JEAN-CHARLES HOGUE
ÉTUDE DES MÉCANISMES ATHÉROGÈNES
ASSOCIÉS À L’INSULINO-RÉSISTANCE ET À
L’HYPERCHOLESTÉROLÉMIE FAMILIALE
Thèse présentée
à la Faculté des études supérieures de l’Université Laval dans le cadre du programme de doctorat en médecine expérimentale
pour l’obtention du grade de Philosophiae Doctor (Ph.D.)
FACULTÉ DE MÉDECINE UNIVERSITÉ LAVAL
QUÉBEC
2008
Résumé
Nous avons observé dans une cohorte de 259 patients atteints d’hypercholestérolémie familiale (HF) hétérozygote et de 208 témoins normolipidémiques que le type de mutation causant l’HF était associé à des changements significatifs dans la taille de la principale sous-population de particules LDL. Les patients porteurs d’une mutation récepteur-négatif présentaient un profil lipidique plus défavorable que les patients porteurs d’une mutation récepteur-défectif et que les témoins. Dans la même cohorte, nous avons associé l’HF avec la présence de plus petites particules HDL, qui sont plus rapidement éliminées de la circulation et qui ne peuvent alors plus assurer correctement leur rôle dans le transport inverse du cholestérol. Finalement, nous avons associé l’HF avec des niveaux élevés de résidus de chylomicrons. Les résidus de chylomicrons participent au développement de l’athérosclérose, tout comme les LDL. De plus, ces résultats soulignent le rôle du récepteur des LDL dans la clairance de ces résidus de la circulation chez l’humain. Dans un second volet, nous avons examiné le métabolisme de l’apoprotéine (apo) B-48 dans le diabète de type 2 (DM2). Nous avons observé que les patients atteints de DM2 présentaient des niveaux élevés d’apoB-48 en circulation, dus à une production intestinale élevée et à un catabolisme ralenti. Nous avons par la suite examiné l’effet d’un traitement au fénofibrate ou à l’atorvastatine sur la cinétique de l’apoB-100 et de l’apoB-48 et sur l’inflammation, le stress oxydatif et l’adhésion monocytaire dans le DM2. L’atorvastatine et le fénofibrate ont été également efficaces pour réduire les triglycérides plasmatiques. L’atorvastatine a principalement augmenté le catabolisme des VLDL et des IDL en augmentant le catabolisme via les récepteurs cellulaires, alors que le fénofibrate a principalement augmenté le catabolisme des VLDL, des IDL et des chylomicrons en augmentant la lipolyse. L’atorvastatine a réduit les niveaux d’apoB-48 en diminuant la production intestinale. L’atorvastatine a été efficace pour réduire l’inflammation, le stress oxydatif et l’adhésion monocytaire dans le DM2, alors que le fénofibrate n’a été efficace que pour réduire un seul marqueur d’adhésion monocytaire. Par contre, le fénofibrate a été associé à une augmentation de cholestérol-LDL et de la phospholipase A2-IIA.
ii
Abstract
We observed in a group of 259 patients with heterozygous familial hypercholesterolemia (FH) and 208 normolipidemic controls that the type of mutation causing FH was associated with changes in the LDL peak particle diameter. Patients carrying a negative-receptor mutation displayed a more deteriorated profile than patients carrying a defective-receptor mutation and than controls. In the same group, we observed that FH was associated to smaller HDL particles, which are more rapidly cleared from circulation and can no longer correctly participate to the reverse cholesterol transport pathway. Finally, we observed that FH is associated to increased fasting levels of chylomicron remnants, which participate to atherosclerosis development, in the same way as LDL do. Moreover, these results underline the role of the LDL receptor in these remnants catabolism. In the second part of the work presented, we examined the metabolism of apoprotein (apo) B-48 in type 2 diabetes mellitus (DM2). We observed that patients with DM2 had increased apoB-48 levels, due to marked increased intestinal production and decreased catabolism. We also observed the effects of a treatment with fenofibrate or atorvastatine on apoB-48 and apoB-100 kinetics and on inflammation, oxidative stress and monocytes adhesion in DM2. Atorvastatin and fenofibrate were equally effective to lower plasma triglyceride levels. Atorvastatin increased VLDL and IDL catabolism by increasing receptor-mediated catabolism while fenofibrate increased VLDL, IDL and chylomicrons catabolism by increasing lipolysis. Atorvastatin decreased apoB-48 levels by decreasing production. Atorvastatin was potent to reduce inflammation, oxidative stress and monocyte adhesion while fenofibrate was potent to reduce only one monocytes adhesion marker. However, fenofibrate increased LDL-C and phospholipase A2-IIA levels.
Avant-Propos
J’aimerais tout d’abord remercier les gens qui ont permis de rendre le présent projet de doctorat possible. Je remercie les Dr Claude Gagné, Jean Bergeron et Daniel Gaudet pour leur implication dans le recrutement des sujets, les infirmières Nicole Roy, Danielle Aubin et Brigitte Brunelle pour les prélèvements et Jean-Marc Gagné et Jacynthe Bernier pour les analyses biochimiques au laboratoire de biochimie médicale du CHUL. Je remercie également Doris Moreau pour m’avoir aidé avec les infinis dédales de la bureaucratie «CHULienne». J’aimerais évidemment remercier mon directeur de recherche, le Dr Patrick Couture, et mon co-directeur de recherche, le Dr Benoît Lamarche, pour la qualité de leur enseignement, de leur support et pour les nombreux à-côtés agréables pendant les congrès, malgré leurs horaires chargés et leurs dizaines (pas tant d’exagération que ça…) de responsabilités. Je remercie ma mère, Colombe, mon père, Normand, mes sœurs, Marie-Odile et Annie-Claude, et mes amis, Élizabeth, Rémi, Martin et Marc-André (pour ne nommer que ceux-là) pour leur support pendant les nombreuses années de mes études et pour ne pas avoir désespéré de me voir terminer un jour mes études… Évidemment, j’aimerais offrir un merci particulier à mes amis/collègues du lab : Andrew, Jeff, Bebelle, Petite, Grande, Julie et Sonia. Leur enseignement et leur support (sans oublier les partys, sorties, etc.) ont été une partie importante de ma motivation et de la complétion de mes études. Finalement, j’aimerais remercier Georges Cousineau, Grand Dieu païen des ultracentrifugations, et Johanne Marin, tenancière du lab, pour leur expertise technique, leur amitié et les partys. Évidemment, je remercie tous ceux que je n’ai pas nommés et qui sont ou ont été à l’INAF.
Tous les résultats présentés dans la présente thèse résultent de mes travaux. J’ai participé au processus de recrutement des sujets et de cueillette des échantillons, j’ai effectué tous les travaux de laboratoire (sauf les mesures standardisées effectuées au laboratoire de biochimie médicale du CHUL), j’ai procédé à la compilation des résultats dans les bases de données, j’ai effectué les tests statistiques et, finalement, la rédaction des articles, incluant la rédaction originale, les corrections et les réponses aux commentaires des pairs. Les analyses de laboratoire que j’ai effectuées sont la mesure des tailles de
iv manipulations menant à l’analyse par GC-MS des apo marquées à l’aide d’un isotope stable et la modélisation mathématique des courbes d’enrichissement. J’ai également déterminé certains polymorphismes et détecté la présence de mutations dans le gène du récepteur des LDL chez certains sujets.
Je dédie cette thèse à la caféine et aux sucres (fermentés ou non) pour leur précieuse contribution à mes travaux de doctorat.
Table des matières
Résumé...i
Abstract... ii
Avant-Propos ... iii
Table des matières ...vi
Liste des tableaux...x
Liste des figures ... xiii
Liste des abbréviations...xv
Introduction...1
1 – Structure des lipoprotéines...5
1.1 – Les apoprotéines ...7 1.1.1 – Les apoprotéines A ...7 1.1.2 – Les apoprotéines B...8 1.1.3 – Les apoprotéines C...9 1.1.4 – L’apoprotéine E ...10 1.1.5 – L’apoprotéine [a] ...10 1.2 – Les lipoprotéines...11 1.2.1 – Chylomicrons...11
1.2.2 – Lipoprotéines de très faible densité (VLDL)...11
1.2.3 – Lipoprotéines de densité intermédiaire (IDL) ...12
1.2.4 – Lipoprotéines de faible densité (LDL)...12
1.2.5 – Lipoprotéines de haute densité (HDL)...13
1.2.6 – Lipoprotéines [a] (Lp[a]) ...13
2 – Métabolisme des lipoprotéines ...15
2.1 – Principales enzymes impliquées dans le métabolisme des lipoprotéines ...15
2.1.1 – Lipase lipoprotéique (LPL)...15
2.1.2 – Lipase hépatique (HL) ...16
2.1.3 – Lipase endothéliale (EL)...16
2.1.4 – Phospholipase A2 sécrétée type IIA (sPLA2)...16
2.1.5 – Protéine de transfert des esters de cholestérol (CETP)...17
2.1.6 – Lécithin:cholestérol acyl transférase (LCAT) ...17
2.2 – Principaux récepteurs impliqués dans le métabolisme des lipoprotéines ...18
2.2.1 – Récepteur des LDL (rLDL) ...18
2.2.2 – Récepteur apparenté au récepteur des LDL (LRP)...20
2.2.3 – Récepteur scavenger de classe B, type 1 (SR-B1)...21
2.2.4 – ATP-binding cassette A1 (ABCA1) ...21
2.3 – Voies de transport des lipides ...22
2.3.1 – Voie des lipides exogènes...22
2.3.2 – Voie des lipides endogènes...23
2.3.3 – Transport inverse du cholestérol...24
2.3.4 – LDL petites et denses et mécanisme de formation ...24
2.3.5 – Mécanisme de formation des HDL petites et denses...26
3 – Développement de l’athérosclérose ...27
3.1 – La dysfonction endothéliale...28
3.3 – Inflammation et développement de l’athérosclérose ...31
3.4 – LDL petites et denses et risque cardiovasculaire...32
3.5 – HDL petites et denses et risque cardiovasculaire ...35
3.6 – Résidus de chylomicrons et risque cardiovasculaire ...36
4 – Études de cinétique ...38
4.1 – Marquage des lipoprotéines ...38
4.2 – Administration du traceur dans le marquage endogène...40
4.3 – Modélisation mathématique...40
5 – Problèmes métaboliques affectant le métabolisme des lipoprotéines...44
5.1 – L’hypercholestérolémie familiale ...46
5.1.1 – Aspects historiques ...46
5.1.2 – Mode de transmission ...47
5.1.3 – Manifestations cliniques ...47
5.1.4 – Prévalence et effet fondateur ...48
5.1.5 – Mutations dans le gène du récepteur des LDL ...49
5.1.6 – Variabilité interindividuelle dans l’expression clinique ...52
5.1.7 – Taille des LDL dans l’hypercholestérolémie familiale...53
5.1.8 – Taille des HDL dans l’hypercholestérolémie familiale ...56
5.1.9 – Résidus de chylomicrons dans l’hypercholestérolémie familiale...56
5.2 – Le diabète de type 2 ...57
5.2.1 – Prévalence et étiologie ...57
5.2.2 – Pathogenèse de l’athérosclérose dans le diabète de type 2...58
5.2.3 – Métabolisme des chylomicrons dans le diabète de type 2 ...60
6 – Traitement des dyslipidémies ...61
6.1 – La diète ...61 6.2 – Les résines...62 6.3 – Les statines...63 6.4 – Les fibrates...64 6.5 – L’ezetimibe ...64 6.6 – L’acide nicotinique ...65 6.7 – L’aphérèse des LDL...65 6.8 – La thérapie génique...66
6.9 – Traitement de la dyslipidémie diabétique...66
7 – Objectifs et hypothèses de travail ...68
7.1 – Chapitre 8 : Taille des LDL selon le type de mutation dans le récepteur des LDL dans l’hypercholestérolémie familiale hétérozygote. ...68
7.2 – Chapitre 9 : Taille des HDL dans l’hypercholestérolémie familiale hétérozygote..69
7.3 – Chapitre 10 : Résidus de chylomicrons dans l’hypercholestérolémie familiale...69
7.4 – Chapitre 11 : Métabolisme de l’apoB-48 dans le diabète de type 2 ...69
7.5 – Chapitre 12 : Effets du fénofibrate seul ou de l’atorvastatine seule sur la cinétique de l’apoB-100 et de l’apoB-48 dans le diabète de type 2 ...70
7.6 – Chapitre 13 : Effets du fénofibrate seul ou de l’atorvastatine seule sur l’oxydation, l’inflammation et l’adhésion cellulaire dans le diabète de type 2...70
8 – Taille des LDL selon le type de mutation dans le récepteur des LDL dans l’hypercholestérolémie familiale hétérozygote...72
viii Abstract...75 Introduction...76 Methods ...77 Results...81 Discussion...84 Acknowledgement ...87 References...88
9 – Taille des HDL dans l’hypercholestérolémie familiale hétérozygote ...100
Résumé...101 Abstract...103 Introduction...105 Methods ...107 Results...110 Discussion...112 Acknowledgement ...115 References...116
10 – Résidus de chylomicrons dans l’hypercholestérolémie familiale...129
Résumé...130 Introduction...134 Methods ...136 Results...139 Discussion...141 Acknowlegements...144 References...145
11 – Métabolisme de l’apoB-48 dans le diabète de type 2 ...153
Résumé...154 Abstract...156 Introduction...157 Methods ...158 Results...162 Discussion...164 Acknowledgements...168 References...169
12 – Effets du fénofibrate seul ou de l’atorvastatine seule sur la cinétique de l’apoB-100 et de l’apoB-48 dans le diabète de type 2 ...180
Résumé...181 Abstract...183 Introduction...184 Methods ...186 Results...190 Discussion...192 Acknowledgements...196 References...197
13 – Effets du fénofibrate seul ou de l’atorvastatine seule sur l’oxydation, l’inflammation et l’adhésion cellulaire dans le diabète de type 2 ...208
Abstract...211 Introduction...212 Methods ...214 Results...217 Discussion...219 Acknowledgements...222 References...223 14 – Conclusion générale...231 Bibliographie ...244
Liste des tableaux
Tableau 1.1 – Principales propriétés physico-chimiques des lipoprotéines. Tableau 3.1 – Facteurs de risque associés au développement des MCV.
Tableau 3.2 – Études portant sur la relation entre la taille des LDL et le risque de MCV. Tableau 5.1 – Quelques problèmes métaboliques affectant le métabolisme des lipoprotéines.
Tableau 5.2 – Prévalence de l’hypercholestérolémie familiale hétérozygote selon les régions du Québec.
Tableau 5.3 – Études analysant les propriétés des particules LDL chez des sujets atteints d’HF.
Tableau 5.4 – Incidence de problèmes de santé chroniques au Canada, en 2000-2001, chez des personnes atteintes de DM2 et chez des personnes non-atteintes.
Tableau 6.1 – Tableau comparatif de la diète recommandée par le Consensus canadien et par le NCEP.
Tableau 8.1 – Demographic, genotypic and biochemical characteristics of participants according to LDL receptor status.
Tableau 8.2 – Electrophoretic properties of LDL particles according to LDL receptor status. Tableau 9.1 – Demographic and genotypic characteristics of participants according to FH/Control status.
Tableau 9.2 – Biochemical characteristics and HDL electrophoretic properties according to FH/Control status.
Tableau 9.3 – Bivariate correlates of metabolic factors to integrated HDL size in FH subjects and in controls.
Tableau 9.4 – Independent contributors to variations in integrated HDL size.
Tableau 10.1 – Demographic and genotypic characteristics of participants according to status.
Tableau 10.2 – Biochemical characteristics of participants according to status.
Tableau 11.1 – Characteristics and lipid/lipoprotein profile of type 2 diabetic patients with marked hypertriglyceridemia before and after treatment with fenofibrate 200 mg/d or atorvastatin 20 mg/d.
Tableau 11.2 – Kinetics of TRL apoB-48 in type 2 diabetic patients with marked hypertriglyceridemia before and after treatment with either fenofibrate 200 mg/d or atorvastatin 20 mg/d.
Tableau 11.3 – Kinetics of VLDL, IDL and LDL apoB-100 in type 2 diabetic patients with marked hypertriglyceridemia before and after treatment with either fenofibrate 200 mg/d or atorvastatin 20 mg/d.
Tableau 12.1 – Baseline characteristics of type 2 diabetic patients according to treatment group.
Tableau 12.2 – Fasting lipid/lipoprotein profile of type 2 diabetic patients before and after treatment with either atorvastatin 20 mg/d or fenofibrate 200 mg/d.
Tableau 12.3 – Plasma levels of inflammation, cell adhesion and oxidation markers in type 2 diabetic patients before and after treatment with either atorvastatin 20 mg/d or fenofibrate 200 mg/d.
Tableau 13.1 – Demographic, anthropometric and biochemical characteristics of subjects according to status.
Tableau 13.2 – Fasting lipid/lipoprotein profile of subjects according to status. Tableau 13.3 – Kinetics of TRL apoB-48 in controls and in type 2 diabetic patients.
xii Tableau 13.4 – Multiple linear regression analysis showing independent contributions of age, BMI and the diabetic/control status to the variance of TRL apoB-48 production rate. Tableau 13.5 – Kinetics of TRL, IDL and LDL apoB-100 in controls and in type 2 diabetic patients.
Liste des figures
Figure 2.1 – Cycle du rLDL.
Figure 4.1 – Modèle utilisé pour la modélisation de la cinétique de l’apoB-100. Figure 4.2 – Modèle utilisé pour la modélisation de la cinétique de l’apoB-48.
Figure 5.1 – Classification des types de mutations dans le gène du rLDL causant l’hypercholestérolémie familiale.
Figure 8.1 – Distribution of LDL-Peak Particle Diameter among controls and FH heterozygotes carrying a defective-receptor (DR) or negative-receptor (NR) mutation in the LDL receptor gene.
Figure 8.2 – LDL-Peak Particle Diameter among controls and FH heterozygotes carrying a defective-receptor (DR) or negative-receptor (NR) mutation according to plasma triglyceride tertiles. Results are listed as mean ± SEM.
Figure 8.3 – Relative risks of having LDL-Peak Particle Diameter <255 Å according to baseline plasma triglyceride levels (above or below median of 1.23 mmol/L) and LDL receptor status.
Figure 9.1 – Comparison of integrated HDL size distribution between FH (A) and controls (B).
Figure 9.2 – Combined impact of FH/Control status and plasma triglyceride levels on integrated HDL size.
Figure 10.1 – Correlations between plasma cholesterol and total apo B levels (A) and apoB-48 levels (B) and between plasma triglyceride and total apo B levels (C) and apoB-apoB-48 levels (D) in all subjects.
xiv Figure 11.1 – ApoB-48 leucine tracer/tracee ratios for TRL in type 2 diabetic patients with marked hypertriglyceridemia before and after treatment with either fenofibrate 200 mg/d (A) or atorvastatin 20 mg/d (B).
Figure 11.2 – ApoB-100 leucine tracer/tracee ratios for VLDL, IDL and LDL in type 2 diabetic patients with marked hypertriglyceridemia before and after treatment with either fenofibrate 200 mg/d (A) or atorvastatin 20 mg/d (B).
Figure 12.1 – Correlation between change in plasma levels of oxLDL and (A) sICAM-1 levels and (B) sE-selectin levels in patients treated with atorvastatin.
Figure 13.1 – Correlation between plasma triglyceride levels and (A) TRL apoB-48 PR and (B) VLDL apoB-100 FCR in patients with type 2 diabetes.
Liste des abbréviations
ABCA1 : ATP-binding cassette A1 apo : apoprotéine
CETP : protéine de transfert des esters de cholestérol C : cholestérol CL : cholestérol libre CM : chylomicron CRP : protéine C réactive DM2 : diabète de type II EC : esters de cholestérol EL : lipase endothéliale
FCR : taux de catabolisme fractionnel FFA : acides gras libres
HDL : lipoprotéine de haute densité HF : hypercholestérolémie familiale HL : lipase hépatique HMZ : homozygote HMG : 3-hydroxy-3-méthylglutaryl HTZ : hétérozygote hyperTG : hypertriglycéridémie
IDL : lipoprotéine de densité intermédiaire IM : infarctus du myocarde
IMT : épaisseur de l’intima-media
LCAT : lécithine:cholestérol acyl transférase LDL : lipoprotéine de faible densité
Lp[a] : lipoprotéine [a] LPL : lipase lipoprotéique
LRP : récepteur apparenté au récepteur des LDL MCV : maladie cardiovasculaire
MMP : métaloprotéinases de la matrice
MTP : protéine microsomale de transfert des triglycérides NO : oxyde nitrique
NOS : synthétases de l’oxyde nitrique oxLDL : LDL oxydées
PL : phospholipides
PPAR : récepteurs des proliférateurs d’activité des péroxisomes PR : taux de production
PS : pool size
rLDL : récepteur des LDL
ROS : composés réactifs d’oxygène RR : risque relatif
sdHDL : particules HDL petites et denses sdLDL : particules LDL petites et denses sPLA2 : phospholipase A2 sécrétée
xvi TG : triglycérides
TTR : ratio traceur au tracé
Introduction
La maladie cardiovasculaire (MCV) est une des principales causes de décès au Canada et dans les pays industrialisés1;2. De nombreux facteurs sont responsables de cette forte prévalence. Plus de 250 facteurs de risque différents ont été identifiés pour le développement des MCV3. Les facteurs de risque traditionnels sont l’hypercholestérolémie, l’hypertension, le tabagisme, le sexe masculin, le diabète et l’histoire familiale. Les habitudes de vie observées dans les pays industrialisés, impliquant un stress important, une mauvaise alimentation et le manque d’activité physique, sont également favorables au développement des MCV, mais leur impact spécifique demeure moins bien connu et varie souvent avec les populations étudiées. Une meilleure compréhension du complexe processus de développement de l’athérosclérose a permis d’identifier les principaux acteurs biologiques de ce développement; les marqueurs de stress oxydatif, d’inflammation et d’adhésion monocytaire sont devenus des marqueurs de risque de MCV. L’étude approfondie du métabolisme et des caractéristiques des lipoprotéines a récemment permis de définir d’autres facteurs de risque non-traditionnels de MCV. La taille des LDL, la taille des HDL, la proportion de cholestérol dans les petites LDL, les taux d’apoprotéine (apo) B-48 et un temps de résidence élevé des LDL en circulation ne sont que des exemples de ces nouveaux marqueurs de risque. Tous ces outils nous permettent donc de mieux cerner les individus à risque et de les prendre médicalement en charge avant qu’un incident cardiovasculaire ne survienne ou pour empêcher l’apparition d’un autre événement. On observe la présence des facteurs de risque traditionnels chez 80 à 90% des patients atteints d’une MCV, laissant tout de même une proportion des patients souffrant de MCV chez qui seuls des facteurs de risque non-traditionnels sont donc observés4. L’utilisation des seuls facteurs de risque traditionnels a été remise en question après que l’on eut observé des patients atteints de MCV présentant des taux normaux de lipides sanguins5;6.
De nombreuses approches sont utilisées pour tenter de faire diminuer le risque cardiovasculaire en prévention primaire et secondaire. Ces approches ne peuvent évidemment que jouer sur les facteurs de risque modifiables et visent habituellement la
2 tabac, diminuer la consommation d’alcool), la perte de poids, le contrôle de la pression sanguine, le contrôle du diabète et le traitement des dyslipidémies. Les traitements pharmacologiques sont souvent nécessaires, car une modification des habitudes de vie n’est souvent pas suffisante7. Les facteurs de risque non-modifiables, tels que le sexe, l’âge et la génétique, doivent être pris en compte dans la planification du traitement et justifieront habituellement une approche plus agressive. Les principales classes de médicaments utilisés dans le traitement des dyslipidémies sont les statines, les fibrates et l’ezetimibe. Les statines sont des inhibiteurs de l’HMG-CoA réductase, l’enzyme-clé de la biosynthèse du cholestérol. L’inhibition de cette enzyme entraîne une surexpression du récepteur des LDL (rLDL), ce qui entraîne une diminution des taux de cholestérol plasmatique8. Les fibrates sont des agonistes des PPAR-α, des récepteurs nucléaires impliqués dans la transcription de gènes pour le catabolisme des lipoprotéines riches en triglycérides (TG) (TRL) et dans la β-oxydation des acides gras9. Toutefois, leur utilisation dans certains types de dyslipidémies est controversée, car ils ont tendance à provoquer une augmentation du cholestérol-LDL (C-LDL) chez les patients avec des taux élevés de TG, ce qui peut augmenter le risque de MCV10;11.
L’hypercholestérolémie familiale (HF) est une maladie autosomique co-dominante causée par la présence d’un ou de deux allèles mutants dans le gène du rLDL. Cette maladie métabolique est caractérisée par une augmentation des taux plasmatiques de C-LDL tout au long de la vie du patient et par des manifestations cliniques telles que l’accumulation de cholestérol au niveau de la peau (xanthélasma), au niveau des tendons (xanthomes) et des artères (athérosclérose)12. L’HF fut la première maladie génétique menant aux MCV à avoir été décrite, elle est la cause la plus fréquente d’hypercholestérolémie primaire monogénique et elle est également une des maladies métaboliques dont la prévalence mondiale est la plus élevée, avec une fréquence de 1:500 pour les hétérozygotes (HTZ)12. Chez les Canadiens-français du Québec, la prévalence moyenne calculée pour les HTZ est de 1:27013. La conséquence clinique majeure de l’HF est le développement précoce et accéléré de l’athérosclérose, un processus complexe et multifactoriel menant au développement des MCV, qui peuvent alors survenir dès l’âge de 35 ans chez les HTZ. La perte totale ou partielle de l’activité du rLDL entraîne une
accumulation des particules LDL en circulation, menant à l’augmentation de leur temps de résidence dans le plasma et à une augmentation des taux de C-LDL de 2-3 fois par rapport à la normale chez les HTZ et de 6-8 fois chez les homozygotes (HMZ).
Le diabète de type 2 (DM2) est associé à un ensemble d’anomalies métaboliques augmentant le risque cardiovasculaire par 2 à 3 fois14. On observe entre autres des taux élevés de TG, des taux abaissés de C-HDL, des taux élevés d’acides gras libres et une prévalence de particules LDL petites et denses (sdLDL). La principale altération se produit au niveau du métabolisme des TRL, qui comprennent les chylomicrons (CM) et les lipoprotéines de très faible densité (VLDL). En effet, dans un état de résistance à l’insuline, le foie produira plus de VLDL et l’intestin produira plus de CM. De plus, le catabolisme de ces particules sera ralenti par la diminution de l’expression des récepteurs pour ces particules et par la saturation des récepteurs restants et des enzymes. Les résidus de TRL partiellement hydrolysés restent donc plus longtemps en circulation. Des études chez l’humain et chez l’animal on montré que ces résidus sont fortement associés au développement de la MCV15. De plus, des études ont montré que les grosses VLDL enrichis en TG sont les précurseurs des particules sdLDL16. Puisque le DM2 est souvent accompagné de surpoids, d’hypertension artérielle et de taux de TG élevés, son traitement est complexe et doit jouer sur plusieurs aspects métaboliques simultanément.
L’apoB-48 est la principale protéine structurale des CM. Elle est strictement produite par l’intestin et elle est indispensable à l’assemblage et à la sécrétion des CM. Les CM sont catabolisés par l’action de la lipase lipoprotéique (LPL), de la lipase hépatique (HL), de l’apo E, de l’apo C-II et des protéoglycans héparan-sulfate17. Les résidus de CM sont retirés de la circulation par un mécanisme impliquant le rLDL et un récepteur apparenté au rLDL (LRP)18;19. Des données récentes suggèrent un rôle non-négligeable des résidus de CM dans le développement de l’athérosclérose. En effet, les résidus de CM seraient moins capturés par les macrophages des plaques athéromateuses que les LDL, mais ils délivreraient environ dix fois plus de cholestérol que le LDL. La masse de cholestérol livrée par les résidus de CM serait donc environ quatre fois plus importante que la masse livrée par les LDL20-24.
4 L’apoB-100 est une grosse protéine d’origine hépatique, essentielle à l’assemblage et à la sécrétion des VLDL. L’apoB-100 possède le domaine requis pour interagir avec le rLDL, qui est responsable à lui seul d’approximativement 70% de la clairance de l’apoB-100 du compartiment plasmatique25. L’apoB-100 est nécessaire à la stabilité structurelle des VLDL, des lipoprotéines de densité intermédiaire (IDL) et des LDL. Le catabolisme des VLDL est assuré par l’action de la LPL, de la HL, de l’apo E, de l’apo C-II et des protéoglycans héparan-sulfates. Au fur et à mesure que les TG des VLDL sont hydrolysés, la particule devient appauvrie en TG et enrichie en cholestérol, la VLDL étant graduellement transformée en IDL, puis en LDL. L’élimination de ces particules de la circulation est assurée par le LRP et le rLDL. Les LDL peuvent être chimiquement modifiées; elles sont alors reconnues comme du non-soi par le système immunitaire et participent au processus d’athérosclérose en s’accumulant au niveau des lésions athéromateuses. L’étude approfondie du métabolisme des lipoprotéines contenant l’apoB-100 et l’apoB-48 permet de mieux comprendre l’effet de certaines pathologies ou les effets de certains médicaments agissant sur les lipides.
En résumé, le présent projet de doctorat permettra d’abord de quantifier certains facteurs de risque non-traditionnels dans l’HF, ainsi que l’impact de différentes classes de mutations du rLDL sur ces facteurs. En deuxième lieu, ce projet permettra d’examiner le métabolisme de l’apoB-48 et de l’apoB-100 dans le DM2. Finalement, nous comparerons les effets de deux médicaments hypolipidémiants fréquemment utilisés pour le traitement de la dyslipidémie diabétique : les statines et les fibrates. Nous examinerons leurs effets sur la cinétique des lipoprotéines contenant l’apoB-48 et l’apoB-100 et sur les paramètres d’inflammation, de stress oxydatif et d’adhésion monocytaire.
1 – Structure des lipoprotéines
Comme leur nom l’indique, les lipoprotéines sont composées de lipides et de protéines. Elles sont des particules globulaires de haut poids moléculaire dont la principale fonction est le transport de composés hydrophobes (les lipides) d’un tissu à l’autre, via un milieu hydrophile (le plasma sanguin). Nous distinguons plusieurs classes de lipoprotéines, qui se différencient sur des bases physico-chimiques (densité, composition) et sur des bases métaboliques. Elles partagent toutes la même structure de base, c’est-à-dire une monocouche de phospholipides (PL) et de cholestérol libre (CL) entourant un cœur fortement hydrophobe composé de TG et d’esters de cholestérol (EC). Les protéines sont soit fortement liées à la particule et ne peuvent s’en dissocier ou sont faiblement liées et peuvent s’en détacher pour aller s’attacher à d’autres lipoprotéines. Les protéines sont les apo, qui déterminent la fonction et le destin métabolique de la particule, et certaines enzymes, qui agissent sur la particule. En ordre croissant de densité, nous retrouvons les CM, les VLDL, les IDL, les LDL et les HDL. Les CM et les VLDL sont fréquemment regroupées sous l’appellation de lipoprotéines riches en triglycérodes (TRL). Le tableau 1.1 présente les propriétés des lipoprotéines, qui seront par la suite décrites plus en profondeur. Les lipoprotéines ne sont toutefois pas des entités distinctes. Par exemple, les lipoprotéines de la classe VLDL n’ont pas toutes une même densité précise, mais couvrent plutôt un spectre de densité. La distribution des HDL est bimodale et elles peuvent être divisées en HDL2 et HDL3. La lipoprotéine [a] (Lp[a]) est une LDL modifiée sur laquelle une molécule d’apo [a] s’est attachée.
6 Tableau 1.1 – Principales propriétés physico-chimiques des lipoprotéines (adapté de Gagné26 et de Gotto27) Classe Densité (g/mL) Diamètre (Å) Mobilité électrophorétique
Principales apo Principaux lipides CM <0.95 800-5000 Origine A-I, A-II, A-IV,
B-48, II, C-III, E TG Résidu de CM <1.006 >500 Origine B-48, E TG, EC VLDL <1.006 300-800 Pré-β B-100, II, C-III, E TG IDL 1.006-1.019 250-350 β étendue B-100, II, C-III, E TG, EC LDL 1.019-1.063 180-280 β B-100 EC HDL2 1.063-1.125
90-120 α A-I, A-II EC, PL
HDL3 1.125-1.210
50-90 α A-I, A-II PL
Lp[a]
1.055-1.120 180-280 Pré-β B-100, apo [a] EC
La composition précise en lipides et les propriétés physico-chimiques des lipoprotéines peuvent varier selon l’alimentation des individus et leur condition métabolique. Par exemple, une diète riche en poisson aura une influence sur les acides gras composant les triglycérides en augmentant la quantité d’acides gras polyinsaturés. De plus, certaines pathologies affectant le métabolisme des lipides peuvent altérer une ou plusieurs classes de lipoprotéines. Par exemple, l’HF provoque une accumulation des LDL dans le plasma, qui provoque un décalage des LDL vers une plus haute densité et une plus petite taille28, et la dyslipidémie de type III provoque l’apparition de β-VLDL, enrichis en EC et en apo E29.
1.1 – Les apoprotéines
Les apo sont un groupe de protéines spécialisées qui s’associent aux lipoprotéines. Elles déterminent l’identité et le destin métabolique des lipoprotéines auxquelles elles sont attachées. Les apo couvrent un large spectre de poids moléculaires, de 6.6 à 550 kDa. La plupart des apo sont solubles dans l’eau et peuvent s’échanger librement entre les lipoprotéines. Seules les apo B demeurent fermement liées à leur particule, à partir de sa sécrétion jusqu’à son internalisation pour catabolisme. Certaines apo sont nécessaires pour interagir avec des récepteurs cellulaires, alors que d’autres agissent comme modulateurs d’enzymes.
1.1.1 – Les apoprotéines A
Chez l’humain, nous retrouvons principalement en circulation trois types d’apo A : l’apo A-I, l’apo A-II et l’apo A-IV. L’apo A-I est une protéine de 28.1 kDa et se retrouve principalement au niveau des HDL. Elle est produite par l’intestin et par le foie. Au niveau intestinal, elle est secrétée avec les CM et elle est transférée au HDL lorsque le CM est hydrolysé. Au niveau du foie, l’apo A-I est secrétée sous forme de HDL naissant. En plus de son importance dans la stabilité des HDL, l’apo A-I est absolument nécessaire à l’activité de la lécithine:cholestérol acyl transférase (LCAT)30-32.
L’apo A-II est un dimère de 17.4 kDa, est la deuxième plus abondante protéine des HDL et son principal site de synthèse est le foie. Mise à part sa présence dans les HDL, le rôle de l’apo A-II dans le métabolisme des HDL n’est pas très clair, d’autant plus que cette apo n’est pas présente chez tous les mammifères. L’apo A-II humaine peut exister sous forme de dimère avec l’apo E, laquelle n’est pas reconnue par les récepteurs hépatiques lorsqu’elle est sous cette forme, laissant entrevoir un rôle potentiel dans la régulation de la clairance des résidus de CM et de VLDL31-33.
8 L’apo A-IV est une protéine de 46 kDa et est abondamment sécrétée avec les CM, mais on ne la retrouve que peu associée aux résidus de CM, aux VLDL et aux HDL. Contrairement à la plupart des apo, la majeure partie de l’apo A-IV est retrouvée libre en circulation et s’échange librement et rapidement entre les lipoprotéines. L’apo A-IV agirait de concert avec l’apo C-II pour activer la LPL et inhiberait la captation des résidus de CM au niveau du foie31;32;34.
1.1.2 – Les apoprotéines B
On retrouve deux formes d’apo B : l’apoB-100, synthétisée par le foie, et l’apoB-48, produite par l’intestin. L’apoB-48 est produite par épissage de l’ARNm du gène de l’apo B et représente 48% de la masse de l’apoB-100. Les apo B ne sont absolument pas solubles dans l’eau en absence de détergents et y forment des agrégats. Elles restent intimement liées à leur lipoprotéine à partir de son assemblage intracellulaire jusqu’à sa captation et sa dégradation intracellulaire.
L’apoB-100, une protéine de 550 kDa, n’est présente qu’en une seule copie dans la VLDL et, par conséquent, dans ses résidus métaboliques, les IDL et les LDL. L’apoB-100 se lie au rLDL. La mesure de l’apoB-100 est utilisée en clinique et en recherche afin de donner une idée de la quantité de VLDL, IDL et LDL en circulation. Les taux d’apoB-100 à jeun ont été associés au développement des MCV35. Le taux de sécrétion de l’apoB-100 par le foie est contrôlé à de nombreux niveaux. Les taux d’acides gras libres (FFA) en circulation et captés par le foie jouent en rôle dans la sécrétion de l’apoB-100, car la molécule d’apoB-100 naissante non-lipidée est rapidement dégradée. Également, les récepteurs LDL et LRP présents à la surface de l’hépatocyte peuvent recapter les VLDL dès leur sécrétion. Ainsi, près de 85% de l’apoB-100 produite ne quitte jamais le foie25;31;36;37.
Comme l’apoB-100, l’apoB-48 n’est présente qu’en une seule copie dans les CM et les résidus de CM. L’apoB-48 elle-même ne se lie pas au rLDL; la captation des résidus de CM se fait donc par l’interaction de l’apo E avec le rLDL et le LRP. Tout comme
l’apoB-100, la sécrétion de l’apoB-48 est finement régulée au niveau co- et post-traductionnel et est dépendante de la quantité de lipides disponibles dans l’entérocyte31;34;37.
1.1.3 – Les apoprotéines C
La famille des apo C comprend trois membres de faible poids moléculaire : l’apo C-I, l’apo C-II et l’apo C-III. Même si leurs fonctions diffèrent, les trois membres de cette famille partagent la caractéristique de se redistribuer entre les lipoprotéines. En effet, à jeun, la majorité des apo C se retrouvent sur les HDL. Lors de la production de CM par l’intestin ou lors de la production active de VLDL par le foie, les apo C se transfèrent à la surface des TRL. À l’inverse, lorsque le cœur des TRL devient pauvre en TG sous l’action des lipases, les apo C retournent aux HDL. Le foie est le site de synthèse majeur des apo C et l’intestin en synthétise une petite partie31;38.
L’apo C-I est la plus petite des apo C avec un poids moléculaire de 6.6 kDa. L’apo C-I participe au remodelage des HDL en activant la LCAT39.
L’apo C-II est une protéine de 8.8 kDa. Sa principale fonction est d’activer la LPL, permettant ainsi l’hydrolyse des TG des CM et des VLDL. L’apo C-II activerait également la LCAT31;34.
L’apo C-III est un peptide de 8.8 kDa et est la plus abondante des apo C dans le plasma humain. Trois isoformes existent dans le plasma, dépendamment du nombre de résidus d’acide sialique présents qui modifient le point isoélectrique de la molécule. Le rôle exact des différents isoformes dans le métabolisme des lipoprotéines n’est pas encore clair, mais des travaux récents suggèrent que l’apo C-III1 et l’apo C-III2 contribueraient d’avantage à l’hypertriglycéridémie (hyperTG) que l’apo C-III0 et que l’apo C-III2 aurait un rôle à jouer dans la formation des sdLDL40. L’apo C-III modulerait également la captation des résidus de TRL par le foie et inhiberait l’activation de la LPL par l’apo C-II31;34.
10
1.1.4 – L’apoprotéine E
L’apo E est un constituant des CM, des résidus de CM, des VLDL, des IDL et de certains HDL; sauf pour les HDL, elle joue un rôle important dans le métabolisme de ces particules en se liant au rLDL. L’apo E est une glycoprotéine de 34.2 kDa et est principalement produite par le foie. Trois isoformes existent couramment dans la population et diffèrent entre elles par le changement d’un seul acide aminé. À la position 112, les apo E3 et E2 possèdent une cystéine, alors que l’apo E4 présente une arginine. L’apo E2 présente une cystéine en position 158 alors que les apo E3 et E4 présentent une arginine. La fréquence de l’allèle ε3 dans la population est de 77%, alors que la fréquence de l’allèle ε4 est de 15% et celle de l’allèle ε2 est de 8%. Ces polymorphismes ont un effet sur la capacité de l’apo E de se lier à son récepteur. En effet, comparativement à l’apo E3, l’isoforme la plus fréquente, l’apo E4 se lie plus facilement au récepteur alors que l’apo E2 se lie moins facilement. Ce niveau d’affinité est l’un des facteurs déterminant le taux de particules IDL qui seront converties en LDL. Ainsi, l’apo E4 favorise une augmentation de la cholestérolémie alors que l’apo E2 favorise une augmentation de la triglycéridémie27;31;41;42.
1.1.5 – L’apoprotéine [a]
L’apo [a] appartient à une famille de protéines impliquées dans la fibrinolyse. Elle est produite par le foie et plus de 95% de l’apo [a] en circulation est associé à une minorité de particules LDL en formant un très fort lien covalent avec l’apoB-100; seule une puissante réduction (qu’on ne peut retrouver en conditions physiologiques) peut briser ce lien. Un des domaines fondamentaux de l’apo [a] est le kringle (du nom d’une pâtisserie scandinave) et ce domaine se retrouve également dans les protéines impliquées dans la fibrinolyse. L’apo [a] est hautement polymorphique et le nombre de kringles varie fortement d’un individu à l’autre. Puisque le kringle est un domaine de 80 acides aminées et que son nombre varie entre 17 et 30 copies selon les individus, cette apo a un poids
moléculaire très variable dans la population. L’apo [a] entre en compétition avec le plasminogène pour la lyse des caillots26;27;43.
1.2 – Les lipoprotéines
1.2.1 – Chylomicrons
Les CM sont synthétisés par l’intestin et sont responsables du transport des lipides d’origine alimentaire vers les tissus périphériques et le foie. L’apoB-48 est présente en une seule copie et est nécessaire à l’assemblage intracellulaire et à la sécrétion de la particule. L’apo E est responsable du captage des résidus de CM par le rLDL et le LRP au niveau du foie. L’apo E n’est pas nécessaire pour la captation des résidus par les macrophages des lésions athéromateuses. Les CM produits suite à un repas riche en lipides contiennent typiquement 75-85% de TG, 0.5-2% d’EC, 10-20% de PL (surtout des phosphatidylcholines), 2% de protéines et 0.5-2% de CL. L’apo C-II permet à la LPL d’hydrolyser les TG et l’apo A-IV présente facilite cette action. L’apo A-I, l’apo C-III et l’apo A-IV présentes inhibent le captage des résidus par le foie. L’apo C-III a aussi comme rôle d’inhiber la LPL. Les CM sont rapidement transformés en résidus de CM qui présentent environ 30% de la masse originale du CM34;44.
1.2.2 – Lipoprotéines de très faible densité (VLDL)
Les VLDL sont produites par le foie et servent au transport des lipides depuis le foie vers les tissus périphériques. L’apoB-100 est présente en une seule copie par particule et est absolument nécessaire à son assemblage et à sa sécrétion. Les VLDL contiennent également l’apo E, l’apo C-II et l’apo C-III. Les VLDL sont hétérogènes : la proportion des lipides peut varier d’une particule à l’autre, de même que la composition en apos. Les TG représentent les principaux lipides présents dans les VLDL avec 55% de la composition du cœur de la particule. Les EC sont présents à 12%, le CL à 7%, les PL à 18% et les
12 protéines à 8%. Ces TG ont été nouvellement synthétisés à partir des FFA captés par le foie ou proviennent des différents résidus de lipoprotéines qui ont été retirés de la circulation. Lorsque la disponibilité des TG est à la baisse, le foie continue de produire des VLDL, mais à un rythme réduit et les VLDL produits sont plus petits et plus denses25;44.
1.2.3 – Lipoprotéines de densité intermédiaire (IDL)
Les IDL sont les résidus métaboliques des VLDL, suite à l’action de la LPL qui a réduit le contenu en TG du cœur de la particule. En conséquent, les apo C, étant hydrosolubles et faiblement attachées aux lipoprotéines, ont alors moins d’affinité pour la particule et s’en détachent pour retourner aux HDL ou aux lipoprotéines plus riches en TG. Ainsi, les IDL ne contiennent que l’apoB-100 et l’apo E, ce qui les distingue des VLDL et des LDL. Les IDL peuvent être directement captés par le foie ou être converties en LDL par l’action de la HL. Par rapport aux VLDL, les contenus en EC et en protéines sont plus importants (29% et 19%, respectivement), alors que le contenu en TG est à la baisse (23%). Les proportions de CL et de PL demeurent relativement inchangés (9% et 19%, respectivement)25;44.
1.2.4 – Lipoprotéines de faible densité (LDL)
Les LDL sont les derniers membres de la cascade métabolique des particules contenant l’apoB-100. Ils sont fortement enrichis en EC (42%) et appauvris en TG (6%). Elles ne contiennent plus que l’apoB-100, qui est nécessaire à l’intégrité structurale de la particule25;44. Les LDL sont hétérogènes en densité et en taille et il a été démontré que ces paramètres avaient une influence sur le risque de MCV, quoique cela ne fasse pas l’unanimité. En effet, mêmes si toutes les LDL sont athérogènes, de nombreuses études associent les sdLDL au développement des MCV. Une étude suggère que les grosses LDL ont la même réduction de la capacité de liaison au rLDL que les petites LDL, les LDL de taille moyenne ayant la capacité optimale de liaison au rLDL, probablement à cause de la
conformation tridimensionnelle de la molécule d’apoB-100 (voir sections 2.3.4 et 3.4 pour plus de détails). Les LDL peuvent être caractérisées selon leur densité par ultracentrifugation séquentielle ou analytique ou selon leur taille sur gradient de gel de polyacrylamide. Dans le dernier cas, elles sont caractérisée selon la taille des populations de LDL en se basant sur un standard de diamètres connus ou selon une classification empirique de prépondérance en petites LDL (pattern B) ou en grosses LDL (pattern A) 16;25;44.
1.2.5 – Lipoprotéines de haute densité (HDL)
Les particules HDL en circulation peuvent être divisées en trois catégories selon leur densité : les HDL naissantes, les HDL2 et les HDL3. Les HDL naissantes ont une forme discoïde stabilisée par les apo. Elles sont sécrétées par le foie et l’intestin et sont dérivées des résidus d’hydrolyse des TRL (CM et VLDL). Leur principal constituant lipidique sont les PL et elles présentent principalement à leur surface l’apo A-I, C-II, C-III et E. Lors de la maturation des HDL, l’apo E est relâchée vers les TRL et les HDL gagnent l’apo A-II et A-IV, ce qui leur permet d’adopter une forme sphérique. Lors de la production de TRL, les apo C-II et C-III se transfèrent aux TRL et reviennent aux HDL lors de la disparition des TRL32;44.
1.2.6 – Lipoprotéines [a] (Lp[a])
La Lp(a) est une lipoprotéine riche en EC qui est associée au développement des MCV et maladies cérébrovasculaires. En fait, il s’agit d’une particule LDL modifiée : une molécule d’apo(a) est fixée par un lien disulfure sur la molécule d’apoB-100. À cause de cette apo supplémentaire, le poids moléculaire et la densité des ces lipoprotéines sont augmentés. L’apo(a) serait synthétisée par le foie indépendamment de la synthèse de l’apoB-100 et serait relâchée en circulation, où elle se grefferait à une LDL. L’apo(a) se lierait de façon non-covalente à l’apoB-100 des LDL, puis il y aurait formation spontanée
14 du lien disulfure. Ce lien ne peut être défait que par une puissante réduction qu’on ne peut retrouver dans des conditions physiologiques43;44.
2 – Métabolisme des lipoprotéines
Tous les tissus ont besoin des différentes sortes de lipides retrouvés en circulation. Le cholestérol est nécessaire à la rigidité des membranes et à la synthèse des sels biliaires et de certaines hormones, alors que les TG sont une source d’énergie et les phospholipides sont nécessaires aux membranes. Les systèmes de transport existent donc pour s’assurer que les différents tissus obtiennent les lipides dont ils ont besoin. Ainsi, de nombreux récepteurs et enzymes existent pour orchestrer ces livraisons.
2.1 – Principales enzymes impliquées dans le métabolisme des
lipoprotéines
2.1.1 – Lipase lipoprotéique (LPL)
La LPL est une glycoprotéine de 448 acides aminées et est responsable de la majeure partie de l’activité plasmatique d’hydrolyse des TG. La LPL est synthétisée par les tissus parenchymateux, sécrétée puis transportée à la surface endothéliale des capillaires sanguins où elle est attachée aux protéoglycans héparan-sulfate. La forme active de la LPL est un hétérodimère composé d’une molécule de LPL et de deux molécules d’apo C-II. La LPL hydrolyse les TG contenus dans le cœur des TRL afin de relâcher les lipides comme source d’énergie. Cette action nécessite l’apo C-II comme cofacteur et est inhibée par l’apo C-III. En perdant la majeure partie de leur TG, les résidus de VLDL (les IDL) et de CM perdent leur apo C-II, empêchant ainsi que la LPL continue son action. L’apo E n’est pas déplacée par l’action de la LPL et reste alors avec le IDL27;45. La LPL joue également un rôle dans le remodelage des particules HDL46.
16
2.1.2 – Lipase hépatique (HL)
La HL est une glycoprotéine de 477 résidus de la même famille que la LPL, synthétisée par les hépatocytes et transportée aux cellules endothéliales du foie, où elle est liée aux protéoglycans héparan-sulfate. L’apo C-II n’est pas nécessaire à son activité, ce qui lui permet d’hydrolyser les TG et les PL des IDL et des HDL. Ainsi, même si ses rôles ne sont toujours pas très bien définis, elle participerait à la formation des LDL et au remodelage des HDL27;47;48. La HL participe à la formation des HDL3 à partir des HDL2, ainsi qu’à la libération d’apo A-I délipidée49. La déficience en HL provoque l’accumulation des lipoprotéines contenant l’apo B et des HDL enrichis en PL et en apo E50;51. L’apo A-II est un inhibiteur de l’activité de la HL, indiquant que les HDL dépourvus de cette apo sont les principaux substrats de la HL52. La HL possède également plus d’affinité pour les HDL enrichis en TG53.
2.1.3 – Lipase endothéliale (EL)
La EL n’a été que récemment découverte et montre une forte homologie avec la LPL et la HL. À la différence des autres lipases, la EL n’est exprimée que par les cellules endothéliales et elle préfère les PL comme substrat, avec une affinité particulière pour les HDL54-56. En hydrolysant les PL des HDL, la EL produit des particules HDL plus petites57. Le rôle de la EL dans le métabolisme des particules contenant l’apo B est beaucoup moins clair.
2.1.4 – Phospholipase A
2sécrétée type IIA (sPLA
2)
La sPLA2 est une enzyme de la famille des phospholipase A2 sécrétées du groupe IIA. Elle est caractérisée par un faible poids moléculaire (14.4 kDa) et par sa capacité à
hydrolyser les phospholipides en position sn-258. Dans le système circulatoire, la sPLA2 est principalement produite par les cellules musculaires lisses des parois artérielles59. Les cytokines pro-inflammatoires augmentent l’expression de la sPLA2 au niveau artériel60. La sPLA2 est un acteur de phase inflammatoire aiguë et les taux plasmatiques de sPLA2 sont élevés dans les cas d’inflammation systémique. En hydrolysant les PL des LDL, la sPLA2 contribue à la formation des sdLDL58;61.
2.1.5 – Protéine de transfert des esters de cholestérol (CETP)
La CETP est une glycoprotéine hydrophobe d’une masse de 74 kDa62;63. Comparativement aux autres protéines plasmatiques et aux apo, cette protéine contient une proportion très élevée d’acides aminées hydrophobes, soit environ 44%63. La CETP est responsable de tous les transferts de lipides neutres dans le plasma (EC, TG et esters de rétinol) et d’une partie des transferts de PL63. Dans le plasma, la CETP permet essentiellement le transfert d’EC des HDL vers les lipoprotéines contenant l’apoB-100 (VLDL, IDL et LDL) en échange de TG et, moindrement, d’EC des LDL vers les TRL en échange de TG61. Il est intéressant de noter que cet échange est approximativement équimolaire64. Aussi, plusieurs études ont montré une association entre la CETP et le remodelage des particules LDL65-68. L’ARNm de la CETP est exprimé principalement au niveau du foie, de la rate et du tissu adipeux et il est exprimé à un moindre niveau dans le petit intestin, les reins, les glandes surrénales et le cœur. Chez la plupart des espèces de mammifère, le tissu adipeux est la principale source de CETP, suivi du foie69.
2.1.6 – Lécithin:cholestérol acyl transférase (LCAT)
La LCAT est une protéine de 416 acides aminés synthétisée par le foie et est la seule enzyme connue chez l’humain responsable de l’estérification du CL à partir de la phosphatidylcholine. En absence de CL, la LCAT présente une activité phospholipase A2 et clive les phospholipides en position sn-2. L’apo A-I est un co-facteur essentiel à la
18 LCAT. L’estérification du CL est nécessaire à la maturation des HDL, au maintien de l’intégrité structurale du HDL et au maintien des taux de C-HDL en circulation27;70;71.
2.2 – Principaux récepteurs impliqués dans le métabolisme des
lipoprotéines
Alors que les enzymes remodèlent les lipoprotéines en circulation, les récepteurs impliqués dans le métabolisme des lipoprotéines sont responsables de l’interaction des lipoprotéines avec les cellules. Dans certains cas, les récepteurs internalisent les lipoprotéines pour dégradation. Dans d’autres cas, les récepteurs ne font qu’attacher la lipoprotéine et médient un transfert de lipides. Les principaux récepteurs sont le rLDL, le LRP, le récepteur scavenger de classe B, type 1 (SR-B1) et l’ATP-binding cassette A1 (ABCA1).
2.2.1 – Récepteur des LDL (rLDL)
Le rLDL est une glycoprotéine de 839 acides aminés (160 kDa) retrouvée à la surface des cellules, découverte en 1974 par Goldstein et Brown72. La protéine mature présente de nombreux liens disulfures qui augmentent grandement sa stabilité. Ce récepteur lie et internalise les lipoprotéines plasmatiques présentant à leur surface l’apo B ou l’apo E. Ainsi, le rLDL reconnaît les LDL, les IDL et les résidus de CM, quoique la clairance des résidus de CM par le rLDL chez l’humain est moins clair73. L’affinité du rLDL est plus grande pour les lipoprotéines contenant l’apo E, simplement parce que l’apo E est habituellement présente en plusieurs copies à la surface des IDL, alors que l’apoB-100 n’est présente qu’en une seule copie à la surface des LDL74. Même s’ils contiennent l’apoB-100, les VLDL et les Lp(a) sont de pauvres ligands du rLDL, probablement à cause de la conformation tridimensionnelle particulière que prend l’apoB-100 sur ces lipoprotéines75. Ce récepteur joue donc un rôle central dans l’homéostasie du cholestérol. Puisque les taux plasmatiques de C-LDL jouent un rôle important dans le développement
de l’athérosclérose, l’activité de ce récepteur a une influence sur la susceptibilité d’un individu à développer une MCV73;76.
Le gène codant pour le rLDL est constitué d’environ 45 kb et est situé sur le bras court du chromosome 19. Il présente 18 exons qui sont édités pour produire un ARN messager d’environ 5.3 kb75. L’exon 1 code pour une suite de 21 acides aminés hydrophobes essentiels à la translocation de la protéine naissante dans le réticulum endoplasmique. Les exons 2 à 6 codent pour le domaine d’arrimage du ligand; les exons 7 à 14 codent pour le domaine d’homologie à l’EGF; l’exon 15 code pour le domaine de glycosylation; l’exon 16 et le début de l’exon 17 codent pour le domaine transmembranaire; et la fin de l’exon 17 et l’exon 18 codent pour le domaine cytoplasmique. Le gène du rLDL présente une homologie avec plusieurs autres protéines, certaines d’entres elles jouant un rôle dans le métabolisme des lipoprotéines, comme le LRP et le gp33075.
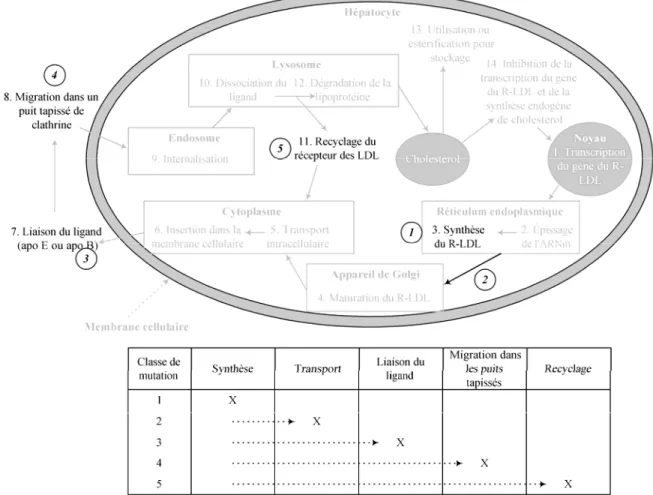
La Figure 2.1 schématise le cycle du rLDL. Le récepteur est synthétisé dans le réticulum endoplasmique rugueux sous la forme d’un précurseur qui subit une maturation dans l’appareil de Golgi. Le récepteur est transporté jusqu’à la surface de la cellule où il est disponible pour lier une lipoprotéine. Lorsqu’une particule se lie au récepteur, le complexe récepteur/lipoprotéine migre dans un puit tapissé de clathrine où il est internalisé. Un lysosome digestif fusionne alors avec l’endosome contenant le complexe, ce qui entraîne une baisse du pH de l’endosome. La particule est alors dissociée du récepteur pour être dégradée. Le récepteur est recyclé à la surface de la cellule, où il peut répéter le cycle d’internalisation. Le cholestérol obtenu par la dégradation de la lipoprotéine peut être immédiatement utilisé ou être estérifié pour entreposage12;75.
La synthèse du rLDL est soumise à un processus de contrôle par rétroinhibition qui a pour principal but de stabiliser la composition lipidique des membranes cellulaires12. Le cholestérol intracellulaire est le principal agent contrôlant l’expression du rLDL. Ainsi, si la cellule manque de cholestérol, elle synthétisera des récepteurs afin d’aller chercher le cholestérol manquant en circulation. C’est par ce mécanisme qu’agissent certains agents hypocholestérolémiant, telles que les statines (voir section 6.3), qui empêchent la synthèse endogène du cholestérol, la forçant à aller chercher le cholestérol en circulation. Ce
20 mécanisme de contrôle impliquant le cholestérol est médié par les «sterol regulatory element binding proteins» (SREBPs), des facteurs de transcription qui activent la transcription du gène du rLDL lors de disette intracellulaire en cholestérol12.
Figure 2.1 – Cycle du R-LDL. (Figure adaptée de Soutar75 et de Goldstein12)
2.2.2 – Récepteur apparenté au récepteur des LDL (LRP)
Le LRP est membre de la famille du rLDL. Il est un dimère composé d’une sous-unité de 515 kDa et d’une sous-sous-unité de 85 kDa, toutes deux encodées par un seul gène, synthétisées en une seule chaîne polypeptidique qui est clivée dans l’appareil de Golgi. Le LRP est synthétisé au niveau du foie, du cerveau et du placenta. Il possède la capacité de lier de nombreux ligands, comme certaines protéinases, certains complexes protéinases:inhibiteur, certaines exotoxines bactériennes, l’apo E, les TRL et certaines lipases dissociées de leur matrice de protéoglycans. Puisqu’il lie l’apo E, le LRP joue un
rôle important dans l’élimination hépatique des résidus de chylomicrons, des VLDL et des IDL77-79.
2.2.3 – Récepteur scavenger de classe B, type 1 (SR-B1)
Le SR-B1 est membre de la grande famille des récepteurs scavengers, dont le rôle premier dans le système immunitaire est de vidanger la circulation d’un grand nombre de ligands indésirables. Dans cette optique, SR-B1 est capable de lier les lipoprotéines modifiées, les ligands anioniques et les cellules en apoptose80. Mais au-delà de son rôle de vidangeur, SR-B1 a été identifié comme étant le récepteur des HDL. Il est principalement exprimé au niveau des hépatocytes, des macrophages et des tissus stéroïdogéniques81. Au contact d’une particule HDL, SR-B1 forme un canal hydrophobe laissant passer les EC dans le sens du gradient de concentration. Ainsi, au niveau des macrophages gorgés d’EC, SR-B1 sert de donneur de cholestérol, alors qu’au niveau du foie et des tissus stéroïdogéniques, grands consommateurs de cholestérol, SR-B1 sert d’accepteur de cholestérol. Il est important de spécifier que SR-B1 n’internalise pas la particule HDL82. Les grosses HDL riches en EC se lient plus facilement à SR-B1 que les petites HDL, contribuant ainsi au renouvellement du pool de petites HDL, qui sont le ligand préféré de ABCA183, un autre système responsable de l’efflux des EC des macrophages. Une surexpression spécifiquement au niveau du foie des souris entraîne une diminution des taux de C-HDL, une augmentation du cholestérol excrété dans la bile et une augmentation du catabolisme de l’apo A1 par le rein84;85.
2.2.4 – ATP-binding cassette A1 (ABCA1)
ABCA1 est un récepteur transmembranaire de 2261 acides aminés exprimé au niveau des macrophages, du foie, du cerveau, des reins et de l’intestin. Les récepteurs de cette famille utilisent l’ATP comme source d’énergie pour transporter des substrats d’un compartiment cellulaire à l’autre ou au travers de la membrane cellulaire86. ABCA1
22 transporte les EC et les PL cellulaires vers les lipoprotéines liées à la surface cellulaire, principalement les HDL87 et préférentiellement les petits HDL83. Au niveau des macrophages, ABCA1 est responsable de l’élimination de l’excès de cholestérol qui est entreposé sous forme de cristaux d’EC. Le mécanisme par lequel ABCA1 transporte cet excès de cholestérol immobilisé vers l’extérieur de la cellule n’est pas encore très bien élucidé, mais impliquerait un transport à l’aide de vésicules contenant ABCA1 qui iraient chercher les EC immobilisés et les transporteraient ensuite vers la surface cellulaire pour être transportés vers une lipoprotéine88.
2.3 – Voies de transport des lipides
Le transport des différents lipides en circulation se fait en suivant trois voies : le transport des lipides exogènes de l’intestin vers les autres tissus, le transport des lipides endogènes du foie vers les autres tissus et le transport inverse du cholestérol, des tissus périphériques vers le foie et les tissus stéroïdogéniques pour élimination. À l’intérieur de ces voies, certains dérèglements peuvent provoquer la formation de lipoprotéines favorisant un état pro-athérosclérotique, telles que les sdLDL et les HDL petites et denses (sdHDL). Les deux états pathologiques dont il sera question dans les prochains chapitres impliquent également un dérèglement de ces voies en favorisant l’accumulation de lipides comme par exemple l’HF, favorisant l’accumulation de particules LDL en circulation et un décalage de ces particules vers les sdLDL.
2.3.1 – Voie des lipides exogènes
La fonction de la voie des lipides exogènes est d’amener les lipides alimentaires (de provenance exogène) aux tissus pour la production d’énergie, le stockage ou la synthèse de molécules. Les lipides alimentaires sont hydrolysés dans le petit intestin et sont absorbés par les cellules épithéliales intestinales. Les lipides y seront réestérifiés et seront assemblés à l’aide de l’apoB-48 pour former les CM. Les CM sont sécrétés dans la lymphe et se
retrouvent dans la circulation sanguine. Au niveau des capillaires des muscles et du tissu adipeux, les TG contenus dans les CM sont hydrolysés en acides gras libres par la LPL pour stockage ou production d’énergie. Le ratio entre l’apo C-II et l’apo C-III présentes dans les CM déterminera la rapidité à laquelle le CM sera transformé en résidu de CM. Puisque seuls les TG ont été hydrolysés, le résidu sera enrichi en EC et en apo E, alors que l’apo A-I, A-II, C-II et C-III seront échangées vers d’autres lipoprotéines en circulation. L’apo A-I relâchée pourra également servir de précurseur de HDL. Les résidus de CM sont captés au foie via le rLDL et le LRP. Habituellement, il ne reste que de très bas niveaux de CM en circulation après environ 12 heures suivant un repas. À jeun, les taux d’apoB-48 sont d’environ 5 mg/L, alors que ces taux augmentent de manière variable suite à un repas, selon la quantité de lipides contenue dans ce repas26;34;89.
2.3.2 – Voie des lipides endogènes
Quelques heures suivant un repas, lorsque la quantité de CM en circulation est faible, les besoins en TG des tissus périphériques sont assurés par les lipides synthétisés par le foie ou transitant par celui-ci (les FFA provenant de la lipolyse), qui sont alors acheminés par les VLDL. De la même manière que les CM, les VLDL seront hydrolysées par la LPL dans les capillaires et l’activité LPL sera modulée par le ratio apo II/apo C-III. Les acides gras libérés par ces lipases serviront alors de source d’énergie aux différents tissus. Les résidus des VLDL, les IDL, subiront l’hydrolyse de leurs TG par l’action de la HL, menant ainsi à la particule LDL fortement enrichie en EC. La HL peut aussi hydrolyser les TG restant dans la particule LDL. La EL est aussi capable d’hydrolyser les TG contenus dans les lipoprotéines contenant l’apoB-100, mais cet aspect de cette lipase n’a été que récemment décrit et demeure nébuleux90. Aussi, en cours de route, les apo des VLDL sont perdues, soit par échange ou soit par libération dans le plasma, et il ne reste alors qu’une seule molécule d’apoB-100, nécessaire au maintien de l’intégrité du LDL. La LCAT peut agir sur les LDL pour estérifier le CL qu’elles contiennent. La CETP peut échanger des EC contre des TG entre différentes classes de lipoprotéines : des EC des HDL contre des TG des LDL, VLDL et IDL; et des EC des LDL contre des TG des IDL et des
24 VLDL. Les LDL seront retirés de la circulation par le rLDL qui reconnaît l’apoB-100. Leur demi-vie moyenne est d’environ 2.7 jours12;26;34;89.
2.3.3 – Transport inverse du cholestérol
Les apo composant la partie protéique du HDL sont synthétisées par le foie et l’intestin et proviennent également de l’hydrolyse des CM et des VLDL par les lipases, qui relâchent alors des constituants en circulation. Les PL composants les HDL proviennent principalement des autres lipoprotéines lors de leur hydrolyse. La HDL reçoit du CL et des EC des autres lipoprotéines lors de leur hydrolyse, mais aussi via SR-B1 et ABCA-1. Le CL est alors estérifié par la LCAT. Au fur et à mesure que le HDL reçoit du cholestérol, sa taille augmente, passant de la classe HDL3 à la classe HDL2. Les EC peuvent par la suite être échangés contre des TG entre les HDL et les lipoprotéines contenant l’apoB-100 par l’action de la CETP. Les EC ainsi transférés aux LDL et aux VLDL retourneront alors au foie via le rLDL. La HDL sera captée par un récepteur SR-B1 du foie ou d’un tissu stéroïdogénique auquel elle donnera son cholestérol. Il est important de noter que la HDL n’est pas internalisée par SR-B1; après avoir livré ses EC, la HDL se retrouve à nouveau en circulation et redevient disponible pour recevoir des EC. La HL est capable d’hydrolyser les TG contenus dans la HDL. Dans le foie, le cholestérol sera transformé en sels biliaires ou sera directement excrété dans la bile, alors que dans les tissus stéroïdogéniques, le cholestérol sera transformé en hormones stéroïdiennes. L’apo A-I dissociée est éliminée par les reins, peut servir de précurseur de HDL ou peut s’associer à un HDL mature32;44;91.
2.3.4 – LDL petites et denses et mécanisme de formation
La formation des particules sdLDL fait partie intégrante des autres voies ci-haut décrites. Ce mécanisme revêt toute son importance dans le fait que les sdLDL sont les particules LDL considérées comme étant les plus athérogènes, quoique cela ne fasse pas l’unanimité (voir section 3.4). Comme mentionné ci-haut, la CETP permet l’échange d’EC
des LDL et des HDL vers les VLDL, en échange de TG et rend alors les particules LDL plus riches en TG, ce qui réduit leur diamètre. Plus les VLDL sont riches en TG, plus il y a de TG pouvant être transférés aux LDL. La particule LDL enrichie en TG est alors un meilleur substrat pour la HL et pour la EL que les particules LDL moins riches en TG. La HL et la EL remodèlent alors la particule LDL en hydrolysant ses TG et en la rendant alors encore plus petite et plus dense61;90. Également, la sPLA2 hydrolyse des PL des LDL et contribue ainsi à la formation des sdLDL. Des taux élevés de sPLA2 ont été associés à la présence de sdLDL et au risque de MCV. Il a été postulé que toute enzyme venant augmenter le ratio TG/EC ou appauvrir les particules LDL en lipides contribuait à la formation des sdLDL58;61.
La conformation 3D de la molécule d’apoB-100 d’une particule LDL est dépendante de la composition du cœur lipidique, de la composition en PL de la surface et de la taille de la particule92;93. Deux sites sur la molécule d’apoB-100, le site A (résidus 3148 à 3158) et le site B (résidus 3359 à 3369), ont été identifiés comme ayant une forte affinité pour les protéoglycans de la média des vaisseaux sanguins94. Il a été démontré que des particules LDL soumises à l’action de la sPLA2 deviennent plus petites, ce qui entraînerait l’exposition du site A qui pourrait alors agir en coopération avec le site B pour lier l’apoB-100, et donc la particule LDL, aux protéoglycans des vaisseaux sanguins, augmentant ainsi leur athérogénécité92;95. Ainsi, il est possible de supposer qu’une diminution de la taille des particules LDL, indépendamment de la cause de la diminution, amènerait une exposition de ces sites et ainsi une plus grande athérogénicité de ces particules.
De plus, des résultats suggèrent que les sdLDL sont plus susceptibles aux modifications chimiques (oxydation, peroxydation, acétylation, glycosylation, etc.) que les grosses LDL96;97. Chez des sujets diabétiques de type II, une relation inverse significative a été montrée entre la taille des particules LDL et leur niveau d’oxydation98. Il a aussi été démontré que certains processus de modification agissaient à des sites particuliers de la molécule d’apoB-10099;100. Compte tenu de la modification de la conformation de l’apoB-100 lorsque la taille de la particule LDL change, il est possible que certains de ces épitopes