Université de Montréal
Implications des spasmes infantiles sur le neurodéveloppement des enfants
Par Jonathan Y Bitton
Département de Neurosciences Faculté de Médecine
Thèse présentée à la Faculté des études supérieures en vue de l’obtention du grade de Ph.D.
en Sciences Neurologiques
Août, 2016
Université de Montréal Faculté des études supérieures
Cette thèse intitulée :
Implications des spasmes infantiles sur le neurodéveloppement des enfants
présentée par : Jonathan Y Bitton
a été évaluée par un jury composé des personnes suivantes :
Dr. Émile Lévy, président-rapporteur Dr. Lionel Carmant, directeur de recherche
Dr. Florin Amzica, membre du jury Dr. François Dubeau, examinateur externe Dr. Stéphanie Fulton, représentante du doyen
Résumé
Le syndrome de West (SW), communément appelé spasmes infantiles (SI), est un trouble épileptique généralement caractérisé par la triade de spasmes infantiles, un modèle d'électroencéphalogramme (EEG) pathognomonique appelé hypsarythmie, et la régression du développement. Alors que des études précédentes ont été en mesure d'obtenir une réponse relativement adéquate par rapport au contrôle des spasmes et la résolution d’hypsarythmie, elles n’ont pas réussi à fournir des options thérapeutiques décisives à l’égard des séquelles neurodéveloppementales souvent associées aux SI.
Notre étude, sur laquelle est basée cette thèse, est la première à utiliser un traitement complémentaire aux médicaments antiépileptiques conventionnels, avec l'intention d'améliorer les résultats neurodéveloppementaux de cette population. Les patients recrutés dans notre essai clinique randomisé (ECR) original ont suivi un protocole de traitement standardisé composé de vigabatrin (VGB) comme traitement de première intention pendant deux semaines, suivi de l'hormone corticotrope (ACTH) chez les non-répondeurs pour une période de deux autres semaines, et le topiramate dans les cas réfractaires. En plus, les patients ont été randomisés pour recevoir soit le traitement expérimental, flunarizine, soit un placebo, pendant six mois. Notre ECR multicentrique consistait à recruter et évaluer 68 patients, la plupart suivis à 8 différentes visites sur une période de cinq ans afin de précisément évaluer leurs progrès neurodéveloppementaux. Notre essai clinique a généré trois études principales qui forment le cœur de cette thèse. Dans une première étude, les données cliniques et cognitives des deux premières années
d’évaluation ont été analysées. Les résultats cliniques à court terme indiquent un taux élevé de cessation de spasmes et de l’hypsarythmie. De plus, cette étude rapporte les premiers résultats cognitifs mesurés par le Bayley Scales of Infant Development (BSID) et le Vineland Adaptive Behavior Scale (VABS).
Notre deuxième étude a essentiellement fourni des données cognitives à plus long terme, 5 ans après le début de son initiation. Les réponses cognitives ont été mesurées par le BSID, le VABS, et aussi par le Stanford-Binet Intelligence Scale (SB5) chez les patients ayant un fonctionnement cognitif plus élevé. Une amélioration significative et progressive des fonctions cognitives a été observée, indépendamment de la thérapie adjuvante. Des facteurs de risque cognitifs à long terme ont également été révélés dans cette étude.
Notre dernière étude a essayé d’élucider la relation entre les SI et les troubles du spectre autistique (TSA). Un test de dépistage avec le Checklist for Autism in Toddlers (CHAT) a été effectué à 24 mois, et un diagnostic a été obtenu par moyen du Autism Diagnostic
Observation Schedule (ADOS) à 30 et 60 mois. L’ADOS a évalué 44 patients, dont 10 ont
été diagnostiqués avec TSA. Une description des facteurs de risque associés aux TSA ont été présentés dans cet article.
Enfin, basé sur nos résultats et les informations à ce sujet dans la littérature, nous avons tenté d'élucider les caractéristiques physiopathologiques de la maladie. Une description des mécanismes biologiques sous-jacents impliqués dans le syndrome de West et des
traitements cibles associés ont été présentés. Bien que le traitement complémentaire, le
flunarizine ne se soit pas avéré être avantageux pour notre cohorte, notre protocole de
traitement a tout de même été en mesure de démontrer des résultats cliniques et
cognitifs supérieurs dans le sous-groupe de patients avec SI dont l’étiologie est inconnue. Ces résultats, ainsi que l’identification de nouveaux facteurs de risque
neurodéveloppementaux potentiels, pourraient être utilisés cliniquement afin d’améliorer le diagnostic et le suivi médical des patients atteints du syndrome de West.
Mots clés :
Spasmes infantiles, syndrome de West, neurodéveloppement, cognition, troubles du spectre autistique, vigabatrin, hormone corticotrope, encéphalopathie épileptique, hypsarythmie, quotient de développement.
Abstract
West syndrome (WS), commonly referred to as infantile spasms (IS), is an epileptic disorder usually characterized by the triad of infantile spasms, a pathognomonic electroencephalogram (EEG) pattern called hypsarrhythmia, and developmental regression. While previous treatment studies were able to achieve relatively adequate spasm control and hypsarrhythmia resolution in this population of patients, they have failed to provide conclusive and definite therapeutic options aimed at improving the poor cognitive outcome often associated to IS.
Our study, on which this thesis is based, was the first to use an add-on treatment to conventional antiepileptic drugs, with the intent to improve long-term cognitive outcome in this population. Patients recruited in our original randomized clinical trial (RCT)
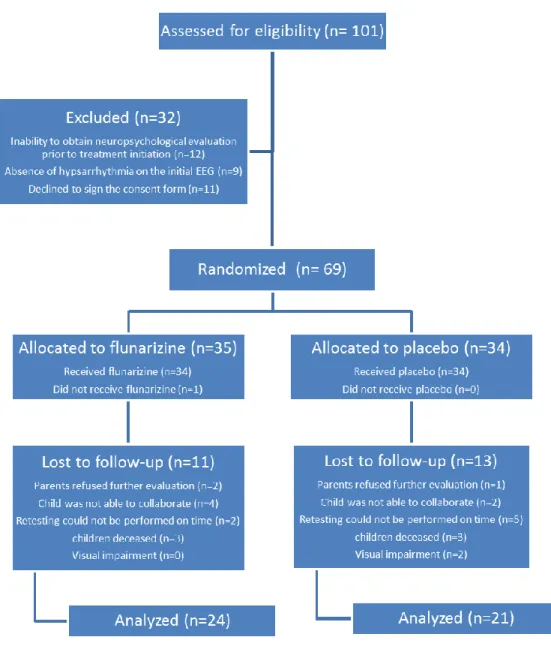
followed a standardized treatment protocol consisting of vigabatrin (VGB) as first-line treatment for two weeks, followed by adrenocorticotropic hormone (ACTH) in non-responders for another two-week period, and topiramate in refractory cases. In addition, patients were randomized to either receive placebo or flunarizine adjunct therapy for six months. Our multi-centric RCT recruited and evaluated 68 patients, most of which were followed at 8 different time points over a five-year period, to precisely evaluate their neurodevelopmental progress.
Our clinical trial generated three main studies which comprise the core of this thesis. In a first study, clinical and cognitive data from the first two years were analyzed. Spasm arrest
and hypsarrhythmia resolution were the short-term clinical endpoint measures, while the Vineland Adaptive Behavior Scale (VABS) and Bayley Scales of Infant Development (BSID) were used as cognitive outcome measures at 2 years. This first study most importantly reports on the superior short-term clinical response rate achieved in our study population. Preliminary cognitive results were also presented in this work.
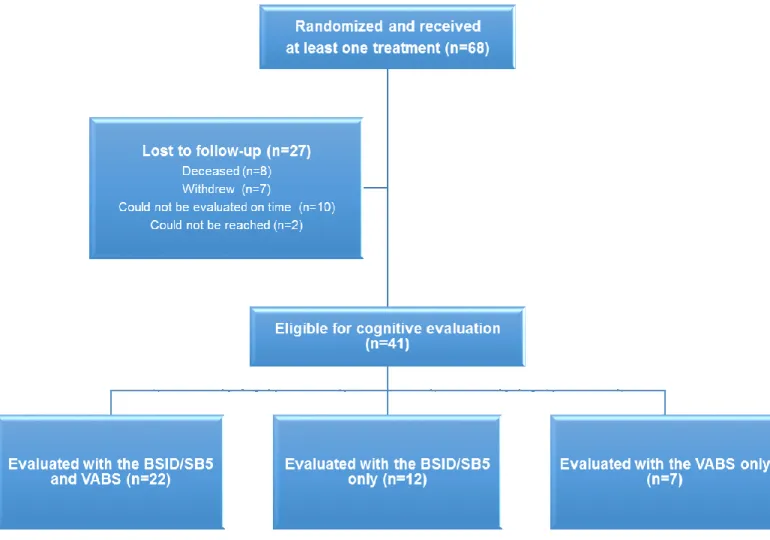
Our second study essentially presented long-term cognitive data 5 years after the start of the study. Cognitive outcome measures were similar to those used at two years with the addition of the Stanford-Binet Intelligence Scale, Fifth Edition (SB5) for higher functioning patients. Most IS patients, particularly those with no known etiology, displayed a
significant and progressive improvement of cognitive functions, irrespective of adjunctive therapy. Risk factors of long term poor cognitive outcome were also revealed in this study.
Our last study tried to understand the relationship between IS and autism spectrum disorders (ASD). Autism was initially screened by means of the Checklist for Autism in Toddlers (CHAT) at 24 months, and formally assessed at the 30-and 60-month follow-up visits using the Autism Diagnostic Observation Schedule (ADOS). ADOS was performed in 44 patients, 10 of which were diagnosed with ASD. A description of risk factors associated with an ASD outcome in the IS population were presented in this article.
Finally, based on our study results and in conjunction with literature information on the topic, we attempted to elucidate the pathophysiological characteristics of the disorder. A
conceivable description of the underlying biological mechanisms implicated in West syndrome and associated target treatments were presented. Although our
complementary treatment, flunarizine, did not prove to be beneficial in our cohort, our treatment protocol was nonetheless able to demonstrate superior clinical and cognitive outcomes in patients with unknown etiologies. These findings, as well as the identification of new potential neurodevelopmental risk factors, could be used clinically to improve the diagnosis and medical follow-up of patients with West syndrome.
Keywords:
Infantile spasms, West syndrome, neurodevelopmental outcome, cognition, autism spectrum disorders, vigabatrin, adrenocorticotropic hormone, epileptic encephalopathy, hypsarrhythmia, developmental quotient.
Table des matières:
Résumé ...iii
Abstract ... vi
Liste des tableaux ... xiii
Liste des Figures ... xiv
Liste de documents spéciaux ... xv
Liste des sigles ... xvi
Liste des sigles (Anglais) ... xvii
Remerciements ... xviii
Partie I - Introduction générale 1. Aperçu historique ... 2 1.1. Épilepsie générale ... 2 1.2. Spasmes Infantiles ... 4 2. Terminologie et classification ... 9 3. Épidémiologie ... 12 3.1. Incidence ... 12 3.2. Prévalence ... 12
3.3. Âge à l’apparition des spasmes ... 13
3.4. Le genre ... 13 3.5. Étiologie ... 14 3.6. Taux de mortalité ... 15 4. Caractéristiques cliniques ... 16 4.1. Spasmes ... 16 4.2. Électroencéphalographie (EEG) ... 17
4.2.1. Enregistrements intercritiques ... 17
4.2.2. Enregistrements critiques ... 19
4.3. Résultats neurodéveloppementaux à long terme ... 20
4.3.1. Délai cognitif ... 20
4.3.1.1. Facteurs de risques pré-SI ... 22
4.3.1.2. Facteurs de risque post-SI ... 23
4.3.2. Autisme ... 25
4.3.3. Crises épileptiques ... 25
4.3.4. Évolution naturelle des SI ... 26
5. Options thérapeutiques ... 28
5.1. Traitements à court terme ... 29
5.1.1. Antiépileptiques ... 29 5.1.1.1. Vigabatrin ... 29 5.1.1.2. ACTH ... 32 5.1.1.3. Topiramate ... 34 5.1.1.4. Pyridoxine ... 34 5.1.1.5. Combinaisons de thérapies ... 35 5.1.2. Diète cétogène ... 36 5.1.3. Chirurgie ... 37 5.1.4. Considérations étiologiques ... 38
5.2. Traitements à long terme ... 38
5.2.1. Traitement antiépileptique ... 39
5.3. Événements indésirables ... 41
5.3.1. ACTH ... 41
6. Physiopathologie ... 43
6.1. Le modèle CRH/stress ... 44
6.2. Les modèles N-méthyl-D-aspartate (NMDA) ... 45
6.3. Le modèle syndrome de Down ... 46
6.4. Le modèle de tétrodotoxine (TTX) ... 47
6.5. Les modèles ARX ... 48
6.6. Le modèle ‘multiple-hit’ ... 50
6.7. Leçons des modèles animaux ... 51
7. Introduction à notre étude ... 53
7.1. Justification de l’étude ... 53
7.2. Étude préliminaire ... 54
7.3. Objectifs ... 56
7.3.1. Objectif principal ... 56
7.3.2. Objectifs secondaires ... 56
Partie II - Présentation d'articles 1. Article 1 ... 58
2. Article 2 ... 86
3. Article 3 ... 120
Partie III - Discussion générale 1. Aperçu global ... 153
1.1. Résultats à court terme ... 154
1.1.1. Interprétations de résultats à court terme ... 155
1.1.1.2. Type de traitement ... 157
1.1.1.3. Ordre et délai de traitement ... 159
1.1.1.4. Polythérapie ... 160
1.1.2. Récurrence ... 162
1.2. Résultats à long terme : Cognition ... 163
1.2.1. Interprétations des résultats à long terme ... 167
1.2.1.1. Étiologie ... 167
1.2.1.2. Type de traitement ... 169
1.2.1.3. Délai de traitement ... 172
1.2.2. Autres facteurs de risque qui pourraient potentiellement influencer la cognition à long terme ... 176
1.2.2.1. Présence d’autres types d’épilepsies avant l’apparition des SI ... 177
1.2.2.2. Présence de retard développemental avant l’apparition de SI... 178
1.2.2.3. Le genre ... 179
1.2.2.4. Âge à l’apparition des SI ... 180
1.2.2.5. Présence de crises épileptiques suivant l’apparition des SI ... 181
1.2.2.6. Anomalies dans l’électroencéphalogramme ... 182
1.3. Résultats à long terme : Autisme ... 183
1.3.1. Facteurs de risque potentiels ... 185
1.3.1.1. Étiologie ... 185
1.3.1.2. Ethnicité... 188
1.3.1.3. Anomalies dans l’électroencéphalogramme ... 190
1.4. Sécurité et innocuité ... 192
2. Orientation future ... 196
Liste des tableaux
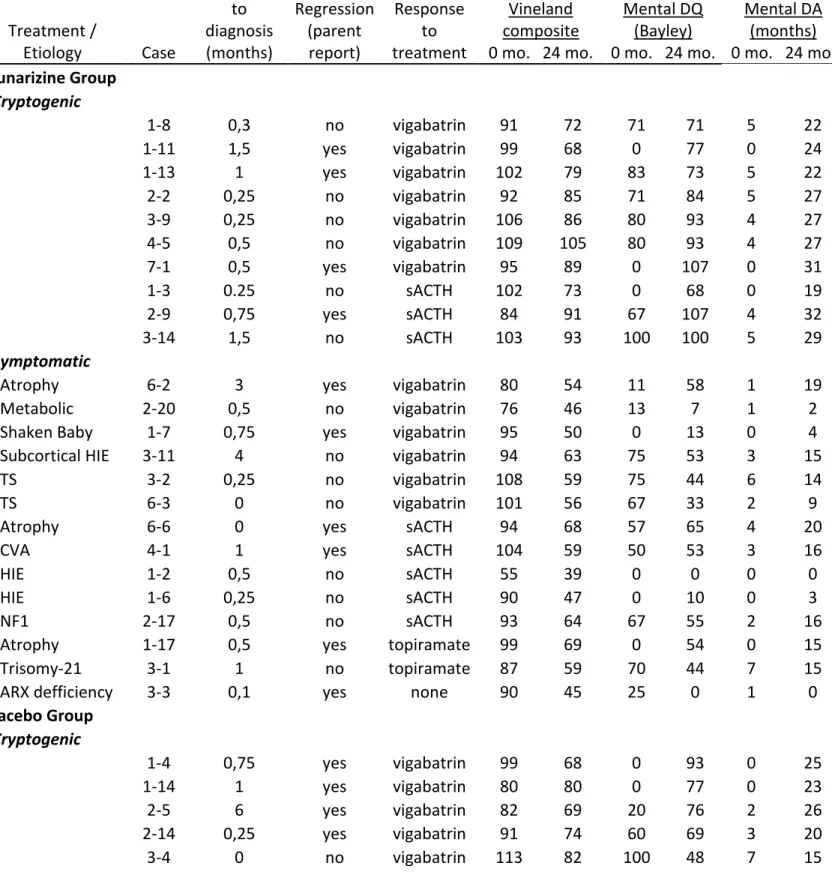
Article 1
Table 1. Clinical characteristics of the study population (N = 68) 78 Table 2. Cognitive outcome and behavioral adaptation in relation to clinical characteristics
(N=69) 79
Table S1 - Detailed clinical characteristics of study population 82
Article 2
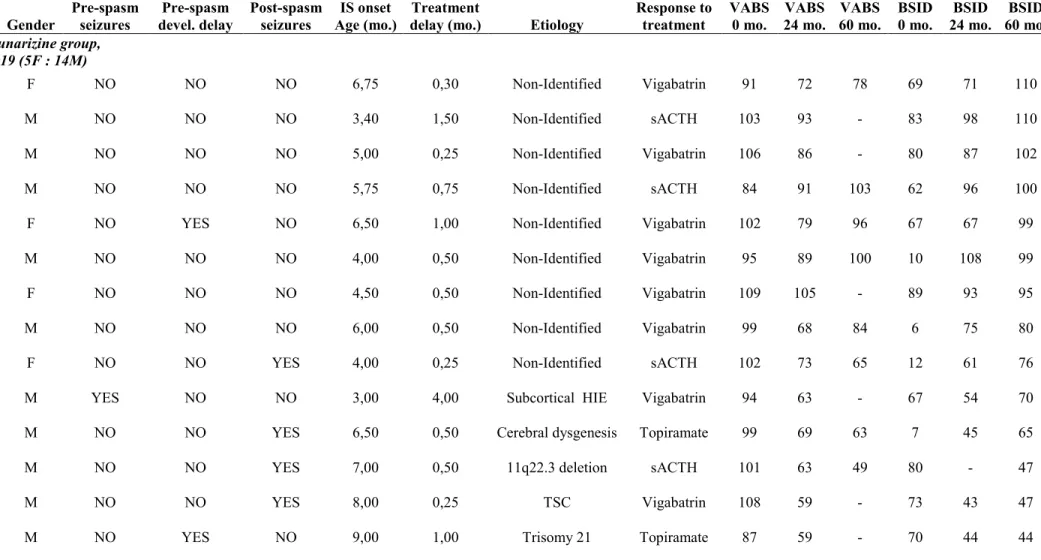
Table 1: Patients characteristics and cognitive evaluations at 60 months 111 Table 2. Patient Clinical data and Cognitive results 113
Article 3
Table 1. ASD outcome in relation to clinical characteristics (N=44) 143 Table 2. Etiology as risk factor for ASD outcome 145 Table S1. Distribution of all focal epileptic discharges post IS treatment for all patients 146
Liste des Figures
Article 1
Figure 1. Flow chart of patients cognitively evaluated at 24 months 84
Figure 2. Response to treatment 85
Article 2 Figure 1. Flow chart of patients cognitively evaluated at 60 months 116
Figure 2 (A-D). Comparison of flunarizine and placebo in the whole cohort (A&B) and in non-identified etiology (NIE) patients (C&D) 117
Figure 3 (A-F). Pre-IS risk factors: Etiology (A&B), pre-IS seizures (C&D) and pre-IS developmental delay (E&F) 118
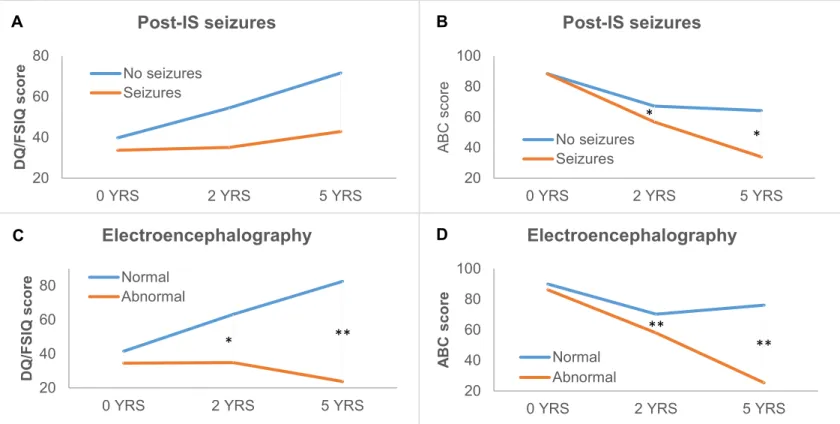
Figure 4 (A-F). Post-IS risk factors: Post-spasm seizures (A&B) and electroencephalography (C & D) 119
Article 3 Figure 1. Flow chart of patients evaluated for ASD with either the CHAT or the ADOS 147
Figure 2. Post-diagnosis epileptiform activity in the 4 cerebral lobes 148
Figure 3. Quantitative EEG improvement over time 149
Figure 4. Difference of cognitive scores between each evaluation 150
Liste de documents spéciaux
Annexe A. Lettre originale du Dr. West au Lancet 216
Annexe B. American Academy of Neurology evidence classification scheme for a
therapeutic article 219
Liste des sigles
AEC = avant l'ère commune ACTH = hormone corticotrope Ca2+ = Calcium
ECR = essai clinique randomisé
EEG = électroencéphalogramme (EEG)
EMIH = Encéphalopathie Myoclonique Infantile avec Hypsarythmie HGC = hybridation génomique comparative
HHS = hypothalamo-hypophyso-surrénalien IRM = imagerie par résonance magnétique NF1 = neurofibromatose de type 1
NMDA = acide N-méthyl-D-aspartique QD = quotient de développement SI = spasmes infantiles
STB = sclérose tubéreuse de Bourneville SW = syndrome de West
TDM = tomodensitométrie
TEP = tomographie par émission de positons TSA = troubles du spectre autistique
VB6 = Vitamine B6
VGB = vigabatrin
Liste des sigles (Anglais)
AAN = American Academy of Neurology ARX = Aristaless-related homeobox
ADOS = Autism Diagnostic Observation Schedule BSID = Bayley Scales of Infant Development CHAT = Checklist for Autism in Toddlers CNS = Child Neurology Society
EDR = Electrodecremental responses FDA = Food and Drug administration
GIRK2 = G-protein–coupled inwardly rectifying potassium-channel subunit 2 HWIS = hypsarrhythmia without infantile spasms
ILAE = International League against Epilepsy ISW = infantile spasms without hypsarrhythmia ISWG = Infantile Spasm Working Group
mTOR = mammalian target of rapamycin NIH = National Institutes of Health
NINDS = National Institute of Neurological Disorders and Stroke SB5 = Stanford-Binet Intelligence Scale, fifth edition
UKISS = United Kingdom Infantile Spasm Study VABS = Vineland Adaptive Behavior Scale WES = Whole exome sequencing
Remerciements
Je tiens tout d’abord à remercier mon directeur de recherche, Dr. Lionel Carmant, qui m’a orienté tout au long de mes études à la maitrise et au doctorat. Il m’a transmis ses
connaissances et m’a offert sa confiance. Il m’a introduit au vaste monde de la recherche médicale et m’a toujours encouragé à m’exprimer dans ce milieu. Je lui en serai toujours reconnaissant. Merci à un maitre et un ami.
Je remercie Dr. Émile Levy et Dr. Florin Amzica qui m’ont parrainé depuis le début de mon doctorat. Chacune de nos rencontres était toujours imbibée de sagesse et de suggestions toujours appréciées. J’étends également mes remerciements au Dr. François Dubeau pour ses conseils soigneux à l’égard de cette thèse. Je remercie aussi Dr. H. Catherine
Sauerwein, qui était non seulement une professeure et une collègue admirable, mais surtout une mentore qui a toujours pris de son temps pour me guider dans mon cheminement académique. Je joins à ses remerciements tous mes collègues et amis du laboratoire Carmant avec qui j’ai eu le privilège de partager des moments mémorables.
J’offre mes plus sincères remerciements à mes parents qui ont depuis toujours investi toutes leurs énergies pour favoriser mon épanouissement et ma réussite. Les sacrifices qu’ils ont fait pour moi sont innombrables. Je leur serai à jamais redevable. À papi Maki et mamie Joëlle, merci pour votre soutien et votre encouragement continu. Maintenant un pas de plus vers le prix Nobel !
À Benny, tu as toujours été un grand frère exemplaire qui m’a tracé et facilité la voie, merci. À Mélissa, merci pour ton fidèle et constant soutien à travers les années. À Moshe et Jennifer, on n’aurait pas pu demander mieux que vous pour se joindre au ‘clan’, merci. À A. Nissim, D. Haim, D. Simha, Akiva et N. Hannah, vous m’offrez tellement sans le savoir. Je suis très fier de vous. Merci.
À Rachel, je ne pourrais jamais te remercier assez pour tout le bien que tu as ajouté dans ma vie. Tu as toujours été là pour m’encourager et me pousser vers tout ce qui est de plus noble dans ce monde. Tu as travaillé tellement dur pour me permettre d’arriver à ce jour. Tu as une part égale dans cette thèse. Merci.
PARTIE 1
1. Aperçu historique 1.1. Épilepsie générale
L'épilepsie est une condition qui a été décrite et étudiée depuis plusieurs millénaires. Des anciens textes akkadiens de la région mésopotamienne datant de l’an 2000 AEC semblent représenter les premiers écrits mentionnant ce désordre (Magiorkinis,
Sidiropoulou, & Diamantis, 2010). D'autres anciens manuscrits célèbres, tels que le texte juridique babylonien, le Code de Hammourabi (1800 AEC) et le papyrus Edwin Smith, un écrit médical égyptien antique (1700 AEC), font également mention de la maladie
(Magiorkinis et al., 2010). Les premiers personnages connus et soupçonnés de souffrir de cette condition incluent le prophète Madianite Bilaam (vers 1300 AEC) et plus tard le roi Saül (environ 1000 AEC), selon certaines interprétations de la Bible (Rosner, 1975). Le Sakikku (1067-1046 avant notre ère), un ancien texte médical babylonien, est le premier manuscrit offrant un compte rendu détaillé de la maladie, décrivant des crises d'épilepsie unilatérales et bilatérales, des crises simples et complexes, le cri épileptique, l'aura épileptique et d'autres caractéristiques cliniques de la maladie (Magiorkinis et al., 2010).
Les savants de ces époques antiques croyaient que l’épilepsie était due à des causes d’origines divine ou supranaturel. En effet, cette condition était habituellement traitée avec des incantations, des rituels et des amulettes contre le ‘mauvais œil’ par les médecins praticiens de l’époque (ILAE, 2005; Magiorkinis et al.; Obladen, 2014). Historiquement, les nourrissons atteints d'épilepsie étaient généralement méprisés et
isolés de la communauté (Obladen, 2014). Le Corpus Hippocraticum daté de l’an 500 AEC et attribué à Hippocrate, est le premier manuscrit connu s’opposant à cette croyance commune et présentant une explication scientifique de la maladie. Dans de morbo sacro, une des œuvres du corpus, l'auteur décrit plutôt un désordre médical traitable prenant source au cerveau (Hippocrates, 1962).
« Voici ce qu'il en est de la maladie dite sacrée : elle ne me paraît avoir rien de plus divin ni de plus sacré que les autres, mais la nature et la source en sont les mêmes que pour les autres maladies… La vérité est que le cerveau est la cause de cette affection, comme des autres très-grandes maladies. » (Page 364)
Malgré la tentative d’Hippocrate de démystifier l'épilepsie, la démonologie restera l'explication privilégiée jusqu'au 18ème siècle (ILAE, 2005). Ce ne sera seulement qu’au
milieu du 19ème siècle, que le premier médicament antiépileptique efficace, le bromure,
sera suggéré par Charles Locock, suivi du premier traitement moderne, le phénobarbital, un demi-siècle plus tard, en 1912 (Brodie, 2010). Depuis, le développement de modèles animaux, a conduit à l'élaboration de dizaines d’autres traitements antiépileptiques (Anderson & Moor, 2010).
1.2. Spasmes Infantiles
Le syndrome de West (SW), aussi connu sous le nom de spasmes infantiles (SI), a été présenté pour la première fois à la communauté scientifique dans la revue médicale The
Lancet, en 1841 (West, 1841). L’auteur de cette publication était le Dr. William James
West, qui en fait, décrivait l’état médical de son propre fils, James Edwin West (West, 1841) (Annexe A).
William James West est né en Angleterre en 1793 ou 1794 (Lux, 2001). Après avoir poursuivi des études de médecine il fut admis en tant que membre du Collège Royal des Chirurgiens en Février 1815 (Lux, 2001). William a épousé Mary Halsey Dashwood en Juin 1828 et ensemble, ils ont eu une fille et deux fils, dont le dernier, James Edwin West, est né en 1840 (Pies & Beardsmore, 1990). Les premiers symptômes de James sont apparus quand il avait quatre mois. Sept mois plus tard, après que le Dr William West eût épuisé toutes les tentatives thérapeutiques qui lui étaient connues, il fit appel à la communauté médicale pour l'aider à trouver un régime de traitement adéquat pour son fils (Duncan, 2001; West, 1841). Sa lettre au Lancet a été écrite le 26 Janvier, 1841 et a été publiée le 13 Février, 1841, correspondant au premier anniversaire de naissance de James Edwin (Lux, 2001; West, 1841). Mary, l'épouse de William, a de son côté également noté une
description minutieuse des symptômes et de l'évolution du désordre de son fils dans son journal personnel (Lux, 2001). Vers 1845, six mois après la mort de son mari, Mary a donné son journal privé à William Newnham, un médecin intéressé par la condition de
James, qui a plus tard publié les détails cliniques de la maladie de James basés sur les écrits de Mary (Newnham, 1849). C’est grâce à ces deux écrits, la lettre du Dr. William West au Lancet et les mémoires de Mary Dashwood transcrites par le Dr. Newnham, que l'histoire de James, ses symptômes et son pronostic, ont été rendus publiques et reconnus comme étant le premier cas documenté de spasmes infantiles (Lux, 2001).
La lettre de West contient une description détaillée de l’état de son fils, et reflète à la fois l'objectivité d'un médecin et la douleur d’un père (West, 1841) :
«The child is now near a year old; was a remarkably fine, healthy child when born, and continued to thrive till he was four months old. It was at this time that I first observed slight bobbings of the head forward, which I then regarded as a trick, but were, in fact, the first indications of disease; for these bobbings increased in
frequency, and at length became so frequent and powerful, as to cause a complete heaving of the head forward towards his knees, and then immediately relaxing into the upright position…these bowings and relaxings would be repeated alternately at intervals of a few seconds, and repeated from ten to twenty or more times at each attack, which attack would not continue more than two or three minutes; he sometimes has two, three, or more attacks in the day; they come on whether sitting or lying; just before they come on he is all alive and in motion, making a strange noise, and then all of a sudden down goes his head and upwards his knees; he then appears frightened and screams out: at one time he lost flesh, looked pale
and exhausted…he neither possesses the intellectual vivacity or the power of moving his limbs, of a child of his age; he never cries at the time of the attacks, or smiles or takes any notice, but looks placid and pitiful…» (Page 724)
À l'âge de six mois la situation de James s’est aggravée lorsqu’il a commencé à éprouver 50 à 60 crises par jour, les attaques de James étaient presque continues depuis leurs premières apparitions à l’âge de quatre mois jusqu'à son premier anniversaire. (West, 1841):
«…the bowing convulsions continued every day, without intermission, for seven months; he had then an interval of three days free; but, on the fourth day, the convulsions returned, with this difference, instead of bowing, he stretched out his arms, looked wild, seem to lose all animation, and appeared quite exhausted. » (Page 725)
Les spasmes se sont finalement arrêtés peu de temps après que James ait eu trois ans bien qu'il eut encore quelques convulsions. À l'âge de quatre ans, il commença à bouger et manger de façon indépendante et à développer une bonne compréhension du langage, sans pour autant communiquer (Lux, 2001). La documentation du Dr. West de crises fréquentes de ‘rire idiot’, et ‘roulages de la tête’ combinée avec son enchantement pour la musique et les ‘couleurs gaies’, ont conduit plusieurs chercheurs à soupçonner que James
a aussi développé des traits autistiques à la suite des spasmes (Lux, 2001; Paciorkowski, Thio, & Dobyns, 2011).
En 1848, James a été envoyé à Park House, un ‘asile pour idiots’ où il avait la réputation d’être un garçon heureux et en bonne santé, pour qui l'équipe médicale avait beaucoup ‘d'espoir’ (Newnham, 1849). Suite à la fermeture de l'asile cinq ans plus tard, James a dû être transféré à l’asile royal d’Earlswood, où en 1858, John Langdon Down (devenu célèbre pour sa description du syndrome de Down) a été nommé directeur médical (Lux, 2001). James est mort quelques années plus tard le 26 Septembre 1860, à l'âge de 20 ans. Une autopsie effectuée par Langdon Down a conclu qu’une tuberculose était responsable de sa mort (Lux, 2001). L'absence de commentaires de Down sur les anomalies du cerveau ont conduit certains chercheurs à spéculer que James avait une forme idiopathique de spasmes infantiles (Lux, 2001).
Au cours des 100 prochaines années, quelques dizaines de rapports médicaux sur le SW ont été publiés, essentiellement décrivant des cas semblables à celui décrit par Dr. West en 1841 (H. Gastaut & Poirier, 1964). Ce n’est que dans les années 1950 que de majeures percées à l’égard de cette condition ont été faites. En 1952, Gibbs et Gibbs ont décrit pour la première fois les caractéristiques électrophysiologiques hypsarythmiques enregistrées sur l'électroencéphalogramme (EEG) généralement associées aux spasmes infantiles (F. A. Gibbs & Gibbs, 1952). Six ans plus tard, en 1958, Sorel et Dusaucy-Bauloye ont dévoilé le premier traitement efficace pour les SI, l'hormone corticotrope, l’adrénocorticotrophine
(ACTH), qui a réussi à contrôler les spasmes chez plusieurs patients (Sorel & Dusaucy-Bauloye, 1958). Depuis, plusieurs autres options thérapeutiques ont été évaluées et approuvées pour le traitement des spasmes infantiles (Go et al., 2012).
L'introduction de la surveillance EEG-vidéo dans l'étude des spasmes infantiles dans les années 1970, a pour la première fois permis d’analyser quantitativement les différentes caractéristiques électrophysiologiques de ce désordre (Frost, Hrachovy, Kellaway, & Zion, 1978). La tomodensitométrie (TDM), l’imagerie par résonance magnétique (IRM) et la tomographie par émission de positons (TEP) développées dans les années 1970 et 1980, ont permis de caractériser les diverses anomalies du cerveau associées aux SI
préalablement indétectables (Hrachovy & Frost, 2003). Ces données ont été utiles pour l’amélioration du diagnostic du SW en identifiant l’étiologie spécifique associée, mais ont aussi servi pour fournir des éclaircissements sur les mécanismes physiopathologiques sous-jacents de ce trouble (Hrachovy & Frost, 2003). Plus récemment, les tests génétiques tel que l'hybridation génomique comparative (HGC) ou le séquençage de l'exome (WES), sont devenus plus répandus et aident à améliorer le diagnostic du SW (Michaud et al., 2014; Paciorkowski, Thio, & Dobyns, 2011).
2. Terminologie et classification
Depuis sa première apparition dans le Lancet au 19ème siècle, la terminologie du désordre
lié aux spasmes épileptiques a connu plusieurs permutations. Au moins 76 synonymes différents ont été enregistrés dans la littérature pour décrire cette condition, y compris les appellations : ‘massive spasms’, ‘bending spasms’, ‘salaam spasms’, ‘nodding convulsions’ et ‘lightning fits’ (Hrachovy & Frost, 2003). En 1960, lorsque Henri Gastaut, fondateur de la division française de la Ligue internationale contre l'épilepsie (ILAE), a organisé le 9ème
Colloque de Marseille, le terme syndrome de West a été officiellement présenté et adopté (H. Gastaut, Roger, Soulayrol, & Pinsard, 1964).
« C’est pourquoi, dans la suite de l’ouvrage et sans préjuger de l’avenir, nous parlerons d’‘Encéphalopathie Myoclonique Infantile avec Hypsarythmie’ pour désigner l’affection en cause et nous utiliserons le sigle : EMIH, excepté lorsque nous nous référerons à l’aspect historique du problème, où nous parlerons alors de syndrome de West. » (Page 19)
À partir de ce congrès, les termes ‘syndrome de West’ et ‘Spasmes Infantiles’ furent le plus souvent utilisés, de façon interchangeable, pour décrire le désordre (Wong & Trevathan, 2001). Cette appellation double pour un même désordre a généré de la confusion dans la littérature, d’autant plus que le terme ‘spasmes infantiles’ était utilisé pour désigner à la fois le syndrome général associé aux spasmes, et à la fois le type de
crise épileptique (Wong & Trevathan, 2001). Cette ambigüité a poussé l’ILAE à consacrer un atelier spécial en 1991 désigné à mieux définir la terminologie associée à ce désordre (ILAE, 1992). Lors de cette rencontre, les experts ont proposé que les termes ‘spasmes’ ou ‘spasmes épileptiques’ soient utilisés pour décrire le type de crise épileptique, et que ‘spasmes infantiles’ et ‘syndrome de West’ servent à représenter le syndrome épileptique en général (Dulac O, 1994). Par ailleurs, certains ont suggéré que le terme ‘syndrome de West’ soit réservé seulement pour les patients démontrant de l’hypsarythmie
accompagnant leurs spasmes épileptiques, alors que ‘spasmes infantiles’ devrait être utilisé plus généralement pour décrire les différentes variétés de syndromes épileptiques qui impliquent des spasmes épileptiques (Wong & Trevathan, 2001). Ces
recommandations ont généralement été retenues par la communauté scientifique internationale. 1
La terminologie étiologique associée aux spasmes infantiles a également causé de la confusion dans la littérature. Pendant longtemps, les termes symptomatiques,
cryptogéniques et idiopathiques ont été utilisés pour décrire l'étiologie associée aux SI, sans pour autant être bien définis (Wong & Trevathan, 2001). Le groupe symptomatique incluait traditionnellement des patients avec des étiologies connues bien que certaines études antérieures englobaient aussi dans cette catégorie des patients sans étiologies
1 Puisque tous les patients de notre étude démontraient aussi de l’hypsarythmie, ce manuscrit utilisera les termes ‘spasmes infantiles’ et ‘syndrome de West’, ainsi que leurs acronymes correspondants ‘SI’ et ‘SW’, de manière interchangeable pour décrire ce désordre.
identifiables mais ayant un retard de développement (Wong & Trevathan, 2001). Les termes idiopathiques et cryptogéniques, représentant des patients avec une étiologie inconnue et un pronostic plus favorable, ont aussi souvent été interchangés (Wong & Trevathan, 2001). Par conséquent, les rencontres de l’ILAE en 1989 et 1992 avaient comme but de clairement définir la classification et la terminologie étiologique associée aux SI (ILAE, 1992). Suite à ces réunions, le terme ‘symptomatique’ fut réservé pour les patients ayant une étiologie connue seulement, ‘cryptogénique’ pour ceux ayant une étiologie indétectable et ‘idiopathique’ pour ceux ayant un examen neurologique et développemental normal au début de l’apparition des spasmes ainsi qu’une absence d’anomalies focales épileptiformes sur l’EEG (ILAE, 1992). Dernièrement, la reclassification des termes étiologiques de l’ILAE en 2010, a présenté les termes ‘génétique,
structural/métabolique et inconnu’, pour définir les 3 nouveaux groupes étiologiques (Berg et al., 2010).2 Le groupe ‘inconnu’ pourrait inclure des patients avec une mutation
génétique probable non détectée (Berg et al., 2010). Dans le but d’améliorer le diagnostic et les traitements, un rapport de l’ILAE en cours de complétion, cherche à étendre ces 3 groupes étiologiques en six catégories: génétique, structurale, métabolique, immunitaire, infectieuse et inconnue (Nelson, 2015).
2 Ce manuscrit se basera sur la plus récente terminologie de l’ILAE. Les termes tels que ‘inconnu’, ‘non identifié’ ou ‘indéterminé’ remplaceront les catégories ‘idiopathique’ et ‘cryptogénique’, et le terme ‘symptomatique’ englobera les étiologies génétiques, structurales et métaboliques.
3. Épidémiologie 3.1. Incidence
L'incidence des spasmes infantiles varie selon les études de 2 à 5 cas par 10 000 naissances (Cowan & Hudson, 1991; Luthvigsson, Olafsson, Sigurthardottir, & Hauser, 1994; Rantala & Putkonen, 1999; R. Riikonen & Donner, 1979; Sidenvall & Eeg-Olofsson, 1995; Trevathan, Murphy, & Yeargin-Allsopp, 1999). Certains considèrent que le taux d’incidence est indépendant de l'emplacement géographique (Wong & Trevathan, 2001) tandis que d'autres ont suggéré un taux d'incidence plus élevé dans les régions nordiques (Rantala & Putkonen, 1999; Shields et al., 1988; Sidenvall & Eeg-Olofsson, 1995). Selon ces études scandinaves, il reste à déterminer si l’incidence élevée est due à des causes
génétiques ou plutôt à des influences environnementales (Hrachovy & Frost, 2003).
3.2. Prévalence
Une étude a rapporté une prévalence de spasmes infantiles estimée à 2 par 10 000 enfants âgés de 10 ans (Cowan, Bodensteiner, Leviton, & Doherty, 1989). Le taux de prévalence plus faible que celui d’incidence s’explique surtout par le développement de spasmes infantiles vers d'autres types de crises (Dulac, 2001), et le taux de mortalité relativement élevé associé aux SI (Wong & Trevathan, 2001). Un taux de prévalence plus élevé dans les régions nordiques a également été suggéré dans certaines études
3.3. Âge à l’apparition des spasmes
Plus de 90% des patients atteints de spasmes infantiles sont diagnostiqués au cours de leur première année de vie (Hrachovy & Frost, 2003; Pellock et al., 2010; Wong & Trevathan, 2001), surtout entre l’âge de 4 et 6 mois (Cohen-Sadan et al., 2009; Cowan & Hudson, 1991; Djuric, Kravljanac, Tadic, Mrljes-Popovic, & Appleton, 2014; Dulac, 2001; Jeavons, Bower, & Dimitrakoudi, 1973; Koo, Hwang, & Logan, 1993; J. Lee, Lee, Yu, & Lee, 2013; Lombroso, 1983; Primec, Stare, & Neubauer, 2006; R. Riikonen, 1982; Sidenvall & Eeg-Olofsson, 1995). Au moins une étude a montré deux pics d'incidences plus élevées correspondant à 3-5 mois et 6-8 mois (Kurokawa et al., 1980). Les spasmes infantiles commencent rarement à un âge inférieur à deux semaines ou supérieur à 18 mois (Cowan & Hudson, 1991; Dulac, 2001), bien que certaines exceptions aient été notées (Kurokawa et al., 1980; Lombroso, 1983; Lux et al., 2005; Rantala & Putkonen, 1999).
3.4. Le genre
Certaines études suggèrent une prédominance masculine significative (Cohen-Sadan et al., 2009; Dulac, 2001; Hrachovy & Frost, 2003; Jeavons et al., 1973; Wong & Trevathan, 2001), ou légère chez les patients avec le SW (R. Riikonen & Donner, 1979; Sidenvall & Eeg-Olofsson, 1995), ou une prédominance masculine que chez les patients avec une étiologie symptomatique (Koo et al., 1993; Lombroso, 1983). Cependant, de nombreuses autres études ne démontrent pas de différence basée sur le sexe (Kurokawa et al., 1980;
Matsumoto et al., 1981; Rantala & Putkonen, 1999; Sharma & Vishwanthan, 2008; Trevathan et al., 1999), et certains investigateurs suggèrent que la prépondérance
masculine observée dans plusieurs études est simplement le reflet du plus grand nombre de garçons participant aux études de SW et qu’en réalité les deux sexes sont affectés de la même manière (Hrachovy & Frost, 2003).
3.5. Étiologie
Plus de 200 étiologies structurales, métaboliques, ou génétiques ont été associées aux spasmes infantiles (Frost & Hrachovy, 2005; Kossoff, 2010). Les étiologies sont souvent subdivisées en 3 catégories : prénatales, périnatales, et postnatales. Les causes prénatales comprennent les infections intra-utérines, des malformations du développement cortical, des syndromes neurocutanés, des troubles métaboliques et des anomalies génétiques ou chromosomiques. Le groupe périnatal se compose principalement d'encéphalopathie hypoxique-ischémique et de traumatismes obstétricaux (Wong & Trevathan, 2001). Les étiologies postnatales comprennent les infections, traumatismes, insultes hypoxiques-ischémiques et tumeurs (Wong & Trevathan, 2001).
Les nouvelles techniques en neuroimagerie ainsi que l’utilisation de récentes techniques de diagnostic génétiques, ont augmenté le rendement de détection de cas
symptomatiques de plus de 20% (Cohen-Sadan et al., 2009; Michaud et al., 2014; Trasmonte & Barron, 1998). En effet, au début des années 1980, les étiologies
symptomatiques représentaient environ 45-60% des cas de SW (Kurokawa et al., 1980; Lombroso, 1983; Matsumoto et al., 1981), tandis que des études plus récentes ont atteint un taux de 70-85% (Cohen-Sadan et al., 2009; Kossoff, 2010; Rantala & Putkonen, 1999; Sidenvall & Eeg-Olofsson, 1995). Les patients sans étiologie connue représentent maintenant une proportion relativement plus faible, estimée à 15-30%, des cas de SW (Cohen-Sadan et al., 2009; Kossoff, 2010; Rantala & Putkonen, 1999; Sidenvall & Eeg-Olofsson, 1995), et sont généralement soupçonnés d’avoir des origines génétiques encore indétectables (Michaud et al., 2014).
3.6. Taux de mortalité
Le taux de mortalité associé aux spasmes infantiles est estimé à 10 à 20% dans la plupart des études (Darke et al., 2010; Hrachovy & Frost, 2003; Matsumoto et al., 1981; R. Riikonen, 1982; Trevathan et al., 1999), un taux provenant majoritairement du sous-groupe symptomatique (Djuric et al., 2014; Hrachovy & Frost, 2003; Jeavons et al., 1973; R. Riikonen, 1996). Toutefois, une étude ayant suivi 214 patients finlandais jusqu’à l'âge adulte de 35 ans, a rapporté un taux de mortalité de 31% chez ces patients, dont le tiers est décédé avant l'âge de 3 ans et presque tous les autres avant l'âge de vingt ans (R. Riikonen, 1996). Les décès chez les patients atteints de SI sont surtout dus à des infections (R. Riikonen, 1996; Wong & Trevathan, 2001).
4. Caractéristiques cliniques
4.1. Spasmes
En 1979, Kellaway et al. ont présenté la première analyse quantitative des caractéristiques des spasmes associés au SW (Kellaway, Hrachovy, Frost, & Zion, 1979). Les spasmes sont généralement constitués de brèves contractions musculaires impliquant le cou, le tronc et les extrémités, de manière bilatérale et symétrique. Ils sont classés en trois catégories: fléchisseurs, extenseurs et mixtes (fléchisseurs et extenseurs) (Hrachovy & Frost, 2003; Kellaway et al., 1979; Wong & Trevathan, 2001).
L'activité musculaire d’un spasme individuel se compose de deux parties: une première composante phasique d'une durée inférieure à 2 s, suivie d'une composante tonique plus soutenue, moins intense, pouvant durer jusqu'à 10 s (Kellaway et al., 1979; Pavone, Striano, Falsaperla, Pavone, & Ruggieri, 2014). Chez certains nourrissons, la phase tonique peut être absente (Pellock et al., 2010). Les spasmes épileptiques ont tendance à se produire en bouffées de deux à plus de 100 spasmes d’une durée de 1 à plus de 10 min par bouffée (Hrachovy & Frost, 2003; Kellaway et al., 1979; Pellock et al., 2010; West, 1841). Les patients peuvent avoir des dizaines de bouffées de spasmes et plusieurs centaines de spasmes à chaque jour (Wong & Trevathan, 2001). Moins souvent, les spasmes peuvent se produire isolément plutôt qu’en groupe, une variante de spasmes connue sous l’appellation infantile spasms single-spasm variant (ISSV) (Pavone et al.,
Les spasmes se produisent à peu près à la même fréquence en journée ou la nuit et se présentent fréquemment à l’éveil (Horita, 2001; Hrachovy & Frost, 2003; Kellaway et al., 1979; Wong & Trevathan, 2001). Certains enfants éprouvent de l'irritabilité ou de
l’hyporéactivité transitoire après que les spasmes se soient atténués (Pavone et al., 2014). Les spasmes peuvent être de nature asymétrique (ex. hémicorps) ou impliquer des
contractions isolées du cou ou des muscles abdominaux entraînant des hochements de tête subtiles ou des légers mouvements du tronc (Wong & Trevathan, 2001). D'autres manifestations cliniques des spasmes peuvent inclure des mouvements oculaires
anormaux, ou des modifications de la respiration et du rythme cardiaque, l’hypersudation et le larmoiement (Wong & Trevathan, 2001). Ces manifestations cliniques plus subtiles rendent le diagnostic souvent plus difficile pour les cliniciens, et d’autant plus pour les parents (Hrachovy & Frost, 2003; Lux & Osborne, 2004; Napuri, E, Dulac, Chaperon, & Riou, 2010; Pellock et al., 2010).
4.2. Électroencéphalographie (EEG) 4.2.1. Enregistrements intercritiques
La manifestation épileptique intercritique la plus commune associée au syndrome de West est appelée hypsarythmie (F. A. Gibbs & Gibbs, 1952; Wong & Trevathan, 2001). Cependant, les spasmes épileptiques ont été rapportés chez des patients avec SI qui ne comprennent pas d’hypsarythmie (Caraballo et al., 2011; Donat & Wright, 1991; Pavone et al., 2014) chez près d'un tiers des patients (Dulac, 2001). En revanche, l’hypsarythmie
n’est pas spécifique aux spasmes infantiles et peut être associée à d'autres conditions non-épileptiques (Friedman & Pampiglione, 1971; Lux & Osborne, 2004). Dans de tels cas, on parlera de infantile spasms without hypsarrhythmia (ISW) ou bien de hypsarrhythmia
without infantile spasms (HWIS) (Pavone et al., 2014).
L’Hypsarythmie a été caractérisée pour la première fois par Gibbs et Gibbs dans les années 1950.
“…random high voltage slow waves and spikes. These spikes vary from moment to moment, both in location and duration. At times they appear to be focal, and a few seconds later they seem to originate from multiple foci. Occasionally, the spike discharge becomes generalized, but it never appears as a rhythmically repetitive and highly organized pattern that could be confused with a discharge of the petit mal variant type. The abnormality is almost continuous, and in most cases it shows as clearly in the waking as in the sleeping record…” (F. A. Gibbs & Gibbs, 1952)
Cette description résume les caractéristiques chaotiques, asynchrones et dynamiques de l’hypsarythmie. Ces caractéristiques électrophysiologiques sont le plus fréquemment observées lors du sommeil à ondes lentes, ensuite lors des périodes d’éveils, et ne se produisent presque pas pendant le sommeil paradoxal (Hrachovy, Frost, & Kellaway, 1984; Watanabe, Negoro, Aso, & Matsumoto, 1993). L’hypsarythmie apparait habituellement au cours de la petite enfance et disparait complètement vers l’âge de cinq ans (Hrachovy & Frost, 2003; Wong & Trevathan, 2001).
Certaines variations du modèle classique d’hypsarythmie ont été décrites et étiquetées ‘hypsarythmie modifiée’ (Hrachovy et al., 1984). Ces variantes peuvent inclurent une augmentation de la synchronisation inter-hémisphérique, des épisodes d'atténuation d’amplitude, la présence d'un foyer constant épileptiforme et une hypsarythmie
asymétrique. Étant donné l’abondance relative de ces types d’hypsarythmie et le fait qu’il ne sont pas nécessairement des dérivés du modèle hypsarythmique classique (Hrachovy & Frost, 2003; Wong & Trevathan, 2001), l'ILAE a conclu que le terme ‘hypsarythmie
modifiée’ ne devrait plus être utilisé, et que les caractéristiques atypiques devraient simplement être précisées le cas échéant (ILAE, 1992).
Bien que l’hypsarythmie et ses variantes soient les plus fréquemment observées sur l’EEG en association avec les spasmes infantiles, d'autres représentations intercritiques peuvent se produire. Ceux-ci peuvent inclure des pointes focales ou multifocales ou des rythmes d’activités électriques anormalement lents ou rapides, qui peuvent apparaître
individuellement ou en combinaisons. Dans certains cas rares, l'EEG peut être normal, mais il est tout à fait probable que dans de tels cas, l’hypsarythmie était présente à un moment donné du désordre (Hrachovy & Frost, 2003).
4.2.2. Enregistrements critiques
Les corrélats EEG critiques des spasmes épileptiques ont été étudiés en détail grâce à la surveillance d’EEG-vidéo (Fusco & Vigevano, 1993; Kellaway et al., 1979). Un premier
rapport à ce sujet a décrit 11 différents types d'enregistrements associés aux SI (Kellaway et al., 1979). Les réponses décrémentielles, electrodecremental responses (EDR),
représentent la caractéristique critique la plus courante, survenant dans plus de 70% des cas enregistrés (Hrachovy & Frost, 2003; Wong & Trevathan, 2001). Par contre, aucune corrélation entre les différents types d’enregistrements critiques à l’EEG et le
comportement clinique des patients avec SI n'a pu être établie (Hrachovy & Frost, 2003).
4.3. Résultats neurodéveloppementaux à long terme
4.3.1. Délai cognitif
La troisième composante traditionnellement inclue dans la triade définissant le syndrome de West, en plus des spasmes cliniques et des enregistrements hypsarythmiques, est la régression neurodéveloppementale (Pellock et al., 2010). Un retard cognitif se produit chez plus de 80% des patients atteints de spasmes infantiles (Hrachovy & Frost, 2003; Jeavons et al., 1973; Koo et al., 1993; Kurokawa et al., 1980; Matsumoto et al., 1981; R. Riikonen, 1982, 1996; Sidenvall & Eeg-Olofsson, 1995; Trevathan et al., 1999), dont
jusqu’à la moitié de ces cas sont considérés graves (Jeavons et al., 1973; R. Riikonen, 1982; Trevathan et al., 1999). Des études longitudinales ont démontré que seulement 1 sur 6 à 8 patients avec SW sont plus tard scolarisés dans des établissements d'enseignement
régulier (Jeavons et al., 1973; Matsumoto et al., 1981; R. Riikonen, 1982), et moins de 5% de cette population se marient et ont des enfants (R. Riikonen, 1996). Plus récemment, le groupe West Delphi a suggéré de supprimer la composante cognitive comme
caractéristique déterminante du SW en raison des difficultés d'évaluations du développement chez les nourrissons (Lux & Osborne, 2004). Néanmoins, un rapport cognitif approprié au moment du diagnostic demeure important en raison de son association avec les résultats développementaux subséquents (Lux & Osborne, 2004).
De nombreuses études ont tenté de révéler les facteurs cliniques associés à un pronostic favorable ou néfaste chez les patients avec SW. Ces marqueurs de risque peuvent
généralement être divisés en deux catégories ; les facteurs potentiels présents avant l’apparition des spasmes et ceux qui se manifestent subséquemment. Les facteurs pré-SI couramment analysés incluent le sexe, l'âge, la présence d'une étiologie symptomatique, le retard mental ou la présence d'autres types de crises. D'autre part, les facteurs post-SI peuvent inclure le type et le délai de traitement, les résultats à l’EEG et l'évolution des spasmes épileptiques vers d'autres types de crises. Bien que certaines études aient tenté de dévoiler les risques associés à un déclin cognitif, elles présentent souvent des résultats contradictoires. Ces résultats peu concluants sont attribués à l'hétérogénéité des
différents protocoles de traitement utilisés, des diverses évaluations cognitives impliquées, et le nombre relativement faible d'enfants analysés dans chaque étude
(Hrachovy & Frost, 2003). Un sommaire des facteurs de risques potentiellement associés à un délai neurodéveloppemental sera présenté dans cette section.3
3 Les données disponibles au début de notre étude sont présentées dans cette section. Une revue de littérature plus récente à l’égard des facteurs de risque cliniques sera abordée dans la discussion générale.
4.3.1.1. Facteurs de risques pré-SI
Le facteur prédictif de risque cognitif à long terme le plus reconnu et universellement accepté est l'étiologie (Glaze, Hrachovy, Frost, Kellaway, & Zion, 1988; Hrachovy & Frost, 2003; Jeavons et al., 1973; Koo et al., 1993; Matsumoto et al., 1981; Rantala & Putkonen, 1999; R. Riikonen, 1982, 1996; Sidenvall & Eeg-Olofsson, 1995; Wong & Trevathan, 2001). Les patients sans étiologie connue associée à leurs spasmes épileptiques ont une
probabilité de 30-50% d'avoir des retards développementaux comparativement à environ 85-100% des patients du groupe symptomatique (Glaze et al., 1988; Koo et al., 1993; Rantala & Putkonen, 1999; Sidenvall & Eeg-Olofsson, 1995). Certains ont suggéré que des étiologies spécifiques comme la neurofibromatose, le syndrome de Down, la leucomalacie périventriculaire et l'hypoglycémie néonatale sont l'exception à cette règle, démontrant des résultats cognitifs relativement meilleurs (Wong & Trevathan, 2001).
D'autres facteurs pré-spasmodiques, tels que la présence de retard mental (Matsumoto et al., 1981; R. Riikonen, 1982, 1996; Wong & Trevathan, 2001), ou la présence d'autres types de crises (Hrachovy & Frost, 2003; Jeavons et al., 1973; R. Riikonen, 1982; Wong & Trevathan, 2001), sont soupçonnés d'avoir un effet négatif sur la cognition. Cependant, certains n’ont pas remarqué de tels effets (Matsumoto et al., 1981).
Un âge plus précoce d’apparition de spasmes, autour de 3-4 mois, a été associé à un risque plus élevé de retard cognitif (Jeavons et al., 1973; Lombroso, 1983; R. Riikonen,
1982; Wong & Trevathan, 2001). Cependant, une quantité comparable d'études n'a
démontré aucun effet de l’âge du début des spasmes sur le développement cognitif (Glaze et al., 1988; Kurokawa et al., 1980; Matsumoto et al., 1981). Aussi, le genre n'a pas été identifié comme facteur de risque lié aux troubles cognitifs (Matsumoto et al., 1981).
4.3.1.2. Facteurs de risque post-SI
Une présence prolongée de spasmes, que ce soit en raison d'un retard de diagnostic ou de traitement des spasmes épileptiques, a aussi été associée à un pronostic inférieur à long terme (Jeavons et al., 1973; Koo et al., 1993; R. Riikonen, 1982, 1996; Wong & Trevathan, 2001). Certaines études ne démontrent pas d'avantages liés à une cessation de spasmes précoce (Askalan et al., 2003; Glaze et al., 1988; Jeavons et al., 1973; Lombroso, 1983) tandis que d'autres l’observent seulement chez les patients sans étiologies identifiables (Kivity et al., 2004; Lombroso, 1983; Matsumoto et al., 1981). Le rapport autoritaire de l’American Academy of Neurology et du Child Neurology Society (AAN/CNS) sur les spasmes infantiles, a conclu que les preuves de bienfaits dus aux traitements précoces étaient ‘contradictoires’ et ‘insuffisantes’ (Mackay et al., 2004).
Une réponse positive au traitement hormonal, ACTH, a été associée à un meilleur
pronostic dans certaines études (Koo et al., 1993; R. Riikonen, 1982, 1996). Par contre, la majorité des essais cliniques n’ont pu démontrer une différence d’efficacité entre l’ACTH et les autres traitements évalués (Glaze et al., 1988; Hrachovy, Glaze, & Frost, 1991;
Jeavons et al., 1973; Kurokawa et al., 1980; Matsumoto et al., 1981). L’AAN/CNS considère qu’il n’y a pas assez de preuves pour endosser un traitement plus qu’un autre pour
améliorer les effets cognitifs à long terme (Mackay et al., 2004). Des études évaluant des doses d’ACTH plus élevées n’ont soit démontré aucun bienfaits (R. Riikonen, 1982) ou soit démontré certains bienfaits limités au sous-groupe de patients sans étiologies connues (Kivity et al., 2004).
Les données électroencéphalographiques ont également été analysées par rapport au neurodéveloppement. Une présence ou non d’hypsarythmie n’a pas démontré d’influence sur les résultats cognitifs (Jeavons et al., 1973; Koo et al., 1993). Cependant, au moins une étude a noté que l’hypsarythmie atypique était liée à des résultats cognitifs néfastes par rapport à l’hypsarythmie classique (Lombroso, 1983). En outre, une étude quantifiant le degré de sévérité d’hypsarythmie a démontré que les enregistrements les plus
‘chaotiques’ étaient liés à des résultats cognitifs inférieurs (Kramer, Sue, & Mikati, 1997). De même, une persistance d’hypsarythmie a démontré une corrélation avec une
détérioration cognitive ultérieure (Dulac, 2001; Gaily et al., 1999). A l’inverse, une
normalisation rapide de l'EEG, de préférence dans les deux semaines suivant le diagnostic, était liée à un meilleur pronostic (Koo et al., 1993).
Des crises récurrentes étaient associées à des résultats cognitifs faibles en général (Hrachovy & Frost, 2003; Matsumoto et al., 1981; R. Riikonen, 1982, 1996) ou du moins chez les patients sans étiologie connue (Koo et al., 1993). D’autres risques de pronostics
négatifs incluaient une présence de déficits neurologiques tels que la paralysie cérébrale (Jeavons et al., 1973; Koo et al., 1993).
4.3.2. Autisme
La relation entre l'épilepsie et l'autisme a été révélée pour la première fois il y a près d’un demi-siècle (Kanner, 1968). Les patients avec des troubles du spectre de l’autisme (TSA) sont plus à risque de développer des troubles épileptiques par rapport à la population générale (Tuchman & Rapin, 2002). Dans de nombreux cas tels que les spasmes infantiles, les troubles épileptiques sont diagnostiqués avant la détection de l’autisme. Des études ont révélé une incidence des TSA de 9 à 35% chez les patients atteints du syndrome de West, une comorbidité généralement exprimée chez les patients SI symptomatiques (Dilber et al., 2013; R. Riikonen & Amnell, 1981; Saemundsen, Ludvigsson, & Rafnsson, 2007, 2008; Sidenvall & Eeg-Olofsson, 1995). Les traits autistiques chez ces patients persistent souvent même après l’arrêt de leurs crises épileptiques (Dulac, 2001).
4.3.3. Crises épileptiques
Plus de la moitié des patients avec SW, en particulier ceux avec des étiologies
symptomatiques, développent d'autres types de crises épileptiques (Glaze et al., 1988; Jeavons et al., 1973; Koo et al., 1993; Lombroso, 1983; Matsumoto et al., 1981; Partikian & Mitchell, 2010; Pavone et al., 2014; R. Riikonen, 1982). Des études longitudinales de
patients ayant des antécédents de SI révèlent la présence d'épilepsie réfractaire
chronique chez la moitié des patients (Koo et al., 1993; Kurokawa et al., 1980; Matsumoto et al., 1981; R. Riikonen, 1982). Les types de crises associés incluent des crises partielles, myocloniques, toniques, et tonico-cloniques (Hrachovy & Frost, 2003; Wong & Trevathan, 2001).
De nombreuses études ont spécifiquement examiné la progression des spasmes infantiles au syndrome de Lennox-Gastaut (SLG) en raison de leurs caractéristiques cliniques, électroencéphalographiques et cognitifs semblables (Donat & Wright, 1991; Wong & Trevathan, 2001). La plupart des études rapportent une prévalence de SLG chez environ 10-25% des patients avec SI (Hrachovy & Frost, 2003; Lombroso, 1983; Rantala &
Putkonen, 1999; R. Riikonen, 1982) alors que certaines études suggèrent que le
pourcentage de cas SLG est aussi élevé que 50% (Ohtahara, Yamatogi, Ohtsukd, Oka, & lshida, 1980; Trevathan et al., 1999; Wong & Trevathan, 2001).
4.3.4. Évolution naturelle des SI
Des données limitées de rémission spontanée sont disponibles grâce aux études pré-ACTH (première moitié du 20ème siècle) où les patients ont reçu des traitements inefficaces
(Mackay et al., 2004), ou à partir de quelques cas plus récents où les parents ont refusé des traitements pour leurs enfants (Hrachovy et al., 1991). Une étude analysant
montré un taux de rémission spontanée de 25% après un an (Hrachovy et al., 1991). La grande majorité des patients de cette étude (91%) ont obtenu des résultats cognitifs anormaux après avoir été suivis pour une moyenne de 80 mois (Hrachovy et al., 1991).
Un autre rapport sur les effets à long terme de 69 patients atteints de SI ayant reçu des traitements considérés inefficaces a été publié dans les années 1950 (E. L. Gibbs, Fleming, & Gibbs, 1954). Dans cette population, à l’âge de 5 ans, 11% des enfants avaient encore des spasmes infantiles, 45% avaient d'autres types de crises, 31% avait une hypsarythmie persistante, 52% avaient des anomalies focales sur leur EEG et seulement 17% avaient un EEG normal. A long terme, 87% des enfants de plus de 1 an avaient un développement intellectuel anormal et le taux de mortalité était de 11% avant l'âge de 2 ans (E. L. Gibbs et al., 1954).
Avec ou sans traitements, la majorité des patients atteints du syndrome de West n’ont plus de spasmes après l’âge de 5 ans (Dulac, 2001; Hrachovy & Frost, 2003; Jeavons et al., 1973; Lux & Osborne, 2004; Pavone et al., 2014; R. Riikonen, 1982; Trevathan et al., 1999).
5. Options thérapeutiques
Depuis la découverte de l’efficacité de l'hormone corticotrope (ACTH) comme traitement pour SI, de nombreux essais cliniques novateurs ont essayé d'élargir l’éventail de
traitements potentiels de ce désordre. Bien que ces tentatives aient conduit à différentes options thérapeutiques, une divergence d’opinions à l’égard du traitement optimal a également surgi (Mackay et al., 2004). Traditionnellement, la pratique américaine était d’administrer l’ACTH initialement, indépendamment de l'étiologie associée (Bobele & Bodensteiner, 1994), tandis que les Britanniques favorisaient le vigabatrin comme première ligne de traitement (Appleton, 1996) et les japonais administraient
généralement, dans l'ordre, la pyridoxine, le valproate, et une faible dose d’ACTH (Ito, Seki, & Takuma). A ce jour, il n'y a pas de consensus clair entre les praticiens quant au traitement optimal pour les spasmes infantiles, sauf dans certains cas d’étiologies bien définis (section 5.1.4. ‘Considérations étiologiques’) (Wilmshurst et al., 2015).
En plus des traitements antiépileptiques, d'autres options thérapeutiques ont été utilisées et évaluées dans le but de dévoiler le traitement optimal pour les spasmes infantiles. Des alternatives aux médicaments pharmaceutiques incluent la diète cétogène, un régime à haute teneur en matières grasses et à faible taux glucidique, ou la chirurgie dans certains cas réfractaires (Kossoff, 2010).
Quel que soit le type de traitement, les chercheurs ont généralement essayé d’atteindre les 3 objectifs suivants dans leur quête d’un traitement optimal (Mackay et al., 2004):
1. Un résultat positif à court terme, défini par une cessation complète de spasmes, une résolution de l’hypsarythmie, et un faible taux de récidive.
2. Une amélioration des résultats à long terme, définie par l'amélioration des capacités cognitives et des taux réduits de crises épileptiques.
3. L'innocuité des traitements.
Un aperçu des options thérapeutiques conventionnelles est présenté dans cette section suivant ces critères, avec une emphase spéciale sur les traitements antiépileptiques utilisés dans notre étude, soit l’ACTH, le vigabatrin, le topiramate et la pyridoxine.4,5
5.1. Traitements à court terme 5.1.1. Antiépileptiques
5.1.1.1. Vigabatrin
Le Gamma-vinyl GABA, vigabatrin, un inhibiteur de l'acide gamma-aminobutyrique transaminase (GABA-T) responsable du catabolisme de GABA, a prouvé son efficacité pour le traitement de spasmes infantiles depuis le début des années 1990 (Chiron et al.,
4 Les données thérapeutiques disponibles au début de notre étude seront présentées dans cette section. Des données plus récentes seront examinées dans la discussion générale.
5 Une attention particulière est donnée au rapport autoritaire de l’AAN/CNS (Mackay et al., 2004) pour établir les preuves d’efficacité des divers antiépileptiques (Annexe B).
1991). Son utilisation a rapidement augmenté, initialement au Canada et en Europe et, par la suite, aux États-Unis après son approbation en 2009 par le Food and Drug
administration (FDA) (Kossoff, 2010; Willmore, Abelson, Ben-Menachem, Pellock, &
Shields, 2009). Le groupe d’étude du AAN/CNS a analysé l'efficacité du vigabatrin dans 14 études; trois essais cliniques randomisés (ECR), six essais ouverts et cinq études
rétrospectives (Mackay et al., 2004).
Dans le seul ECR contrôlé par placebo (preuve de classe I)6, le vigabatrin ou placebo a été
utilisé pour les 5 premiers jours de traitement avant de passer à une étude ouverte de vigabatrin en monothérapie (Appleton, Peters, Mumford, & Shaw, 1999). Dans cette étude, 35% des patients traités avec vigabatrin n’avaient plus de spasmes dans un délai de 5 jours par rapport à 10% dans le groupe placebo, et 42% de tous les patients entrant dans la phase ouverte de l’étude ont atteint par la suite un arrêt complet des spasmes (Appleton et al., 1999). Dans les deux autres ECR (preuve de classe III), une étude a montré un taux d'arrêt des spasmes de 23% à 2 semaines, et de 65% à 3 mois (Elterman, Shields, Mansfield, Nakagawa, & Group, 2001), tandis que l'autre étude a montré un taux d’arrêt des spasmes et une résolution de l’hypsarythmie de 48% au bout de 2 semaines (Vigevano & Cilio, 1997). Les patients avec étiologies inconnues ont eu un taux de réponse légèrement meilleur dans ces trois ECR (27-57% vs. 21-44%) (Appleton et al., 1999;
Elterman et al.; Vigevano & Cilio, 1997).
Les six études prospectives ont eu un taux de réponse de 0 à 59% pour les enfants avec des étiologies symptomatiques et de 50 à 100% pour les patients sans étiologies connues (Covanis, Theodorou, Lada, Skiadas, & Loli, 1998; Fejerman et al., 2000; Granstrom, Gaily, & Liukkonen, 1999; Siemes, Brandl, Spohr, Volger, & Weschke, 1998; Vles, van der
Heyden, Ghijs, & Troost, 1993; Wohlrab, Boltshauser, & Schmitt, 1998). Des doses variées de vigabatrin ont été utilisées dans ces neuf études (3 ECR et 6 essais ouverts) où une cessation de spasmes a été obtenue en 12 à 35 jours, une résolution d’hypsarythmie atteinte en 7 à 35 jours chez 11 à 83% des enfants, et un taux de rechute enregistré dans 0 à 29% des cas.
Le vigabatrin s’est avéré être très efficace dans les cas où la sclérose tubéreuse de Bourneville (STB) était associée aux SI. Quarante-et-un des 45 patients analysés dans le rapport du groupe AAN/CNS avec un diagnostic de STB ont vu un arrêt de leurs spasmes grâce au vigabatrin (Aicardi, Mumford, Dumas, & Wood, 1996; Covanis et al., 1998; Elterman et al., 2001; Fejerman et al., 2000; Granstrom et al., 1999; Vigevano & Cilio, 1997; Vles et al., 1993).
Basé sur ces travaux, le rapport du groupe AAN/CNS a conclu que le vigabatrin était ‘possiblement efficace pour le traitement à court terme des spasmes infantiles’ et ‘possiblement efficace pour le traitement à court terme des spasmes infantiles chez la majorité des enfants atteints de STB’ (Mackay, 2004).
5.1.1.2. ACTH
L'hormonothérapie a été le premier traitement crédité à avoir un effet rapide sur les spasmes et demeure le traitement de choix de nombreux médecins (Mackay et al., 2004; Sorel & Dusaucy-Bauloye, 1958). Quatorze études évaluant l'efficacité de l'ACTH sur la cessation des spasmes et la résolution d’hypsarythmie ont été inclues dans le rapport de l’AAN/CNS (Mackay et al., 2004).
Les études d’ACTH à doses élevées ou faibles ont démontré un arrêt des spasmes chez 42 à 100% des patients, généralement en 1-2 semaines (Mackay et al., 2004). Parmi ces études, cinq étaient des ECR, dont une (preuve de classe I) qui a démontré un taux d'arrêt de spasmes de 87% avec une dose élevée (Baram et al., 1996) et un autre (preuve de classe II) qui a atteint 42% avec une faible dose d’ACTH (Hrachovy, Frost, Kellaway, & Zion, 1983). Toutefois, deux études d'ECR (preuve de classe III) n’ont noté aucune différence dans la réponse clinique entre les différentes doses d’ACTH administrées (Hrachovy, Frost, & Glaze, 1994; Yanagaki et al., 1999). Dans quatre des cinq ECR, les patients sans
étiologies connues ont démontré une meilleure réponse clinique (Hrachovy et al., 1994; Hrachovy et al.; Vigevano & Cilio, 1997; Yanagaki et al., 1999). Des rechutes sont
survenues dans 15-33% de ces cinq études. (Baram et al., 1996; Hrachovy et al., 1994; Hrachovy et al.; Vigevano & Cilio, 1997; Yanagaki et al., 1999).
Quatre autres études ouvertes ont fourni des preuves de classe III (Hrachovy, Frost, Kellaway, & Zion, 1980; Lombroso, 1983) ou classe IV (Kusse et al., 1993; Snead et al., 1989). Une étude (n=5) a observé une réponse clinique chez tous ses patients avec une faible dose d’ACTH (Hrachovy et al., 1980) tandis qu’une autre étude (n=15) a eu un succès de 93% avec une dose élevée (Snead et al., 1989). Les taux de rechute étaient de 20 et 36%, respectivement. Les autres études rétrospectives ont fourni des preuves de classe IV (Cossette, Riviello, & Carmant, 1999; R. Riikonen & Simell, 1990; Sher & Sheikh, 1993; Singer, Rabe, & Haller, 1980; Snead, Benton, & Myers, 1983). L’ensemble de tous ces essais cliniques ont illustré un taux de cessation de spasmes entre 59 à 100%, une résolution de l’hypsarythmie entre 57 et 97%, et un taux de rechute entre 9 et 62%.
Deux des études, dont un ECR, ont comparé l'efficacité du vigabatrin et de l’ACTH (Cossette et al., 1999; Vigevano & Cilio, 1997). Bien que la normalisation du EEG était supérieure pour les enfants traités avec l’ACTH dans les deux études, le taux de rechute était plus élevé (Cossette et al., 1999; Vigevano & Cilio, 1997). Les effets secondaires étaient également plus fréquents chez les patients traités avec l’ACTH (27-37% contre 6-13%) (Cossette et al., 1999; Vigevano & Cilio, 1997).
Basé sur ces études, le rapport AAN/CNS a conclu que l'ACTH était ‘probablement efficace dans le traitement à court terme des spasmes infantiles et dans la résolution de
l’hypsarythmie'. Toutefois, la dose et la durée de traitement optimale n’ont pu être déterminées (Mackay et al., 2004).