 8 mutations déjà décrites………...………...131
Tableau 3 : Séquences des amorces utilisées pour la technique du minigène pour les mutations
d’ACVRL1………..………....131
Tableau 4a: Résultat du site HSF pour l’étude de la mutation c.1048+1G>A………..………….89 Tableau 4b : Résultat du site HSF pour l’étude de la mutation c.1048+5G>A………..…….90 Tableau 5 : Résultat du site HSF pour l’étude de la mutation c.733A>G……….………..92 Tableau 6: Tableau obtenu par le site HSF pour l’étude de certaines mutations faux-sens au niveau de
l’exon 8 d’ACVRL1……….95
Tableau 7: Tableau obtenu par le site HSF pour l’étude de certaines mutations faux-sens au niveau de
l’exon 3 d’ACVRL1……….……….………..……….…97
Tableau 8: Significativité fonctionnelle de 22 mutations faux-sens d’ACVRL1………..….99
Tableau 9 : Séquences des amorces utilisées pour la mutation dirigée de c.1088G>A (p.Cys 363Tyr)
d’ENG………...……….…………...136
Tableau 10 : Séquences des amorces utilisées pour la technique du minigène des mutations
d’ENG……….…136
Tableau 11: Tableau obtenu par le site HSF pour l’étude des 4 mutations faux-sens au niveau de
XIII ACVRL1 (ALK1): Activin Receptor Like Kinase 1) ADN : Acide désoxyribonucléique
ARNsn : small nuclear RNA
BAEC : cellules endothéliales aortiques bovines BH4 : tétrahydrobipterine 4
BMP : Bone Morphogenetic Protein BPS : Branch-Point Sequence BRE : BMP Response Element BSA : Albumine de Sérum Bovin CAM : membrane chorioallantoîdienne CBP80-CBP20 : Cap Binding Protein CML : cellules musculaires lisses ddNTPs : didésoxyribonucléotides DECID : Decay inducing complex
DPC4: Deleted in Pancreatic Carcinoma locus 4 dpp : decapentaplegic
E 14 : âge embryonnaire 14 ECM: Extra cellular matrix
EDHFs : Endothelium-derived hyperpolarizing factors EJC : Exon-Jonction-Complex
ENG : Endogline
eNOS : endothelial nitric oxyde synthase
ERAD : Endoplasmic-Reticulum-Associated protein Degradation ESE : Exonic Splicing Enhancer
XIV GDF2 : Growth Differentiation Factor 2
GS : domaine riche en glycine et sérine HeLa : Henrietta Lacks
HHT ou THH : Télangiectasie Hémorrhagique Héréditaire HMEC-1 : Human Microvascular Endothelial Cell
HMVECd : Human MicroVascular Endothelila Cell from the dermis HMVECl : Human Microvascular Endothelial Cell from lungs hnRNP : heterogenous nuclear ribonucleoproteins
HPAEC : cellules endothéliales pulmonaires humaines HSF : Human Splice Finder
Hsp90 : heat shock protein 90
HUVECs : Human Umbelical Vein Endothelial Cells Id : Inhibiteurs de la différenciation
ISE : Intronic Splicing Enhancer ISS : Intronic Splicing Silencer
JPHT : Syndrome de polypose juvénile associé à HHT KO : Knock Out
L1cre : Cre recombinase sous le contrôle d'un promoteur L1 L-ENG : Long ENG
LOH :①Loss①of①Hétérozygotie①ou①perte①de①l’hétérozygotie① MADH4 : Mothers Against dpp Homolog 4
MAPKs: Mitogen-Activated Protein Kinase MAV : Malformations artério-veineuses
MEEC : cellules embryonnaires endothéliales murines MH2 et MH1: domaine d'homologie Mad
MMPs : métalloprotéases MR : Réponse myogénique
XV mRNP : messenger ribonucleoprotein
NES: signal d'exportation nucléaire NMD : Non-sens mediated decay
PAH : Hypertension Artérielle Pulmonaire PAI : Plasminogen Avtivator Inhibitor PBS : Phosphate Buffered Saline
PDGF-B : Platelet-Derived Growth Factor B
PDZ : postsynaptic density 95/Drosophila disk large/zonula occludens-1 binding motif PPP2R2B : sous unité B-beta de la protéine phosphatase PP2A
PTC : codons-stop prématurés RGD : Arg-Gly-Asp
R-SMAD: Recepteur Smad SE : splicing enhancers S-ENG : Short ENG
shRNA : short hairpin RNA siRNA : short interferent RNA Smad : Small+ Mad
Smurfs : Smad Ubiquitin Regulatory Factors SNO-Hb : S- nitroso-hemoglobin
snRNP : small nuclear ribonucleoproteins SOD : Superoxyde dismutase
SS : splicing silencers
TGFβ : Transforming Growth Factor β
THH1 : Télangiectasie Hémorrhagique Héréditaire du type 1 THH2 : Télangiectasie Hémorrhagique Héréditaire du type 2 TNF-α : Tumor Necrosis Factor-α
XVI vbg : violet beauregarde
VEGF : Vascular Endothélial Growth Factor VSMCS : cellules musculaires lisses vasculaires ZP : Zona Pellucida
XVII 1- Publications :
Cette thèse a mené à la soumission d’un article et envisage la rédaction d’un deuxième article :
Ferdos Alaa el Din, Sylvie Patri,Vincent Thoreau, Montserrat Rodriguez-Ballesteros,
Eva Hamade, Sabine Bailly, Brigitte Gilbert-Dussardier, Raghida Abou Merhi*, Alain Kitzis*. “ Functional and splicing analysis of 23 ACVRL1 mutations in a cohort of patients affected by Hereditary Hemorrhagic Telangiectasia (ROW)” (Résumé dans l’annexe).
Un deuxième article est envisagé pour la deuxième partie du travail mais des expériences ultérieures sont nécessaires pour confirmer les résultats.
2- Participation dans les conférences :
- Présentation orale dans la 21ème Conférence scientifique annuelle de LAAS en 2015, « Functional and splicing analysis of 23 ACVRL1 mutations in a cohort of patients affected by Hereditary Hemorrhagic Telangiectasia (ROW)» à l’Université de Saint- Joseph de Beyrouth.
- Présentation de poster dans la 20ème Conférence scientifique annuelle de LAAS en 2014 « Identification and characterization of 23 mutations of ACVRL1 gene in a cohort of patients affected by Hereditary Hemorrhagic Telangiectasia » à l’Université Libanaise de Beyrouth.
- Présentation de poster dans la 7ème assise de génétique humaine et médicale en 2014 au Palais des congrès, Bordeaux. « Identification and characterization of 12 mutations of ACVRL1 gene in a cohort of patients affected by Hereditary Hemorrhagic Telangiectasia »
- Présentation orale dans la 3ème Rencontre Internationale en immunologie cellulaire et moléculaire en 2013 « Identification and characterization of 12 mutations of
XVIII
ACVRL1 gene in a cohort of patients affected by Hereditary Hemorrhagic Telangiectasia » à l’Université Libanaise de Beyrouth.
3- Formations pédagogiques à l’école doctorale de Poitiers pendant les quatre
années de thèse (Attestation ci-joint).
4- Assistance au cours de Master 1 (Master Biologie Santé, Sciences du Médicament, Université de Poitiers), intitulé Mécanismes Moléculaires et Cellulaires des Maladies Génétiques au cours de l’année universitaire 2012/2013.
XIX
Je, soussigné, Monsieur BERGES Thierry, co-responsable de l’Unité d’Enseignement de M1 intitulée Mécanismes Moléculaires et Cellulaires des Maladies Génétiques (Master Biologie Santé, Sciences du Médicament, Université de Poitiers), certifie que Mademoiselle ALAA EL DIN Ferdos a bien assisté aux enseignements dispensés dans cette UE au cours de l’année universitaire 2012/2013.
Les interventions en cours portaient sur les thèmes suivants :
- Maladies mitochondriales: modes de transmission, manifestations physiopathologiques, dysfonctions moléculaires et cellulaires
- Génétique et transport vésiculaire - Maladies à Prion
- Mécanismes épigénétiques des maladies
Pour faire valoir ce que de droit.
1
2 Avant propos
La recherche fondamentale en génétique des maladies rares comme c’est le cas du Rendu-Osler-Weber se base sur des études épidémiologiques après l’établissement des manifestations cliniques de la maladie chez les patients et leur famille. Ces études permettent de trouver une histoire naturelle de la maladie et donnent une raison commune à l’apparition de la maladie dans certaines zones géographiques. Rendu-Osler-Weber est bien détecté à Poitiers et la recherche d’effet fondateur probable est donc logique.
L’étape suivante est la caractérisation des gènes impliqués. Trois gènes sont actuellement identifiés par études de liaison : ACVRL1, ENG et Smad4. Les transcrits et les protéines produits par ces gènes sont également étudiés pour identifier les bases moléculaires de la maladie. La découverte des voies de signalisation canoniques empruntées par les protéines permet d’établir des outils fonctionnels de diagnostic pour vérifier l’implication des mutations des gènes dans la maladie. Cette étape était le point central de mon projet de thèse. Notre laboratoire étant spécialisé dans le diagnostic moléculaire de la maladie était particulièrement intéressé par un test fonctionnel pour étudier un grand nombre de mutations d’ACVRL1 détectées chez les patients.
Dans ce contexte, l’objectif de ce travail est d’étudier les conséquences physiopathologiques des mutations d’ACVRL1. Sicertains types de mutations comme les mutations non sens (stop) ou des insertions/délétions entraînant un décalage du cadre de lecture ou des mutations d’épissage semblent à priori être pathologiques, mon étude fonctionnelle cible les mutations faux-sens et les mutations introniques en dehors des sites d’épissage. Le mécanisme moléculaire de ces mutations est testé au niveau de l’activité des protéines et/ou couplé à l’analyse de l’ARNm afin d’étudier l’expression et la stabilité de ce dernier. La combinaison des deux niveaux d’analyse (ARN et protéine) est bénéfique pour expliquer l’expressivité variable des manifestations cliniques chez les patients d’une même famille.
Avant d’aborder notre travail, la partie introductive de ce manuscrit détaille en premier la maladie de Rendu-Osler-Weber du point de vue clinique et génétique. Dans un second temps, les voies de signalisation impliquées et leur intérêt dans le diagnostic moléculaire de la pathologie ensuite la physiopathologie de la maladie qui ouvre vers des approches thérapeutiques. La seconde partie présente les résultats obtenus, la discussion et des perspectives envisagées pour la suite d’étude de Rendu-Osler-Weber.
Chapitre 1
I- LA MALADIE DE RENDU OSLER WEBER (HHT)2 1. Définition
Cette maladie a été décrite pour la première fois par un dermatologue français, Henri RENDU, en 1896. La description a été complétée par un médecin canadien, William OSLER, en 1902 puis par un dermatologue anglais, Frederick Parkes WEBER, en 1907. Elle a été immédiatement rebaptisée HHT pour Hereditary Hemorrhagic Telangiectasia, par l’américain Hanes en 1909. La maladie est due à un trouble de la fabrication des vaisseaux (vasculogenèse) et à une perturbation de l’entretien (angiogenèse) et de la régulation des vaisseaux (homéostasie). La lésion de base est une dilatation des vaisseaux distaux (télé-angio-ectasie). Cette lésion se manifeste par une tendance hémorragique lorsqu’elle est cutanée ou muqueuse. Ce même type de lésion, lorsqu’elle est située dans un organe, se traduit par l’installation d’un shunt artérioveineux entre les artérioles et les veinules qui s’exprime de façon dépendante du viscère où il s’installe (Plauchu, Brunet et al. 1992).
2. Symptômes et diagnostic clinique
Les symptômes de la maladie n'apparaissent pas à la naissance, mais se développent avec l'âge. Les critères diagnostiques ont été établis lors d’une conférence de consensus à Curaçao (Shovlin, Guttmacher et al. 2000). Quatre critères sont observés : 1) la survenue d'épistaxis (présents chez 95 % des patients), 2) la présence de télangiectasies (dilatations des petits vaisseaux périphériques) au niveau de la peau ou des muqueuses buccales (présentes chez 84 % des patients), 3) l'atteinte viscérale (télangiectasies pouvant être hémorragiques du tube digestif et/ou fistules artério-veineuses dans certains organes), 4) l'histoire familiale (positive chez 98 % des patients) (Plauchu, de Chadarevian et al. 1989). Un patient est déclaré atteint de la maladie s'il est positif pour au moins trois des critères, et suspect s'il est positif pour deux critères (Plauchu, Brunet et al. 1992). Le test génétique est disponible et permet de confirmer le diagnostic par identification de la mutation familiale devant des signes d'appel, y compris viscéraux.
Les épistaxis sont spontanées, irrégulières, récidivantes. Elles entraînent souvent une anémie. L'anémie résultante de ces saignements oblige le patient à une reconstitution continuelle des réserves de fer (parfois via des transfusions). Elles sont dues à des télangiectasies disséminées dans la tache rouge nasale. Les télangiectasies cutanées et muqueuses, souvent localisées au
3 niveau du visage, des doigts et des muqueuses buccales (lèvres, langue), atteignent ensuite l’estomac et l’intestin. Une télangiectasie peut aussi évoluer en grossissant à cheval sur la jonction capillaire qu’elle élargit, créant ainsi un shunt veineux : c'est la fistule artério-veineuse (Figure 1). Elles se manifestent principalement dans l’intestin (25-33%) (Kjeldsen and Kjeldsen 2000), le foie (30%) (Dupuis-Girod, Chesnais et al. 2010), les poumons (15-30%) (Cottin, Blanchet et al. 2006) ou le système nerveux central (10-15%) (Post, Letteboer et al. 2005).
En règle générale, les patients ont une espérance de vie normale mais celle-ci dépend fortement des complications viscérales. Quelques cas de mortalité pendant la grossesse ont été rapportés chez des patientes non dépistées pour les fistules artério-veineuses pulmonaires. On note une expressivité de la maladie très variable entre les patients et même au sein d’une même famille (Govani and Shovlin 2009) ; (Plauchu, Brunet et al. 1992).
Fistule pulmonaire. Pointe blanche: artère, Flèche: fistule Pointe noire: veine
Fistule cérébrale (flèche) avec dilatation d'une veine (pointe) par IRM
Fistules artério-veineuses de la moelle épinière Artère hépatique dilatée et tortueuse
A
B
C
Figure 1 : Symptômes de la maladie de Rendu-Osler-Weber. A : Imageries des fistules artério-veineuses. D’après (Jaskolka, Wu et al. 2004). B : Télangiectasie cutanée sur un patient. D’après (Srinivasan, Hanes et al. 2003).C : Représentation schématique d’une évolution d’une télangiectasie cutanée. représente la dilatation des veines, illustre la disparition progressive des capillaires et montre la connexion directe entre artères et veines. D’après(Guttmacher, Marchuk et al. 1995).
Chapitre 1
I- LA MALADIE DE RENDU OSLER WEBER (HHT)4 3. Epidémiologie
La prévalence minimale de la Télangiectasie Hémorragique Héréditaire est estimée à 1 sur 10000 (Guttmacher, Marchuk et al. 1995) mais elle est plus élevée dans certaines zones géographiques isolées (Abdalla and Letarte 2006). Ainsi, au nord du Japon elle est estimée de 1 sur 5000 à 1 sur 8000 comparable à sa prévalence au Danemark, ainsi que pour d’autres populations américaines ou européennes. La prévalence la plus élevée dans le monde, 1 sur 1331, est connue aux Antilles Néerlandaises dans la population Afro-Caraîbes (Abdalla and Letarte 2006).
En France, cette maladie était méconnue avant les années 1980 et répertoriée comme très rare (moins de 1/50 000 sujets). Le regain d’intérêt est dû à la découverte de concentrations géographiques de la maladie en France en 1970, 1/1 3345 habitants dans l’Ain, 1/5062 dans le Jura et 1/4287 dans les Deux Sèvres (Figure 2) dans la région Poitou-Charentes où dans certains cantons, la prévalence peut atteindre 1/1000 (Plauchu, Bideau et al. 1978) ; (Bideau, Plauchu et al. 1980). Une enquête épidémiologique développée en France dans les années 1980 sur l’ensemble du pays a permis d’affirmer une prévalence de la maladie supérieure à 1/8 460 en région Rhône-Alpes (pour les naissances entre 1910 et 1920) (Plauchu and Dupuis-Girod 2009). Plus tard, une étude nationale large a été réalisé entre 1982 et 1986 et a montré une grande différence d’incidence entre les départements en France variant entre un maximum de 1/3375 habitants à un minimum de 1/126000 (Bideau, Plauchu et al. 1989). Le recrutement actuel permet d’évaluer l’incidence en France entre 1/5 000 à 1/8 500, soit un nombre de malades probables autour de 10 000. En outre, Cette grande prévalence du Rendu- Osler -Weber en Poitou-Charentes fait suspecter un (des) effet(s) fondateur(s) (Plauchu and Dupuis-Girod 2009).
L’identification des gènes responsables de cette maladie a permis d’en établir la grande hétérogénéité génétique et d’identifier un grand nombre de mutations différentes présentes sur le territoire français. Toutefois, de nombreux malades résidant dans le principal pôle de concentration étudié précédemment (Rhône-Alpes) partagent une mutation unique associée à un haplotype spécifique. Au terme de cette étude, exceptionnelle par son aspect interdisciplinaire et sa durée, les différentes démarches convergent pour affirmer l’existence d’un effet fondateur local, et s’accordent sur une datation approximative de cette mutation (Brunet, Lesca et al. 2009).
5 Certaines mutations familiales largement répandues, chez des patients français et italiens, ont été étudiées en 2008 par analyse d’haplotype. L’âge de l’ancêtre commun le plus récent « Most Recent Common Ancestors (MRCAs) » est estimé pour cinq mutations entre 100 et 500 années. La plus intéressante parmi ces mutations était une insertion de guanidine, c.1112dupG (p.Gly371fsX391) du gène ACVRL1 qui est responsable de l’incidence élevée de HHT dans le Haut-Jura et l’Aine. Cette mutation est associée à un effet fondateur et a probablement été introduite dans la population par un patient qui habitait une vallée de montagnes des Haut-Jura il y a 300 ans et qui s’est diffusée au cours des générations principalement dans la région Rhône-Alpes mais aussi ailleurs (Lesca, Genin et al. 2008) ; (Lesca, Plauchu et al. 2004).
Au LIBAN, cette maladie est mal étiquetée et nous n’avons pas réussi à avoir des patients
pour étudier la présence de certaines mutations dans la population libanaise.
Figure 2 : Epidémiologie de la maladie de Rendu-Osler-Weber en France en A et le département de Deux- Sèvres en B. D’après (Bideau, Plauchu et al. 1989).
7 La HHT est une maladie vasculaire génétique, de transmission autosomique dominante. Plusieurs études sur les enfants dont les deux parents sont affectés avec un même gène muté, mais dont la mutation n’est pas forcément identique, soutiennent une létalité homozygote in utéro ou infantile dans la maladie HHT (Snyder and Doan 1944) ; (El-Harith, Schmidtke et al. 2006) ; (Wooderchak-Donahue, McDonald et al. 2007).
La pénétrance dépasse 50% à 15 ans et est quasi complète vers 50 ans. Une des caractéristiques de cette pathologie est la grande variabilité des symptômes et de leur gravité à l’intérieur même d’une famille, suggérant que d’autres facteurs génétiques inconnus ou que des facteurs environnementaux influencent le phénotype de HHT (Plauchu, de Chadarevian et al. 1989).
La HHT est une pathologie hétérogène, plusieurs gènes ont été identifiés responsables de la maladie. Les études de liaison ont premièrement identifié le gène ENG sur le chromosome 9 (locus q33-34), codant pour l’endogline responsable de la forme HHT1 (OMIM 131195) (Fernandez-Ruiz, St-Jacques et al. 1993) ; (McAllister, Lennon et al. 1994). Puisque certaines familles ne sont pas porteuses de mutations de ce gène (Heutink, Haitjema et al. 1994) ; (Shovlin, Hughes et al. 1994), en 1995, l’équipe de Henri Plauchu a trouvé un locus du chromosome 12 (locus q11-14) où des mutations dans le gène ACVRL1 ont été identifiées, responsables de la forme HHT2 (OMIM 600376) (Johnson, Berg et al. 1995) ; (Johnson, Berg et al. 1996). Cette deuxième forme de la maladie, très similaire à HHT1 mais due à un gène différent, a été nommée HHT2 pour la distinguer de la première. La forme HHT1 est plus fréquente en Amérique du Nord, dans les Caraïbes et dans les populations du nord de l’Europe tandis que la forme HHT2 est répandue en Italie, en Espagne et en France.
Un troisième gène est identifié, SMAD4, localisé sur le chromosome 18 (Gallione, Repetto et al. 2004). Sa mutation est liée à la présence du syndrome de polypose juvénile (causé aussi par des mutations d’ALK3 et de HHT (JPHT).
Il existe au moins deux autres gènes non identifiés qui peuvent causer des HHT classiques, HHT3 identifié sur le chromosome 5q entre D5S2011 et D5S2490 (Cole, Begbie et al. 2005) et HHT4 sur le chromosome 7p entre D7S2252 et D7S510 (Bayrak-Toydemir, McDonald et al. 2006).
Une autre étude en 2007 a révélé que les mutations dans le gène de la protéine morphogénétique osseuse 9 « BMP9 » (également connu sous le nom de GDF2), provoquent un syndrome vasculaire chevauchant phénotypiquement avec l’anomalie de la télangiectasie hémorragique héréditaire (Wooderchak-Donahue, McDonald et al. 2007).
Chapitre 1
II- LA GÉNÉTIQUE DE HHT8 Une découverte récente par Xu et al. a particulièrement montré que la sous- unité régulatrice B-beta de la protéine phosphatase PP2A (PPP2R2B) interagit avec ENG, ACVRL1 et TGFβ receptor type 2 et que l’ENG régule l’activation de ces protéines cibles par l’intermédiaire de cette sous-unité. Selon leurs travaux la PPP2R2B pourrait être le gène HHT3 (Xu, Barrios-Rodiles et al. 2014).
1. Le gène Endogline (ENG)
1.1 La structure de l’ENG
L’ADNc de l’endogline, codant pour une glycoprotéine membranaire intégrale du type I a été isolé en 1990 (Gougos and Letarte 1990).
Le gène ENG est composé de 14 exons, les 12 premiers codent pour un large domaine extracellulaire de 140 kDa (561 aa) sous forme d’un dôme comprenant deux monomères orientées antiparallèlement et enfermant une cavité (Llorca, Trujillo et al. 2007).
Ce domaine extracellulaire est composé d’un peptide signal du côté N-terminal, un domaine orphelin ne montrant aucune identité avec des domaines protéiques connus et un domaine zone pellucide (ZP) dans la région juxtamembranaire de 260 aa, composée de deux sous-domaines organisés en un monomère ouvert en forme de U contenant des résidus de cystéines conservés et constituant 30% de la région extracellulaire. Le ZP est impliqué dans l’interaction de l’endogline avec les récepteurs au TGFβ en absence de celui-ci (Guerrero-Esteo, Sanchez-Elsner et al. 2002) (Figure 3).
La structure primaire de ce large domaine extracellulaire suggère qu’il y a 5 motifs de N- glycosylation et un motif de O- glycosylation mais le domaine ne possède pas de chaîne de glycosaminoglycanes (Figure 4). Ce domaine extracellulaire contient aussi dans une zone dégagée une séquence peptidique Arg-Gly-Asp (RGD). RGD est une structure clée de reconnaissance de protéines adhésives présente dans la matrice extracellulaire (ECM) comme la fibronectine (Gougos and Letarte 1990) ; (Lopez-Novoa and Bernabeu 2010).
L’exon 13 code pour un domaine transmembranaire hydrophobe de 25 aa et l’exon 14 code pour un court domaine intracellulaire de 47 aa riche en sérines et thréonines. Ce domaine est sans activité kinase, il est fortement phosphorylé surtout sur les résidus sérines par les récepteurs sérine-thréonine kinase de TGFβ (TBRI et TBRII) (Abdalla and Letarte 2006) ; (Gregory, Xu et al. 2013). Il contient un motif consensus PDZ (pour postsynaptic density 95/Drosophila disk large/zonula occludens-1) du côté C- terminal responsable de l’interaction
9 de l’endogline avec les protéines contenant des domaines PDZ et la phosphorylation de l’endogline sur les résidus thréonine distaux (Lopez-Novoa and Bernabeu 2010).
Le domaine extracellulaire de l’ENG peut être clivé par protéolyse à l’aide de la métalloprotéinase 14 et circulé sous forme soluble, qui peut servir comme antagoniste d'origine naturelle de la signalisation TGFβ et donc jouer un rôle dans les maladies cardiovasculaires (Venkatesha, Toporsian et al. 2006). Cette forme circulante est un mauvais pronostic dans les cancers ou à une pré-éclampsie dans la grossesse si son taux est élevé (Li, Hampson et al. 2000) ; (Calabrò, Fonsatti et al. 2003) ; (Venkatesha, Toporsian et al. 2006). L’ENG également connue sous le nom de CD105 est exprimée tôt durant le développement sur l’endothélium vasculaire et sur le tissu mésenchymateux dérivé de l’endocardium (Kapur, Wilson et al. 2012). Elle est présente à des niveaux élevés sur l’endothélium vasculaire chez l’adulte (Wong, Hamel et al. 2000).
L’ENG est aussi exprimée dans les cellules stromales d'origine mésenchymateuse, les cellules musculaires lisses, le syncytitrophoblaste du placenta, les cellules souches mésenchymateuses et hématopoétiques, les pré-erythroblastes, les cellules leucémiques des lignées lymphoïdes et myéloïdes et les monocytes activés. Dans le tissu cardiaque, l’endogline est exprimée dans l’endocardium et les fibroblastes et faiblement exprimé par les cardiomyocytes (Kapur, Wilson et al. 2012).
Chapitre 1
II- LA GÉNÉTIQUE DE HHT10 Figure 4 : Structure de l’endogline. A : Représentation schématique de la structure de l’endogline sous forme d’un dimère. B : Les séquences en acides aminés du domaine cytoplasmique des deux formes S et L d’endogline. C : Modèle prédictif in silico de la structure du domaine extracellulaire. Les nombres d’aa représentent les bornes des domaines globulaires « vu de côté » D : Microscopie électronique montrant l’organisation des domaines extracellulaires d’un dimère d’endogline « vus du haut ». D’après (Lopez-Novoa and Bernabeu 2010).
11 1.2 Fonction de l’ENG
Il existe deux isoformes d’ENG suite à un épissage alternatif : une forme courte (S-ENG pour Short ENG) et une forme longue (L-ENG pour Long ENG), différentes de 30 acides aminés dans le domaine intracellulaire (Figure 4B) (Bellon, Corbi et al. 1993).
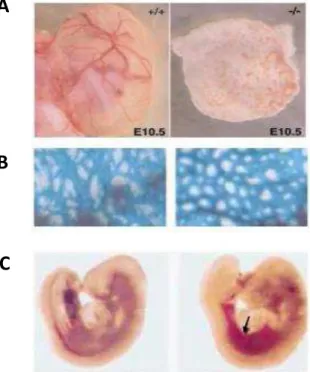
La L-endogline étant l’isoforme principal, les études fonctionnelles de l’ENG se référent à cette forme. Les souris déficientes en ENG meurent au cours du développement embryonnaire (E10.5) du fait d’un défaut d’angiogenèse. Le sac vitellin des souris invalidées ne présente pas de vaisseaux sanguins bien que la vasculogenèse soit bien établie (Figure 5) (Bourdeau, Dumont et al. 1999) ; (Arthur, Ure et al. 2000).
Figure 5 : Invalidation totale de l’ENG chez les souris. A : Les souris invalidées pour l’ENG ne présentent pas de vaisseaux sanguins au niveau du sac vitellin. D’après (Li, Sorensen et al. 1999). B : Visualisation des vaisseaux à E 9.0 au niveau du sac vitellin. Les souris invalidées présentent un plexus vasculaire primitif mais pas de vaisseaux bien différenciés. D’après (Bourdeau, Dumont et al. 1999). C : Présence d’hémorragies internes chez les embryons à E 9.0 (flèche noire). D’après (Bourdeau, Dumont et al. 1999).
Les souris hétérozygotes pour l’ENG, Eng+/-, ont été décrites en 1999, elles sont viables et se développent normalement. Certaines de ces souris montrent des signes cliniques relatifs à HHT : saignements de nez et télangiectasies (Bourdeau, Dumont et al. 1999). Ces souris présentent une réponse défectueuse aux substances vasodilatatrices comme l’acétylcholine (Jerkic M, Rivas-Elena JV. 2004). De même, il a été montré que ces souris présentent un défaut de synthèse de l’oxyde nitrique (NO) et/ou une diminution d’expression de la NO
A
C
B
Chapitre 1
II- LA GÉNÉTIQUE DE HHT12 synthase endothéliale (eNOS) (Jerkic, Rivas-Elena et al. 2004) ; (Jerkic, Rivas-Elena et al. 2006) ; (Toporsian, Jerkic et al. 2005).
Suite à ces observations, Toporisan et al. ont émis l’hypothèse que l’haploinsuffisance d’ENG dans les cellules endothéliales induit un découplage de l’association de l’eNOS avec Hsp90 (pour heat shock protein 90) puis une dimintion de libération de NO et une augmentation de la production de l’O2- (Toporsian, Gros et al. 2005) (Figure 6).
Figure 6: Modèle d’activation de l’eNOS en condition normale et en cas d’absence de l’endogline (HHT1).
Dans le cas normal, un pool d’Endogline réside dans les cavéoles d’une cellule endothliale d’une artériole pulmonaire, où il agit comme un échafaudage moléculaire pour faciliter l’association de l’eNOS et Hsp90 lors de l'activation endothéliale. Dans HHT1, la diminution du taux d’endogline induit un découplage de la réaction de production du NO entrainant une vasomotricité défaillante dans les artérioles. Il en résulte une augmentation de la production de l’O2- par l’eNOS et la formation de H2O2 par le superoxyde dismutase (SOD) et l’ONOO, qui
hyperpolarisent le muscle lisse et altèrent les contractions vasculaires. Les composés O2 ou ONOO- peuvent oxyder le cofacteur essentiel de l’eNOS, la tétrahydrobioptérine (BH4) en dihydrobioptérine (BH2), découplant l’eNOS dans HHT1. SNO-Hb : source privilégiée de NO dans les microvaisseaux. D’après (Toporsian, Gros et al. 2005)
2. Le gène ACVRL1 (ALK1)
2.1 La structure d’ALK1
Le deuxième locus HHT2 a été localisé sur le chromosome 12q13 où le gène candidat identifié est ACVRL1. Le gène ACVRL1 s’étend sur plus de 15 kb d’ADN génomique. L’ADNc fut cloné en 1993 à partir du tractus urogénital d’un embryon de rat (He, Gustafson et al. 1993). La même année, l’ACVRL1 humain fut cloné par deux équipes (Attisano, Cárcamo et al. 1993) ; (ten Dijke, Ichijo et al. 1993). Le gène d’ACVRL1 comprend 10 exons.
13 Tous les introns suivent la règle GT-AG de l’épissage, sauf pour l'intron 6, qui a une jonction d'épissage AGgcaag (Abdalla and Letarte 2006). La région codante est contenue dans neuf exons, le codon d’initiation se trouve dans l'exon 2 et le codon de terminaison dans l'exon 10 (Figure 7). Le gène ACVRL1 code pour la protéine ALK1 (pour Activin A receptor, type II-like kinase 1). Elle est constituée d’un petit domaine extracellulaire codé par les exons 2, 3 et une partie de l’exon 4, un court domaine transmembranaire codé par le reste de l’exon 4 et une partie de l’exon 5 et un large domaine intracellulaire codé par les exons 5 à 10 (Ricard, Bidart et al. 2010).
La protéine ALK1 comprend 503 aa. Les résidus 1 à 21 forment une séquence signale qui cible la protéine vers la membrane. Les résidus 22 à 118 forment le domaine extracellulaire ou l’éctodomaine qui se lie aux ligands. Ce domaine est suivi par un domaine transmembranaire de 23aa (résidus 119 à 141). Le domaine intracellulaire comprend les résidus 142 à 503 avec un domaine GS (résidus 172-201) et le domaine protéine kinase (résidus 202 à 492) (Figure 8) (Townson, Martinez-Hackert et al. 2012).
ALK1 contient 10 résidus cystéine conservés et un site de N- glycosylation potentiel dans le domaine extracellulaire. La partie intracellulaire d’ALK1 est presque entièrement constituée d'un domaine kinase contenant 12 sous-domaines avec des résidus hautement conservés au cours de l’évolution (Abdalla and Letarte 2006).
Le récepteur ALK1 a été fortement détectée dans les cellules endothéliales des vaisseaux sanguins du poumon, de l’aorte et la veine cave (Panchenko, Williams et al. 1996). ALK1 est exprimée aussi dans le placenta et les sites d’interaction des cellules épithéliales-mésenchymateuses (Abdalla and Letarte 2006) (Roelen, van Rooijen et al. 1997).
Chapitre 1
II- LA GÉNÉTIQUE DE HHT14 Figure 8: Structure de la protéine d’ALK1. D’après(Mahmoud, Upton et al. 2011).
2.2 Fonction d’ALK1
L’invalidation complète d’ALK1 est létale pour les souris autour de E10.5 suite à des défauts d'angiogenèse (Urness, Sorensen et al. 2000) ; (Oh, Seki et al. 2000). Les embryons invalidés pour ALK1 présentent un retard de croissance. Ces souris forment leur plexus vasculaire mais le réseau vasculaire ne se développe pas que ce soit dans l'embryon ou dans le sac vitellin et ils présentent des fistules artério-veineuses avec une fusion de l'aorte dorsale et de la veine cardinale (Figure 9) ce qui rappelle le phénotype observé dans l’invalidation de l’ENG décrit précédemment.
L'invalidation hétérozygote d'ALK1 a été faite en 2003 (Srinivasan, Hanes et al. 2003). Sur 47 souris étudiées, 23% ont montré des lésions vasculaires sous forme de vaisseaux dilatés à paroi fine au niveau cutané et des extrémités, 6% au niveau des muqueuses orales et 28% sur des organes internes sous forme d’hémorragies (Srinivasan, Hanes et al. 2003). Ces observations sont similaires aux symptômes de la maladie de Rendu-Osler. A noter que ces manifestations apparaissent sur des souris âgées (1 an minimum), ce qui corrèle bien avec l'apparition tardive des symptômes de la maladie.
15 Figure 9 : Invalidation totale d’ACVRL1 chez la souris. Comparaison entre un embryon sauvage (+/+) et un embryon invalidé pour ACVRL1 (-/-).A : La vascularisation de la tête à E9.5 montre une absence des capillaires chez les souris ACVRL1 (-/-). D’après (Oh, Seki et al. 2000) B : Absence de vaisseaux sanguins dans le sac vitellin d’un embryon à E10.5 ACVRL1 (-/-). C : L’injection de l’encre montre le plexus vasculaire dans le sac vitellin sans détection de vaisseau alors que la vasculogenèse est intacte ACVRL1 (-/-). D’après (Urness, Sorensen et al. 2000).
L’invalidation d'ACVRL1 dans les cellules endothéliales a été faite en 2008 (Park, Lee et al. 2008). Ils ont exprimé la Cre recombinase sous le contrôle d'un promoteur L1 qui est activé après E 10.5 pour éviter qu’il interfère avec la formation cardiaque. Les souris obtenues présentent les caractéristiques HHT (dilatation de lumen, amincissement des parois vasculaires...). Le sac vitellin de ces souris invalidées pour ALK1, L1cre(+) ; Alk13lox /3loxp, forme des vaisseaux tortueux et dilatés et dans certaines régions il y a des veines et des artères qui se connectent directement sans le biais de capillaires (Park, Lee et al. 2008). A noter que cette invalidation complète est létale.
Une étude réalisée en 2009 avec une délétion inductible d’ACVRL1 dans les cellules endothéliales a permis d'étudier le rôle d'ALK1 dans la vascularisation adulte (Park, Wankhede et al. 2009). Ce groupe a utilisé la recombinase Cre sous le contrôle d'un promoteur inductible par le tamoxifène. L'invalidation à l'âge adulte conduit aussi à une létalité par hémorrhagie chez les souris adultes (âgées de plus de 2 mois) en 9 à 21 jours. Dès 8 jours, les souris montrent une perte de poids, une anémie, des hémorragies intestinales et pulmonaires. Leurs veines et leurs artères pulmonaires sont dilatées, des fistules
artério-A
B
Chapitre 1
II- LA GÉNÉTIQUE DE HHT16 veineuses sont visibles dans le tractus digestif et l'utérus. Les femelles meurent plus tôt (9 à 14 jours après l'invalidation d'ACVRL1) que les mâles (10 à 21 jours après l'invalidation).
3. Le gène MADH4 ou SMAD4
3.1 La structure de SMAD4
En 1998, un locus a été identifié sur le chromosome 18q21 par une étude de liaison dans une large famille et des mutations germinales dans le gène MADH4 (mothers against dpp homolog 4) ont été ultérieurement trouvées (Howe, Ringold et al. 1998). Le nom de ce gène a été donné par les auteurs qui ont montré que des mutations du gène Mad chez la mère induisaient une perte de l’activité de dpp (decapentaplegic) dans l’embryon, un analogue de BMP2 et BMP4 (deux membres de la famille BMP pour Bone Morphogenetic Protein) chez la drosophile (Sekelsky, Newfeld et al. 1995) . MADH4 est un membre de trois homologues de Mad qui ont été trouvés chez Caenorhabditis elegans et dénommés sma (sma2/3/4) car des mutations dans ces gènes donnent des corps de petites tailles ( small ) (Savage, Das et al. 1996). Les gènes des mammifères furent ensuite décrits et dénommés Smad (condensé de Sma et Mad). En outre, SMAD4 est l’une des premières SMAD humaines identifiées et connue alors sous le nom DPC4 pour Deleted in Pancreatic Carcinoma locus 4 (Hahn, Hoque et al. 1996).
Le gène SMAD4 est constitué de 11 exons. L'ARNm code pour une protéine constituée de 552 acides aminés (Howe, Ringold et al. 1998). Comme tous les récepteurs SMAD, SMAD4 possède deux domaines bien conservés: MH1 et MH2 (domaine d'homologie Mad). Les deux domaines sont connectés par une « région lien » ou linker (Figure 10).
Du cȏté N-terminal de SMAD4, il existe le domaine MH1 caractérisé par une structure en épingle à cheveux qui présente une activité de liaison à l'ADN (Shi, Wang et al. 1998). Ce domaine MH1 porte une séquence d’importation nucléaire (Xiao, Latek et al. 2003). Du côté C-terminal, le domaine MH2 est responsable de l'interaction avec des protéines qui participent au complexe de translocation dans le noyau, ainsi qu’au recrutement des co-facteurs de liaison à l'ADN. Le domaine MH2 de Smad4 apparait aussi responsable de l’homo- oligomérisation des trimers de Smad4 et l’hétéro-oligomerisation entre Smad4 et les trimères R-SMAD. Le « linker » de SMAD4 a un signal d'exportation nucléaire riche en leucine (NES) reconnu par CMR1. Les interactions de SMAD4 avec les R-SMADs phosphorylés masquent NES,
17 protégeant ainsi SMAD4 contre sa reconnaissance par le facteur nucléaire d'exportation, CMR 1 et son exportation du noyau. L’exportation de SMAD4 ne devient possible qu'après déphosphorylation de R-SMAD et la dissociation du complexe (Gallione, Repetto et al. 2004). Cette région contient également des sites de phosphorylation notamment pour les MAPKs (Mitogen-Activated Protein Kinase) (Kretzschmar, Doody et al. 1999), ainsi qu’un motif PY qui permet des interactions avec les Smurfs (Smad Ubiquitin Regulatory Factors), E3 ubiquitine liguase spécifique des Smads (Zhu, Kavsak et al. 1999).
Figure 10: Structure typique des Smads (en se basant sur la structure de Smad3). Représentation schématique des différents domaines de R-Smads. Structure cristallographique du domaine MH1. Structure cristallographique du domaine MH2. D’après (Makkar, Metpally et al. 2009).
3.2 Mutations de SMAD4
SMAD4, est responsable d’une forme syndromique de Rendu-Osler puisqu’il réalise une association avec la polypose juvénile. La polypose juvénile (PJ) est une maladie autosomique dominante de malformations vasculaires (Gallione, Repetto et al. 2004). C’est un syndrome de prédisposition au cancer caractérisé par des anomalies congénitales, des polypes dans le tractus gastro-intestinal et le développement de tumeurs dans ces tissus (Hampel and Peltomaki 2000). Il existe actuellement 88 mutations de SMAD4 dans la base de données « http://www.arup.utah.edu/database/smad4/SMAD4 ». Les mutations de SMAD4 trouvées DNA binding site
Cofactors binding site and DNA binding
Linker region ձ
ղ
Chapitre 1
II- LA GÉNÉTIQUE DE HHT18 chez des patients qui ont le syndrome combiné Polypose Juvénile/Télangiectasie Hémorragique Héréditaire (JP/HHT) représentent 2 à 3% du nombre total des mutations trouvées dans SMAD4. Les mutations trouvées sont de tous types, des délétions (28,4%), des mutations faux-sens (27,27%) et des mutations non-sens (21,6%).
3.3 Fonction de SMAD4
SMAD4 est un gène suppresseur de tumeur (Howe, Ringold et al. 1998) ; (Hahn, Schutte et al. 1996). Le produit protéique de ce gène est un constituant particulièrement important du complexe de transcription de nombreux gènes (Zhang, Musci et al. 1997). SMAD4, contrairement aux autres SMAD, n'est pas activée par phosphorylation, mais agit comme un médiateur intracellulaire (Chiao, Hunt et al. 1999).
Ainsi SMAD4 forme des complexes cytoplasmiques avec d’autres membres de la famille des SMADs qui ensuite se déplacent vers le noyau et régulent la transcription des gènes du cycle cellulaire comme partie de la voie de signalisation de TGFβ (de Caestecker, Piek et al. 2000). SMAD4 est délété ou muté pendant la tumorigenèse dans de nombreuses tumeurs humaines. Quelques mutations se produisent dans la partie N-terminale de la protéine au niveau de la région MH1 pourvue de séquences spécifiques de liaison à l’ADN (Wu, Chen et al. 2000).
4. Les théories de « Second Hit » et « LOH »
L’apparition tardive des MAVs au niveau des artérioles distales et les capillaires dans certains organes comme les poumons chez les patients HHT1 et pas dans tout le corps a mené les chercheurs à postuler le besoin d’un déclencheur externe autre que le caractère génétique ou « second hit » (Botella, Sánchez-Elsner et al. 2002) ; (Docherty, Lopez-Novoa et al. 2006) ; (Sánchez-Elsner, Botella et al. 2002) ; (Gougos and Letarte 1988) ; (Mahmoud, Allinson et al. 2010). Ce déclencheur peut être une inflammation, une infection ou autre qui accompagne la perte d’un allèle de l’ENG. Ces conditions empêchent que l’ENG atteigne un niveau d’expression minimum pour réaliser sa fonction optimale.
Concernant HHT2, afin de mieux comprendre le développement tardif de ces lésions vasculaires, Park S. O. et al ont réalisé une blessure cutanée au niveau du derme sur des souris adultes (2 mois) après invalidation d'ALK1. Ils ont observé des jonctions directes entre les artères et les veines dans la zone proche de la blessure avec une dilatation des vaisseaux qui