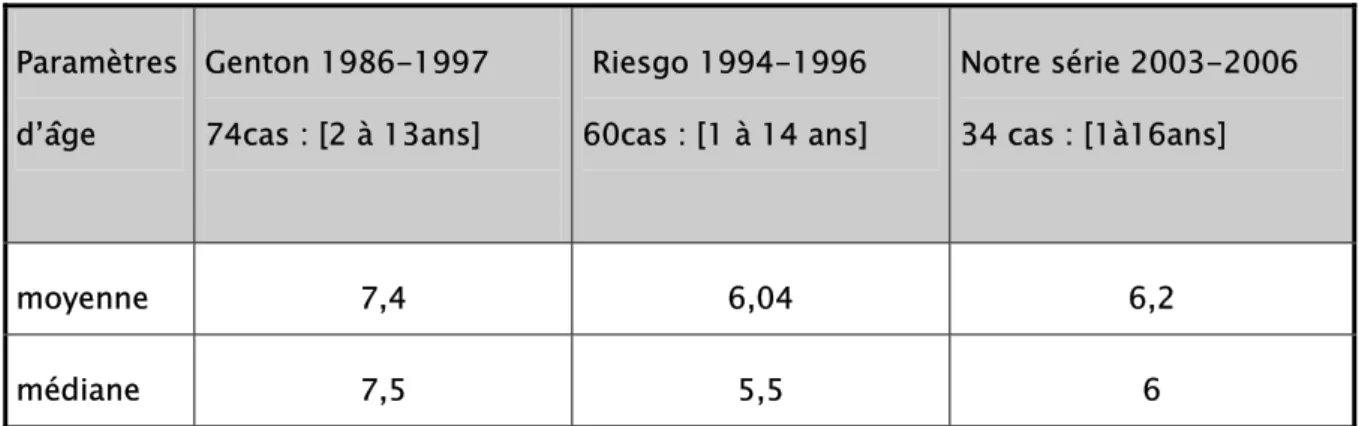
Epilepsie rolandique bénigne : à propos de 34 cas
Texte intégral
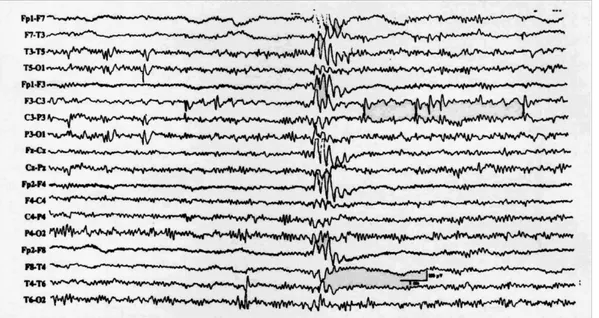
Figure
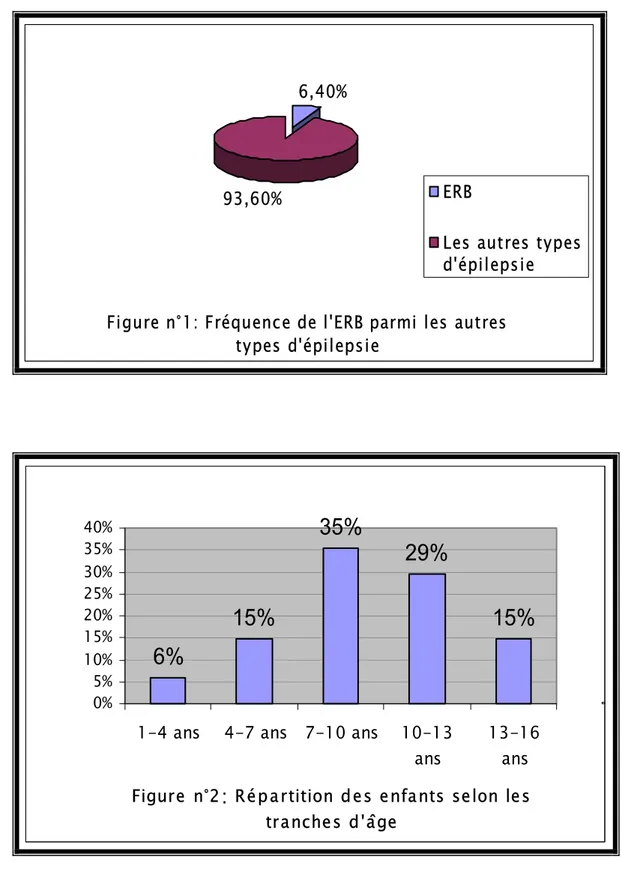
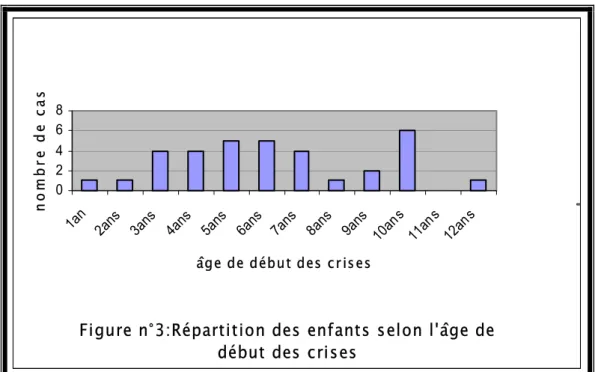
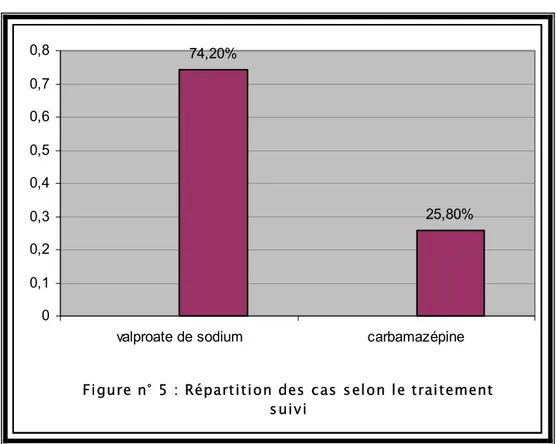
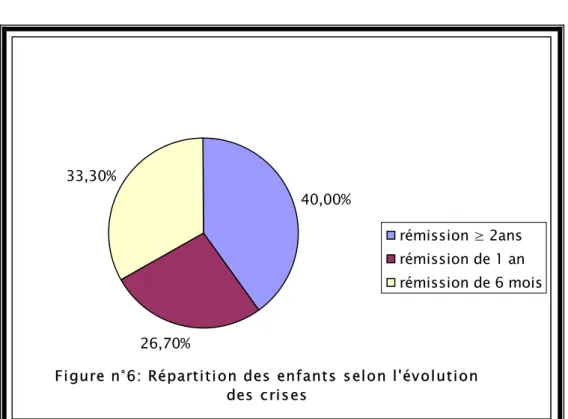
Documents relatifs
In a previous note [3] the second author introduced a new approach which follows completely the spirit of the modern theory of linear elliptic boundary value problems (weak
The results show that the two metals have strikingly different effects on monocytes and T cells because Be induces cell death in monocytes while leaving T cells alive, whereas
In a similar tone to the theatre-centered essays, the authors discuss the body as a subversive site and an important medium of expressing social criticism.. The first of these
2. New proposed classification of pulmonary arterial hypertension in the setting of congenitally malformed hearts as based on circulatory pat hophysiology. Sign ificant shu nting
Using these assays we discovered that SPC3649 (miravirsen) invades the stem structure of miR-122 precursors and inhibits Drosha and Dicer processing of pri-miR-122 and
The test fish preferred the side where odor from the heavier shoal was supplied; this shows that they could appreciate odor cues from conspecifics in our apparatus and should
خامت ـ ـ ـ ـ ـ ـ ـ ـ ـ ـ ـ ــة خلصت هذه الدراسة إىل جمموعة من النتائج هي: التقنيات الزمنية الواردة يف الرواية شكلت قادة أساسية انبثق عنها
A small hardware and an Android application will allow the user to connect easily on a GPIB bus to send and receive data and commands.. The device developed must offer a new and
