Nouvelle méthode de synthèse de matériaux
bidimensionnels riches en carbone à partir de
polydiacétylènes
Mémoire
Isabelle Levesque
Maîtrise en chimie
Maître ès sciences(M. Sc.)
Québec, Canada
© Isabelle Levesque, 2014
III
Résumé
L’intérêt pour les nanomatériaux, tel que les fullerènes, les nanotubes de carbone et les nanoparticules, est en constante croissance depuis le milieu des années quatre-vingt. Parmi ces nanomatériaux, un qui attire particulièrement l’intérêt des chercheurs est le nanoruban de graphène, soit une feuille de graphène ayant une largeur de moins de 50 nm. Étant donné les faibles dimensions de ce matériau, qui créent un confinement quantique, et du motif de ses parois, les nanorubans de graphène ont des propriétés électroniques qui peuvent être différentes de celles du graphène. De plus, celles-ci sont modulables, c’est-à-dire que la valeur de la bande interdite peut être contrôlée en variant différents facteurs. Les nanorubans de graphène peuvent donc être des semi-conducteurs avec différentes valeurs de bande interdite alors que le graphène est un matériau qui a une valeur de bande interdite nulle. Malgré le grand intérêt pour les nanorubans de graphène, il n’existe encore aucune méthode de synthèse, physique ou chimique, qui permet, à grande échelle, le contrôle des propriétés électroniques.
Dans le cadre de ce projet, nous avons voulu développer une méthode alternative, appelée méthode hybride, afin d’obtenir un nanoruban de graphène. La stratégie principale se base sur la réactivité de l’unité 1,3-diarylbutadiyne qui peut polymériser par réaction topochimique sous irradiation UV pour former un nanoruban de polydiacétylène (PDA). Pour permettre ce type de réactions, des paramètres très stricts doivent être respectés et il est possible de les obtenir en auto-assemblant des précurseurs linéaires, contenant des unités 1,3-diarylbutadiyne, sous forme de gel. Nous avons donc développé ces précurseurs, en choisissant avec soin les fonctions présentes sur ceux-ci, afin de permettre l’auto-assemblage sous forme de gel. Par la suite, ces précurseurs auto-assemblés ont permis d’obtenir, après irradiation, des feuillets de PDA. Finalement, les PDAs ont été graphitisés par voie thermique. Afin de mieux comprendre la réactivité et l’influence des unités aryles présentes dans nos PDAs, nous avons étendu notre étude à une nouvelle famille de précurseurs. Ceux-ci n’étaient pas linéaires, ils avaient plutôt un motif en forme « V », c’est-à-dire avec les butadiynes en position 1,3 sur un phényle central. De plus, nous avons évalué l’effet des différentes fonctions des précurseurs sur les propriétés de gélification.
V
Table des matières
Résumé ... III Table des matières... V Liste des tableaux ... IX Liste des figures ... XI Liste des schémas ... XV Liste des abréviations ... XVII Remerciements ... XIX Avant-Propos ... XXI
Préambule ... XXII
Introduction ... 1
1.1. Le graphène et les nanorubans de graphène ... 1
1.1.1. Le graphène ... 1
1.1.2. Le nanoruban de graphène ... 3
1.1.2.1. Type des parois ... 3
1.1.2.2. Largeur et longueur de nanorubans ... 4
1.1.2.3. Défauts dans la structure ... 5
1.1.3. Intérêt des nanorubans de graphène ... 5
1.2. Méthodes de synthèse ... 6
1.2.1. Méthode physique ... 6
1.2.2. Méthode chimique ... 9
1.2.3. Méthode hybride ... 11
1.3. Polymérisation topochimique ... 14
1.3.1. Auto-assemblage sous forme de cristaux et co-cristaux ... 15
1.3.2. Polymérisation topochimique de diacétylènes substitués par des aryles ... 17
1.4. Objectif ... 19
1.5. Méthodologie ... 20
VI Chapitre 2 ... 25 2.1. Résumé ... 26 2.2. Abstract ... 26 2.3. Introduction ... 27 2.4. Résultats et discussion ... 30 2.5. Conclusion ... 37 2.6. Annexes ... 38 2.6.1. Informations générales ... 38 2.6.2. Appareillage ... 38 2.6.3. Test de gélification ... 39
2.6.4. Images de microscopie électronique à balayage ... 39
2.6.5. Images de microscopie électronique à transmission ... 39
2.6.6. Procédures expérimentales ... 40 2.6.7. Caractérisation ... 43 2.7. Références ... 51 Chapitre 3 ... 55 3.1. Résumé ... 56 3.2. Abstract ... 56 3.3. Introduction ... 57 3.4. Procédures expérimentales ... 59 3.4.1. Informations générales ... 59 3.4.2. Appareillage ... 59 3.4.3. Test de gélification ... 60 3.4.4. Microscopie ... 60 3.4.5. Synthèse ... 61 3.5. Résultats et discussion ... 65 3.6. Conclusion ... 76 3.7. Remerciements ... 77 3.8. Annexes ... 78 3.8.1. UV-Visible Spectroscopy ... 78 3.8.2. Fluorescence spectroscopy ... 79
VII
3.8.3. Differential scanning calorimetry (DSC) ... 80
3.8.4. Powder X-rays diffraction (PXRD) ... 82
3.8.5. NMR 1H and 13C spectra ... 85 3.9. Références ... 89 Conclusion ... 95 4.1. Nouveaux oligomères ... 98 4.2. Étude du mécanisme ... 100 4.3. Polyynes ... 103
IX
Liste des tableaux
Tableau 2.1 Gelation properties of BD3 at 10 mg/mL ... 43
Tableau 3.1 Gelation properties of OPBDs T1 to T4 in commonly used organic solvents. ... 69
Tableau 3.2 Properties of OPBDs-based organogels . ... 70
XI
Liste des figures
Figure 1.1 Représentation d’une partie d’un feuillet de graphène ... 1
Figure 1.2 Schématisation de la définition d’un conducteur et d’un semi-conducteur 2 Figure 1.3 Nanoruban de graphène de type zig-zag (a) et de type chaise (b) ... 3
Figure 1.4 Exemples de défauts pouvant être présents dans la structure ... 5
Figure 1.5 Illustration schématique d’oxydation chimique de graphène pour l’obtention de nanorubans de graphène ... 7
Figure 1.6 Schéma représentant l’ouverture de nanotube de carbone par réactions d’oxydation ... 8
Figure 1.7 Exemple de synthèse chimique de nanoruban de graphène à partir de précurseurs moléculaires du groupe du professeur Müllen ... 10
Figure 1.8 Schématisation de la synthèse d’une nanotige ou d’un nanotube (a) et du nanoruban (b) par méthode hybride ... 12
Figure 1.9 Macrocyle synthétisé (a) pour l’obtention d’une nanotige par méthode hybride (b) ... 13
Figure 1.10 Schéma de la polymérisation topochimique... 14
Figure 1.11 Structure cristalline de la glycine d’oxalamide ... 16
Figure 1.12 Co-cristaux de l’acide oxalamide dicarboxylique et de diacétylène ... 17
Figure 1.13 Représentation de la double conjugaison dans les PDAs avec groupements aryle ... 18
Figure 1.14 Synthèse de PDAs à partir d’un oligomère auto-assemblé sous forme gel ... ... ... 19
Figure 1.15 Schéma de rétrosynthèse d’un nanoruban de graphène ... 21
Figure 2.1 UV/visible (a) and photoluminescence (b) of PDA-BD3 in dilute THF solution as a function of temperature. The photoluminescence peak at 533 nm is attributed to the formation G-BD3. The green curve represents the photoluminescence spectrum of G-BD3 once it is has been heated to reflux in THF for few minutes. Inset: color change in THF solution from PDA-BD3 (left) to G-PDA-BD3 (right). ... 33
Figure 2.2 Raman spectra of G-BD3 films. ... 34
Figure 2.3 SEM (a,b), HRTEM (c,d), SAED pattern (e) and zoom on a folded edge (f) of G-BD3. ... 35
Figure 2.4 Plausible mechanisms for the cycloaromatization reaction of PDA-BD3 to yield G-BD3 based on Fukuzawa’s report ... 37
Figure 2.5 Ethyl acetate-based gel (10 mg/mL). ... 44
Figure 2.6 SEM images of ethyl-acetate-based xerogel of BD3 (10 mg/mL). Scale bars are 10 µm (a), 10 µm (b) and 1 µm (c). ... 44
Figure 2.9 Comparative Raman spectra of BD3 and PDA-BD3. ... 46
XII
Figure 2.12 HRTEM images of G-BD3. Scale bars are 20 nm (a), 5 nm (b), 100 nm (c),
200 nm (d), 50 nm (e) and 100 nm (f). ... 47
Figure 2.12 HRTEM images of G-BD3. Scale bars are 20 nm (a), 5 nm (b), 100 nm (c), 200 nm (d), 50 nm (e) and 100 nm (f). ... 47
Figure 2.13 AFM (Tapping mode, left) and height profile of G-BD3 deposited on a glass substrate. ... 48
Figure 2.14 Dynamic light scattering (DLS) data for PDA-B ... 48
Figure 2.15 Comparative IR spectra of PDA-BD3 (blue) and G-BD3 (red) ... 49
Figure 2.16 Fluorescence confocal microscopy of G-BD3 suspended in methanol. Bright spot are aggregates of G-BD3. Excitation at 560 nm ± 20 nm and emission at 630 nm ± 35 nm. ... 50
Figure 3.1 General approach toward 2D PDAs from meta-linked OPBD precursors. 66 Figure 3.2 Synthetic pathway for OPBDs T1 and T2. ... 68
Figure 3.3 Synthetic pathway toward meta-linked OPBD T3 and T4. ... 69
Figure 3.4 SEM images of a 10 mg/mL xerogel of a) T1 (octane), b) T2 (octane), c) T3 (toluene) and d) T4 (toluene). Scale bars are 5 µm (a to c) and 10 µm (d). ... 71
Figure 3.5 Raman spectra for a) T3 and PDA3 and b) T4 and PD4. ... 75
Figure 3.6 UV-visible spectra of a) G3 and PDA3 and b) G4 and PDA4. ... 76
Figure 3.7 TEM images of PDA3 a) before heating and b) after heating and PDA4 c) before heating and d) after heating. Scale bars are a) 2 µm and b) to d) 500 nm. ... 77
Figure 3.8 UV−visible spectrum of T3 and PDA3 in THF ... 78
Figure 3.9 UV−visible spectrum of T4 and PDA4 in THF ... 78
Figure 3.10 Photoluminescence spectrum of PDA3 and G3 in THF ... 79
Figure 3.11 Photoluminescence spectrum of PDA4 and G4 in THF ... 79
Figure 3.12 DSC curves of T1 gel in n-octane ... 80
Figure 3.13 DSC curves of T2 gel in n-octane ... 80
Figure 3.14 DSC curves of T3 gel in toluene ... 81
Figure 3.15 DSC curves of T4 gel in toluene ... 81
Figure 3.16 PXRD of octane-based xerogel (10 mg/mL) of T1 ... 82
Figure 3.17 PXRD of octane-based xerogel (10 mg/mL) of T2 ... 82
Figure 3.18 PXRD of toluene-based xerogel (10 mg/mL) of T3 ... 83
Figure 3.19 PXRD of toluene-based xerogel (10 mg/mL) of T3 with controlled cooling of the gel ... 83
Figure 3.20 PXRD of toluene-based xerogel (10 mg/mL) of T4 ... 84
Figure 3.21 PXRD of toluene-based xerogel (10 mg/mL) of T4 with controlled cooling of the gel ... 84
Figure 3.22 1H NMR of compound 1 in CDCl3... 85
XIII Figure 3.24 1H NMR of compound T1 in CDCl3 ... 86 Figure 3.25 13C NMR of compound T1 in CDCl3 ... 86 Figure 3.26 1H NMR of compound 2 in CDCl3... 86 Figure 3.27 1H NMR of compound T2 in CDCl3 ... 87 Figure 3.28 13C NMR of compound T2 in CDCl3 ... 87
Figure 3.29 1H NMR of compound T3 in DMSO ... 87
Figure 3.30 13C NMR of compound T3 in DMSO ... 88
Figure 3.31 1H NMR of compound T4 in CDCl3 ... 88
Figure 3.32 13C NMR of compound T4 in CDCl3 ... 88
Figure 4.1 Oligomère BD3 synthétisé pour la formation de nanoruban ... 95
Figure 4.2 Nanoruban de polydiacétylènes formé à partir de l’oligomère BD3 avec le motif arylènyne mis en évidence dans l’encadré vert ... 96
Figure 4.3 Oligomères liés en méta pour la formation de matériaux graphitiques ... 97
Figure 4.4 Oligomère linéaire avec une nouvelle unité gélificatrice faite à partir de l’ester L-Alanine octadécyle ... 99
Figure 4.5 Oligomère linéaire avec une nouvelle unité gélificatrice faite à partir d’un dérivé du cholestérol ... 99
Figure 4.6 Motif -ène-yne-ène inspiré de la structure du PDA mis en évidence en bleu ... 101
Figure 4.7 Molécules modèles inspirées du motif -ène-yne-ène pouvant former le chrysène ou le fulvalène ... 101
Figure 4.8 Motif -yne-ène-yne inspiré de la structure du PDA mis en évidence en bleu ... 102
Figure 4.9 Molécules modèles inspirées du motif -yne-ène-yne pouvant former le chrysène ... 103
XV
Liste des schémas
Schéma 2.1 Synthesis of the reactive molecular precursor BD3 and the general strategy for the preparation of graphitic materials from assembled butadiynes-containing molecular precursors. The green region shows the arylenyne motif. Side groups on PDA-BD3 have been omitted for clarity. ... 29
XVII
Liste des abréviations
a.u. Unité arbitraire
AFM Microscopie à force atomique
CGC Concentration critique de gélification
cm Centimètre
DLS Diffusion dynamique de la lumière
DMSO Diméthylsulfoxyde
DSC Calorimétrie différentielle à balayage EELS Spectroscopie des pertes d'énergie
eV Électron-volt
eq. Équivalent
g Gramme
h Heure
HRMS Spectroscopie de masse à haute résolution
HRTEM Microscopie électronique en transmission haute résolution
Hz Hertz Hν Onde lumineuse IR Infrarouge J Constante de couplage (RMN) kDa Kilodalton M Molaire mg Milligramme mL Millilitre mmol Millimole
Mn Masse moléculaire moyenne en nombre
mol Mole
nm Nanomètre
NMR Résonance magnétique nucléaire
o-DCB ortho-dichlorobenzène
OPBD Oligophénylènebutadiynylène
XVIII
PI Indice de polydispersité
PXRD Diffraction des rayons-X sur poudre SAED Diffraction des électrons sur surface SEM Microscopie électronique à balayage TBAF Fluorure de tétra-n-butylammonium TBDMS tert-Butyldimethylsilyl
Tc Température critique de fonte du gel TEM Microscopie électronique en transmission Tgel Température critique de gélification
THF Tétrahydrofuranne
TLC Chromatographie sur couche mince TMEDA Tétraméthyléthylènediamine TMS Triméthylsilyle UV Ultra-violet ° Degré °C Degré Celsius µL Microlitre µm Micromètre Å Ångström δ Déplacement chimique (RMN)
XIX
Remerciements
Je tiens en premier lieu à remercier mon directeur de maîtrise, le professeur Jean-François Morin, pour l’opportunité qu’il m’a donné d’effectuer ma maîtrise au sein de son laboratoire. Sa passion et son dévouement pour son laboratoire nous poussent à continuellement vouloir faire progresser nos projets et à toujours donner le meilleur de nous-mêmes. Sa disponibilité, sa patience et son ouverture d’esprit face à nos idées font de lui une personne avec qui, il est des plus agréable et motivant de travailler.
Je souhaiterais également remercier tous les membres du groupe présent ou passé pour l’ambiance et la bonne humeur qui règnent au laboratoire, mais également pour toutes les discustions qui ont permis de faire avancer le projet. Le désir de voir les autres membres du laboratoire réussir autant que soi-même fait de vous .des collègues extraordinaires. J’aimerais également remercier plus spécifiquement deux de mes collègues actuels au laboratoire, soit Jules Roméo Neabo ainsi que Simon Rondeau-Gagné. J’ai vraiment apprécié travailler en étroite collaboration avec vous sur ces différents projets, vous m’avez été d’une aide inestimable et je vous en suis extrêmement reconnaissante.
De plus, je me dois de remercier Philippe Dufour, professionnel de recherche du laboratoire Morin, pour m’avoir prise sous son aile dès mon premier stage au laboratoire. C’est avec patience qu’il a pris le temps de me former au début de mon parcours en chimie et qu’il a su me transmettre sa passion pour la science.
Finalement, je voudrais remercier ma famille ainsi que mes amis pour leur support et encouragement continus, mais plus particulièrement mon copain Mathias. Il a toujours été là pour me soutenir et me pousser à persévérer. Sa patience et sa confiance en moi m’ont permis de réaliser ce projet.
XXI
Avant-Propos
Article Chapitre 2
Matériau Graphitique Multicouche à partir
d’un Précurseur Moléculaire
Layered Graphitic Materials From a Molecular Precursor
Isabelle Levesque, Jules Roméo Néabo, Simon Rondeau-Gagné, Cécile Vigier-carrière,
Maxime Daigle, and Jean-François Morin
Département de Chimie and Centre de recherche sur les matériaux avancés (CERMA), 1045 Ave. de la médecine, Université Laval, Québec, Canada G1V 0A6.
XXII
Préambule
Ma contribution dans cette partie du projet est significative. J’ai effectué la synthèse complète de l’oligomère, du polydiacétylène ainsi que du matériau graphitique. J’ai également participé à la caractérisation des différents matériaux. De plus, j’ai contribué à la rédaction et à la correction de l’article et j’ai également fait la section « Informations supplémentaires » de l’article dans son entièreté.
XXIII
Article Chapitre 3
Synthèse, Gélification et Polymérisation topochimique de
Dérivés Oligophénylènebutadiynylènes liés en position meta
Synthesis, Gelation and Topochemical Polymerization of meta-
linked Oligophenylenebutadiynylene Derivatives
Isabelle Levesque, Jules Roméo Néabo, Simon Rondeau-Gagné,Maxime Daigle, and Jean-François Morin
Département de Chimie and Centre de recherche sur les matériaux avancés (CERMA), 1045 Ave. de la médecine, Université Laval, Québec, Canada G1V 0A6.
XXIV
Préambule
J’ai grandement contribué à cette deuxième partie du projet. Dans un premier temps, j’ai grandement contribué à la synthèse des quatre différents monomères. J’ai fait l’étude des propriétés de gélification ainsi que des propriétés de polymérisation et de graphitisation. De plus, j’ai participé activement à la caractérisation des différents matériaux. Finalement, je me suis impliquée pour la rédaction de l’article ainsi que pour sa correction. J’ai également écrit toute la section « Informations supplémentaires » de l’article.
Introduction
1.1. Le graphène et les nanorubans de graphène
1.1.1. Le graphène
Le graphène est un allotrope du carbone. Il est constitué d’un réseau bidimensionnel de dimension infini fait entièrement d’atomes de carbone qui sont tous hybridés sp2. C’est en fait une monocouche de graphite. Connu depuis de nombreuses années, le graphène a été isolé pour la première fois en 2004 par les professeurs Andre K. Geim et Konstantin S. Novesolev, qui, tous deux, étaient professeurs à l’Université de Manchester au Royaume-Uni au moment de cette réalisation. C’est d’ailleurs le premier matériau bidimensionnel à avoir été isolé. C’est pour cette grande avancée scientifique que le prix Nobel de physique de 2010 leur a été remis puisqu’ils ont été les premiers à avoir isolé, identifié et caractérisé le graphène.1
Figure 1.1 Représentation d’une partie d’un feuillet de graphène
Le graphène possède des propriétés physiques et mécaniques intéressantes tels que sa très faible épaisseur, sa solidité, sa flexibilité ainsi que son imperméabilité. Puisque la distance entre deux atomes de carbone opposés dans une même maille n’est que de 0,142 nm, rien ne peut passer au travers.2 Par contre, compte tenu du but de ce projet de maîtrise concerne les propriétés électroniques du graphène, les propriétés physiques et mécaniques ne seront pas exposées davantage.
2
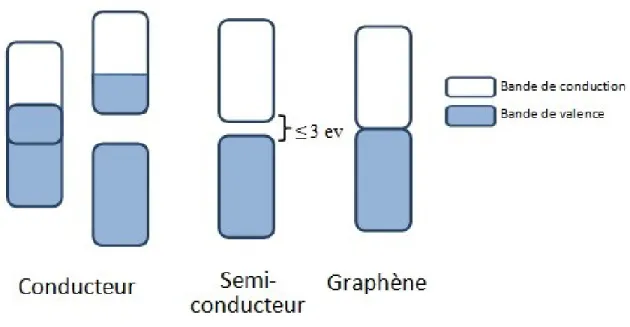
Les propriétés électroniques du graphène en font un matériau de très grand intérêt. Étant donné qu’il est entièrement fait d’atomes de carbone hybridés sp2, le graphène se présente sous forme de motif alvéolé. Il reste donc, pour chaque atome de carbone, un électron qui est délocalisé sur toute la surface et qui est responsable des propriétés électroniques du graphène. Le graphène est souvent considéré comme étant un matériau conducteur, mais cette notion est quelque peu erronée. Le graphène est, en fait, un matériau semi-conducteur dont la valeur de la bande interdite est nulle. De façon générale, il existe deux façons de décrire un matériau conducteur. Il est possible de dire qu’un matériau est conducteur lorsqu’il y a un chevauchement de la bande de conduction et de la bande de valence ou, encore, lorsque la bande de valence et celle de conduction sont séparées par une certaine valeur de bande interdite, mais que la bande de conduction est peuplée en électrons. Quant à un matériau semi-conducteur, tous les électrons sont dans la bande de valence et la valeur de la bande interdite est de moins de 3 eV. Dans le cas du graphène, tous les électrons sont dans la bande de valence, mais la valeur de la bande interdite est de 0 eV. Le graphène respecte donc la définition d’un semi-conducteur. Il serait plus juste de décrire le graphène comme un semi-conducteur qui possède les propriétés électroniques d’un conducteur.1
3 1.1.2. Le nanoruban de graphène
Un second matériau de la famille du graphène, et tout aussi intéressant d’un point de vue électronique, est le nanoruban de graphène. Ce matériau est défini comme étant un feuillet de graphène dont la largeur est de tout au plus 50 nm, plutôt que d’être de dimension infinie. Cette différence dans les dimensions a un grand effet sur les propriétés électroniques. Il est possible d’obtenir un matériau qui peut être semi-conducteur et dont la valeur de la bande interdite peut être modulée lorsque certains paramètres, tels que le type des parois du nanoruban, la longueur et la largeur du nanoruban, ainsi que les défauts présents dans la structure, sont contrôlés.
1.1.2.1. Type des parois
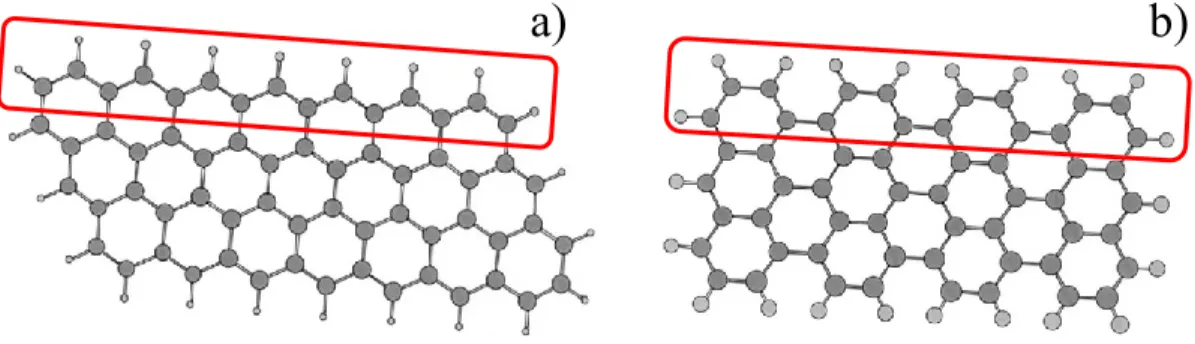
Les nanorubans de graphène existent sous deux différentes formes qui sont nommées forme zig-zag et forme chaise. Il est possible de facilement déterminer le type de nanoruban en regardant la structure de la paroi la plus longue d’un nanoruban de graphène. Lorsque la paroi est similaire à la structure qui est encadrée à la Figure 1.3 a) le nanoruban est de type zig-zag. Si la paroi est plutôt comme celle encadrée sur le nanoruban illustré à la Figure 1.3 b), il est de type chaise.3
Figure 1.3 Nanoruban de graphène de type zig-zag (a) et de type chaise (b) 3
Dépendant si le nanoruban est de type chaise ou zig-zag, les propriétés électroniques vont différer. Les nanorubans de type zig-zag sont toujours des matériaux conducteurs.
4
Pour ce qui est des nanorubans de type chaise, ils peuvent être soit conducteurs ou semi-conducteurs. Ce qui va déterminer leur conductivité électronique est la largeur du nanoruban.
1.1.2.2. Largeur et longueur de nanorubans
La variation de la largeur d’un nanoruban de graphène peut modifier les propriétés électroniques du matériau. Comme mentionné précédemment, les nanorubans de type zig-zag sont toujours conducteurs, donc la variation de leur largeur n’a aucune influence sur les propriétés électroniques. Par contre, pour ceux de type chaise la largeur va avoir une influence sur les propriétés électroniques. Lorsque la largeur diminue, la délocalisation des électrons ne se fait plus sur une aussi longue distance sur cet axe. Cette diminution de la distance de délocalisation crée un confinement quantique. Ce confinement se traduira par une diminution de la valeur de la bande interdite. Il peut y avoir un changement de conductivité de conducteur à semi-conducteur et plus le nanoruban sera étroit, plus la valeur de la bande interdite sera grande. Il est donc possible de moduler les propriétés électroniques en contrôlant la largeur du nanoruban.
La longueur peut également être un facteur qui influence les propriétés électroniques. Pour les nanorubans de type zig-zag, tout comme la modification de la largeur, la modification de la longueur n’a aucun effet puisqu’ils sont toujours conducteurs. Pour ceux qui sont de type chaise, la diminution de la longueur diminue la valeur de la bande interdite. Plus un nanoruban est court, moins les électrons sont délocalisés sur une grande distance dans cet axe. Il y a ainsi création d’un confinement quantique. Par contre, cet effet est notable que pour des nanorubans ayant une longueur de 10 nm ou moins. Dans le but d’utiliser les nanorubans de graphène dans des dispositifs électroniques, des nanorubans de moins de 10 nm de longueur ne sont pas utilisés. Il n’y a donc pas d’intérêt à modifier la valeur de la bande interdite par la modification de la longueur, car les nanorubans utilisés en nano-électronique sont toujours de plus de 10 nm.
5 1.1.2.3. Défauts dans la structure
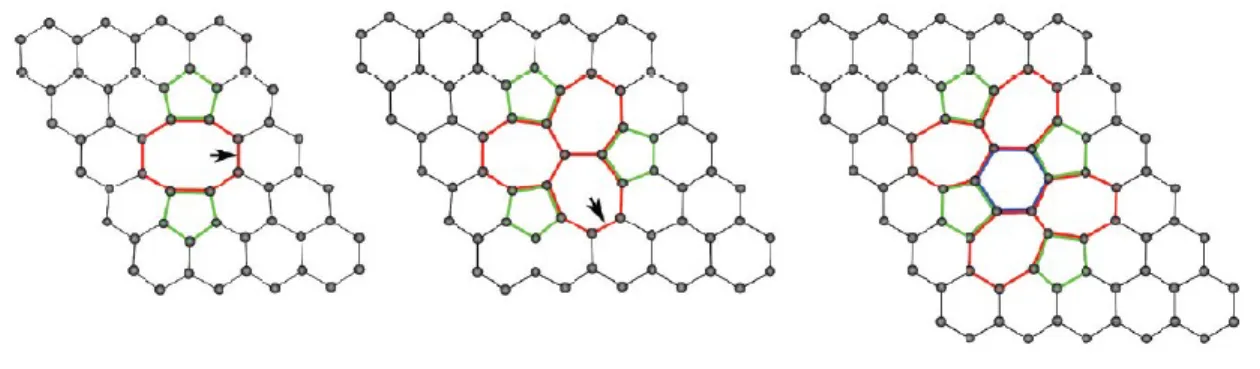
Les propriétés électroniques peuvent également être modifiées par la présence de défauts dans la structure. Différents défauts, comme ceux illustrés à la Figure 1.4, peuvent être présents dans les nanorubans de graphène, mais également dans le graphène. Lorsque la structure n’est pas parfaite, il y a une modification du recouvrement orbitalaire. Le recouvrement devenant moins bon, la délocalisation des électrons ne se fait pas aussi facilement que dans les nanorubans de graphène ou dans le graphène exempts de défaut. Cette délocalisation moins efficace des électrons vient diminuer la valeur de la bande interdite. Dépendant du nombre de défauts et de la nature de ceux-ci, la variation de la bande interdite sera différente.4
Figure 1.4 Exemples de défauts pouvant être présents dans la structure4
1.1.3. Intérêt des nanorubans de graphène
Le principal intérêt des nanorubans de graphène est dans le domaine de la nano-électronique. Comme ils peuvent être semi-conducteurs, il pourrait être envisageable de les utiliser comme matériau actif. Les nanorubans de graphène pourraient servir d’électrode dans différents dispositifs tels que dans des transistors à effet de champs, des cellules solaires, des supercapacitances et des capteurs.5 , 6 Bien que ces différents dispositifs nécessitent des semi-conducteurs n’ayant pas tous la même valeur de bande interdite, les nanorubans de graphène pourraient être utilisés pour chacun d’entre eux puisque la valeur de leur bande interdite peut être modulée. De plus, il pourrait être
6
envisageable de fabriquer des dispositifs tout carbone en utilisant les nanorubans comme matériaux actifs et le graphène comme matériau conducteur.
1.2. Méthodes de synthèse
Il existe différentes méthodes de synthèse permettant d’obtenir des nanorubans de graphène. Les deux plus connues sont la méthode physique et la méthode chimique. La méthode physique peut être associée à l’approche de synthèse qui est qualifiée comme étant descendante, tandis que la méthode chimique peut-être associée à la méthode ascendante.7 De plus, il existe depuis quelques années une nouvelle façon de penser la synthèse de matériaux. Cette façon combine à la fois la méthode physique et la méthode chimique. Cette nouvelle méthode de synthèse fait depuis peu son apparition dans la littérature comme étant une méthode de synthèse hybride.8,9
1.2.1. Méthode physique
Les méthodes physiques de synthèse de nanorubans de graphène sont généralement des méthodes qualifiées comme étant descendantes. À partir d’un matériau plus complexe, tel que le graphène ou les nanotubes de carbone, il est possible par irradiation ou oxydation d’obtenir des nanorubans de graphène.
Un premier exemple d’une façon d’obtenir des nanorubans par méthode physique est par oxydation chimique du graphène à l’aide d’un plasma à l’oxygène. Un exemple du groupe du professeur Xiangfeng Duan et de celui du professeur Yu Huang permet de bien illustrer cette méthode (Figure 1.5).10 La première étape de cette procédure consiste à peler mécaniquement sur un substrat de silicone des feuillets de graphène de deux ou quelques couches d’épaisseur de graphène (Figure 1.5 a). Bien qu’ils utilisent une méthode physique pour obtenir leurs feuilles de graphène sur le substrat de silicium, il serait possible d’utiliser cette méthode de synthèse de nanorubans pour des feuillets de graphène obtenus par une méthode de synthèse chimique. Par la suite, la deuxième étape consiste à aligner des nanotiges de silicone sur le graphène qui serviront de masques lors
7 du découpage. Une fois le masque en place, un plasma à oxygène est envoyé sur la surface (Figure 1.5 c) et il découpe que le graphène qui n’est pas protégé, ce qui laisse des nanorubans de graphène sous les nanotiges. Si le plasma est envoyé verticalement au substrat, les nanorubans obtenus auront une largeur similaire au diamètre des nanotiges. Par contre, il est possible de diminuer la largeur des nanorubans si le plasma est envoyé avec un angle par rapport à la surface (Figure 1.5 e). Finalement, il est possible de retirer les nanotiges de silicone grâce aux ultrasons. Grâce à cette méthode, ils sont en mesure d’obtenir les nanorubans sur un substrat de silicium.
Figure 1.5 Illustration schématique d’oxydation chimique de graphène pour l’obtention de nanorubans de graphène10
Une seconde façon d’obtenir des nanorubans de graphène par méthode physique est par ouverture de nanotubes de carbone. La méthode ici présentée est un exemple du groupe du professeur Tour11 qui consiste à ouvrir des nanotubes de carbone multicouches. Lorsque ceux-ci sont traités avec du permanganate de potassium (KMnO4) et de l’acide sulfurique (H2SO4),une série de réactions d’oxydation permet l’ouverture des nanotubes. La première étape proposée est la formation de l’ester de manganate sur les nanotubes (Figure 1.6 c). C’est d’ailleurs cette première étape qui est limitante dans la méthode. Par la suite, une seconde oxydation est possible, ce qui forme les deux cétones (Figure 1.6 d). Ces deux cétones qui sont juxtaposées engendrent de l’encombrement et, par le fait même,
8
une déformation des liens β,γ-alcènes (illustré en rouge sur la Figure 1,6 c). C’est d’ailleurs la déformation de ces liens qui les rend très réactifs à l’oxydation subséquente. Ils seront donc à leur tour facilement oxydés pour former, chacun d’eux, deux nouvelles cétones. L’oxydation se propagera de cette façon jusqu’à ce qu’il y ait ouverture complète du nanotube de carbone. Ces réactions ne procèdent pas de façon aléatoire sur les nanotubes de carbone. Une fois qu’il y a un début d’ouverture, elle continue de se propager de façon linéaire. De plus, comme la première étape est celle qui est limitante, il n’y aura pas plusieurs sites de début d’ouverture sur les nanotubes. L’ouverture peut se faire de façon longitudinale sur le nanotube de carbone, tel qu’illustré à la figure 1.6 a) ou encore en spirale, dépendamment du lieu de la première oxydation et également du vecteur de chiralité du nanotube.
Figure 1.6 Schéma représentant l’ouverture de nanotube de carbone par réactions d’oxydation11
a)
b)
c)
d)
9 Les méthodes physiques de synthèse de nanorubans possèdent de nombreux avantages. Ce type de synthèse permet d’obtenir des nanorubans très rapidement et il est possible d’en obtenir une grande quantité. Par contre, les désavantages sont également nombreux. Premièrement, il est impossible d’éliminer la présence d’impuretés dans les matériaux produits. Deuxièmement, il est très difficile de moduler avec précision les dimensions des nanorubans qui sont obtenus. De plus, dans une même synthèse, tous les nanorubans ne sont pas uniformes, c’est-à-dire que des nanorubans de différentes largeurs et longueurs seront obtenus. Également, d’une fois à l’autre, les résultats de synthèse seront différents, ces méthodes sont donc non-reproductibles.8 Tous ces désavantages font que le contrôle des propriétés électroniques est difficile, ce qui limite l’utilisation des nanorubans de graphène faits par cette méthode dans des dispositifs électroniques.
1.2.2. Méthode chimique
Les méthodes chimiques de synthèse de nanorubans de graphène sont des méthodes considérées comme étant ascendantes. À partir de précurseurs moléculaires, grâce aux méthodes de synthèses modernes et aux techniques permettant la transformation de groupements fonctionnels, il est possible d’élaborer la synthèse de matériaux avec un contrôle précis sur les dimensions, la forme et les fonctions chimiques présentes.
Le professeur Müllen est l’un des pionniers dans le domaine de la synthèse chimique de nanorubans de graphène. Depuis les dix dernières années, il a publié bon nombre d’articles dans lesquels il est possible de trouver des exemples de synthèse de nanorubans de graphène où il est en mesure de contrôler la forme des parois, les groupements fonctionnels présents, ainsi que la largeur du matériau. L’exemple illustré à la Figure 1.7 est l’un des plus récents de ce groupe de recherche.12 La technique employée pour l’obtention des nanorubans dans cet article est similaire à celles de ces autres matériaux publiés.13,14,15,16 Le matériau se fabrique à partir d’un précurseur riche en carbone qu’ils ont synthétisé, dans cet article le précurseur est un para-terphényle oligophénylène qui contient des groupements solubilisants, des chaînes alkyle longues de douze atomes de carbone (Figure 1.7 4b). Ensuite, par polymérisation de Yamamoto, ils obtiennent le
10
large polymère 5 (Figure 1.7). Finalement par réaction de cyclodéshydrogénation de Scholl, en utilisant chlorure de fer (III) (FeCl3) comme oxydant, ils obtiennent le nanoruban 6 (Figure 1.7).
Figure 1.7 Exemple de synthèse chimique de nanoruban de graphène à partir de précurseurs moléculaires du groupe du professeur Müllen12
Bien que les méthodes chimiques permettent d’obtenir des nanorubans purs avec les dimensions désirées, donc avec des propriétés électroniques spécifiques, elles possèdent certains désavantages. Premièrement, la réaction de Scholl est très difficile à contrôler. Il est tout simplement impossible de prédire le produit qui sera obtenu, car les réactions de cyclodéshydrogénation ne s’effectuent pas nécessairement sur tous les sites possibles du matériau. Cette réaction est, également, très sensible aux groupements fonctionnels. De plus, les méthodes de synthèse chimique nécessitent de nombreuses étapes, ce qui fait qu’il peut être relativement long et ardu d’obtenir le nanoruban souhaité. Également, étant donné les différents réactifs, les solvants et les purifications nécessaires afin d’obtenir un nanoruban de graphène pur, cette voie est plutôt dispendieuse et elle permet d’obtenir qu’une très faible quantité du matériau souhaité.8 Compte tenu de tous ces
11 inconvénients, la méthode chimique ne semble pas être une bonne alternative pour l’obtention de matériaux aux propriétés électroniques contrôlées à l’échelle industrielle.
1.2.3. Méthode hybride
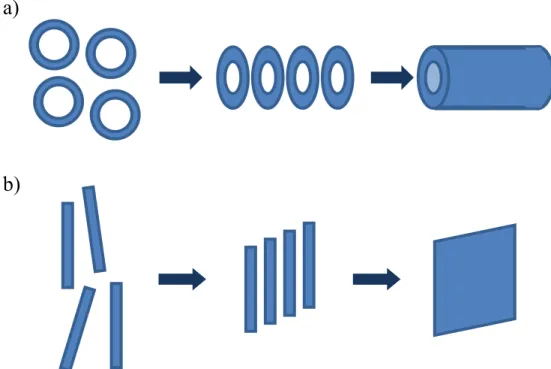
Afin d’outrepasser les désavantages des méthodes physiques et chimiques, mais tout en conservant leurs avantages, une nouvelle méthode de synthèse a commencé à faire son apparition dans la littérature scientifique depuis quelques années. Puisque cette nouvelle méthode combine à la fois une partie de l’approche physique et une partie de l’approche chimique, certains chercheurs l’ont nommé méthode hybride. Celle-ci consiste à synthétiser en un minimum d’étapes un précurseur riche en carbone. Celui-ci doit être en mesure de s’auto-assembler d’une façon ou d’une autre, par exemple sous forme de cristaux, sous forme de gel ou encore sur une surface. Finalement une fois assemblé, le précurseur est exposé à un stimuli physique, tel que la lumière ou la chaleur. Sous l’effet de ce stimuli, les précurseurs polymérisent entre eux pour former un matériau riche en carbone.8 Cette méthode peut s’appliquer à la synthèse de nanorubans de graphène, mais également à une grande gamme de matériaux riches en carbone, tels que les nanotubes17, les nanotiges18 et les nanoparticules19. Le matériau final obtenu dépend directement du choix du précurseur. Si le précurseur est un macrocycle riche en carbone, le matériau final devrait être un nanotube ou une nanotige (Figure 1.8 a) alors que si le précurseur est un oligomère linéaire riche en carbone, le matériau final devrait être un nanoruban (Figure 1.8 b).
12
Figure 1.8 Schématisation de la synthèse d’une nanotige ou d’un nanotube (a) et du nanoruban (b) par méthode hybride
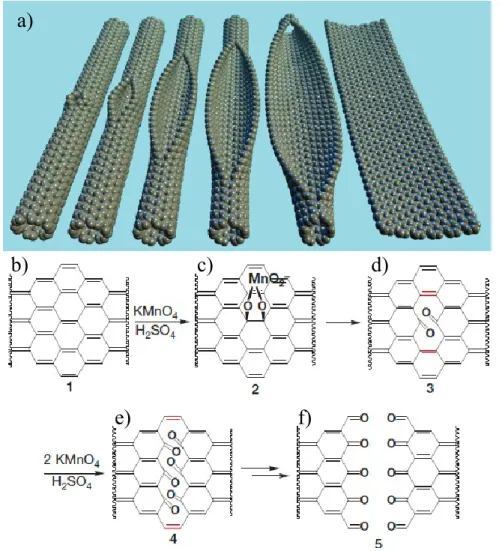
Jusqu’à ce jour, il n’existe aucun exemple dans la littérature de synthèse de nanoruban de graphène par méthode hybride. Par contre, il est possible de bien illustrer cette méthode de synthèse avec un exemple du groupe du professeur Morin où nous avons été en mesure de synthétiser une nanotige à partir d’un macrocycle auto-assemblé.20 La première étape consiste à synthétiser un macrocycle riche en carbone par voie organique. Ce macrocyle a été conçu afin de pouvoir s’auto-assembler sous forme de gel (Figure 1.9 a). Les fonctions amides portant une longue chaine alkyle facilitent la gélification, puisque les amides permettent la formation de ponts hydrogène alors que les longues chaines alkyle augmentent les interactions de van der Waals intermoléculaires. De plus, les unités butadiyne peuvent réagir entre elles pour former des polydiacétylènes (PDAs), par polymérisation topochimique, lorsqu’elles sont soumises à un stimulus physique (Figure 1.9 b) ce qui permet la création du nouveau matériau riche en carbone.
b)
a)
13
Figure 1.9 Macrocyle synthétisé (a) pour l’obtention d’une nanotige par méthode hybride (b)20
a)
14
La synthèse de matériaux riches en carbone par voie hybride permet d’obtenir les matériaux souhaités avec un contrôle sur les paramètres structuraux, et ce, grâce au fait qu’il est possible de synthétiser le précurseur souhaité. De plus, cette méthode de synthèse se fait dans des conditions douces, ce qui rend possible la présence d’un grand nombre de différentes fonctions chimiques. Également, le matériau souhaité s’obtient assez rapidement. Par contre, ces méthodes sont encore relativement nouvelles et il reste encore beaucoup de travail à faire dans le but de les rendre plus efficaces. Les deux principaux points à améliorer sont de maximiser l’auto-assemblage des précurseurs et de trouver les meilleures conditions dans lesquelles la polymérisation va s’effectuer afin d’augmenter les quantités de matériau produit.
1.3. Polymérisation topochimique
Une grande partie de la stratégie de la méthode hybride repose sur les unités butadiyne présentes sur notre précurseur de départ. En 1968, Gerhard Wegner a découvert la capacité de polymérisation des unités butadiyne sous forme solide pour former un polydiacétylène. Cette polymérisation, qui se nomme polymérisation topochimique, se fait par addition 1,4 entre les différentes unités 1,3-butadiyne lorsque celles-ci sont chauffées ou irradiées avec de la lumière UV ou des rayons gamma (Figure 1.10). 21,22
15 Par contre, pour que la polymérisation topochimique puisse avoir lieu, il ne suffit pas qu’il y ait présence d’unités butadiyne. Certains paramètres doivent être strictement respectés (Figure 1.10), sans quoi aucune réaction n’a lieu. Premièrement, la distance intermoléculaire entre les unités butadiyne doit être de 4,9 Å et elles doivent avoir un angle d’inclinaison de 45 ° les unes par rapport aux autres. Étant donné la tension engendrée lors de la polymérisation, de longues chaines alkyle flexibles sont habituellement ajoutées à chacune des extrémités des alcynes. Cet ajout permet le maintien des paramètres et ainsi permet à la polymérisation d’avoir lieu sur une plus grande distance. De plus, le polymère qui est obtenu est généralement soluble dans les solvants organiques usuels. Cette réaction permet d’obtenir des polymères conjugués très intéressants compte tenu de leurs propriétés thermochromique et solvatochromique.23
1.3.1. Auto-assemblage sous forme de cristaux et co-cristaux
Comme mentionnée précédemment, la polymérisation photochimique nécessite un respect de certains paramètres de distance et d’angle. Il faut donc que les unités 1,3-butadiyne soient bien placées afin que la polymérisation puisse avoir lieu. L’une des façons utilisées pour s’assurer d’avoir un arrangement parfait pour la polymérisation est d’auto-assembler les molécules sous forme de cristaux.24 Par contre, le principal problème de ce mode d’assemblage est qu’il peut être long et ardu de faire croitre des cristaux. Il se peut même qu’après de nombreux essais, aucun cristal ne soit obtenu. De plus, lorsqu’il est possible d’obtenir des cristaux, il se peut que l’arrangement spatial des molécules ne respecte pas les critères requis pour permettre la polymérisation topochimique et donc que le PDA ne puisse tout simplement pas être obtenu. L’organisation dans l’espace des molécules sous forme de cristal s’avère très difficile à prédire.
Afin de pallier au problème de l’orientation non prévisible des molécules contenant des unités butadiyne dans l’espace sous forme de cristaux dans le but d’obtenir des PDAs, certains groupes, tel que celui du professeur Lauher, ont développé une méthode d’auto-assemblage faisant appel à des co-cristaux.25 Ils ont tout d’abord dû trouver une molécule
16
qui, lorsqu’elle cristallise, possède la distance et l’angle entre les molécules similaires à ceux nécessaires pour la polymérisation topochimique. Un tel processus est très ardu, car il n’existe rien qui puisse permettre de prédire parfaitement l’organisation d’une molécule dans l’espace lorsqu’elle est sous forme de cristaux. Après essais et erreurs, ils ont déterminé que la molécule de glycine d’oxalamide serait une des molécules qui respecterait ces critères (Figure 1.11).
Figure 1.11 Structure cristalline de la glycine d’oxalamide 25
Par la suite, en faisant croitre des co-cristaux de diacétylènes substitués à leurs extrémités par des groupements pouvant faire des ponts hydrogène avec la glycine d’oxalamide, les groupements 1,3-butidiyne devraient avoir la distance et les angles nécessaires pour la polymérisation topochimique (Figure 1.12). De cette façon, ils ont réussi à obtenir des PDAs. De plus, il est possible de récupérer que le PDA et d’éliminer la molécule qui a servi à la formation du co-cristal, soit l’acide oxalamide dicarboxylique.
17
Figure 1.12 Co-cristaux de l’acide oxalamide dicarboxylique et de diacétylène25
1.3.2. Polymérisation topochimique de diacétylènes substitués par des aryles Un type de diacétylène qui est particulièrement intéressant serait un diacétylène avec des substituants aryle aux deux extrémités. Avec ce type de substitution, les PDAs seraient non seulement conjugués dans l’axe de la polymérisation, mais également perpendiculairement à celle-ci (Figure 1.13). Une telle conjugaison permettrait d’obtenir de nouvelles propriétés électroniques qui seraient très intéressantes. Malgré le grand intérêt pour ce type de PDA, il existe très peu d’exemples dans la littérature où les groupements diacétylène avaient comme substituants des groupements phényle. Théoriquement, il semble improbable que des diacétylènes possédant des groupements aryle puissent polymériser étant donné la tension engendrée lors de la polymérisation. Comme mentionné précédemment, les unités diacétylène possèdent généralement de longues chaines alkyle afin de minimiser cette tension et d’éviter l’arrêt de la polymérisation. Par contre, si les diacétylènes possèdent des groupements phényle, l’augmentation de la tension, qui ne peut être atténuée par des groupements phényle, vient briser le cristal et par le fait même l’arrangement. La polymérisation ne peut donc pas avoir lieu.
18
Figure 1.13 Représentation de la double conjugaison dans les PDAs avec groupements aryle
Une alternative afin de pallier au problème de rigidité lors de la polymérisation topochimique de diacétylènes substitués par des aryles sous forme de cristaux a été proposée par le groupe du professeur Morin.26 Nous proposons d’effectuer l’auto-assemblage sous état gel plutôt que sous état solide. Puisqu’un gel est beaucoup moins rigide qu’un cristal, il y a de bonnes raisons de croire que la polymérisation topochimique pourrait avoir lieu, malgré la présence de groupements aryle. Par contre, les gels sont également moins ordonnés que les cristaux, ce qui fait que l’auto-assemblage se ferait probablement sur une plus courte distance. Afin de permettre la formation d’un gel, nous avons soigneusement choisi le précurseur à synthétiser. Premièrement, le précurseur choisi (Figure 1.14 a) possède deux fonctions amides portant chacune une longue chaine alkyle. Les groupements amide sont très souvent utilisés pour orienter correctement des unités butadiyne sous forme de cristaux ou de gel. La distance intramoléculaire entre deux groupements amide placés face à face est d’environ 4,8 Å, ce qui est très près des distances nécessaires afin de permettre la polymérisation topochimique. De plus, les fonctions alcool facilitent la gélification et les chaines alkyle augmentent la solubilité ce qui facilite également la gélification.
19
Figure 1.14 Synthèse de PDAs à partir d’un oligomère auto-assemblé sous forme gel26
Ce précurseur s’est avéré être une excellente molécule pour s’auto-assembler sous forme de gel. Nous avons été en mesure d’obtenir des gels dans de nombreux solvants chlorés ainsi que dans le toluène. Le gel a été déposé sur une plaque de verre et séché afin d’obtenir un xérogel. Finalement, la polymérisation topochimique a été faite grâce à une source de lumière UV de 254 nm. Après purification, nous avons obtenu des PDAs de masse molaire de 27,9 kDa avec un indice de polydispersité de 1,8.
1.4. Objectif
Étant donné que jusqu’à ce jour aucun exemple de synthèse de nanorubans de graphène par méthode hybride n’a été rapporté dans la littérature, l’objectif de ce projet est de développer une méthode permettant de le faire. Pour obtenir ces nanorubans de graphène, certaines étapes devront être suivies. Premièrement, il faudra développer la synthèse d’un précurseur linéaire riche en carbone qui pourra s’auto-assembler grâce aux
hν (254
nm)
20
fonctionnalités présentes sur la molécule. Deuxièmement, les meilleures conditions d’auto-assemblage devront être déterminées pour, troisièmement, mettre le précurseur auto-assemblé en présence d’un stimuli physique qui permettra à la polymérisation topochimique d’avoir lieu entre les différentes molécules. Le produit, après cette polymérisation, sera un nouveau matériau riche en carbone qui, une fois graphitisé, deviendra un nanoruban de graphène. Les résultats de cet objectif sont présentés au chapitre 2 de cet ouvrage.
La seconde partie du projet est d’appliquer cette même stratégie, mais cette fois à une nouvelle classe de précurseurs. Ceux-ci, plutôt que d’être linéaires sont en forme de « V », c’est-à-dire avec un phényle au centre de la molécule qui sera substitué en position 1,3. Le but de cette modification est de mieux comprendre le processus qui permet l’obtention de matériaux graphitique lorsque les molécules polymérisées sont chauffées dans le but d’être graphitisées. Le chapitre 3 de cet ouvrage décrit en profondeur les résultats de cet objectif.
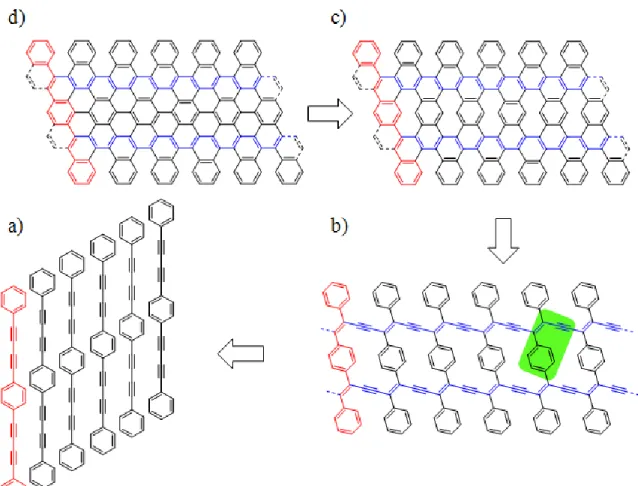
1.5. Méthodologie
C’est en s’inspirant d’un schéma de rétrosynthèse d’un nanoruban de graphène (Figure 1.14) que nous avons développé notre stratégie. Il semble être possible de synthétiser un nanoruban de graphène à partir d’un monomère linéaire possédant des unités butadiyne comprises en deux groupements phényle en position 1,4 (Figure 1.15 a). En auto-assemblant sous forme de gel ces précurseurs et en les soumettant à un stimuli physique, les unités butadiyne pourront réagir entre elles pour former un large polymère de PDA comme illustré à la Figure 1.15 b). De plus, ce nouveau polymère possèdera des motifs arylényne (encadré vert de la Figure 1.15) qui sont reconnus pour cycloaromatiser lorsque chauffsé27 , 28 ou irradiés29 , 30. De cette façon, il serait possible d’obtenir un nanoruban de graphène, mais celui-ci comportera des défauts au centre de sa structure (Figure 1.15 c). Finalement, un nanoruban de graphène pourra être obtenu par réaction de cyclodéshydrogénation. Avec cette approche, il est possible de moduler la largeur du nanoruban en choisissant des précurseurs de différentes largeurs. Plus il y aura d’unités
21 butadiyne entre deux phényles sur le monomère, plus le ruban sera large et donc plus bas sera la valeur de sa bande interdite.
22
1.6.
Références
1 Geim, A. K.; Novoselov, K. S. Nat Mater 2007, 6, 183.
2 "The Nobel Prize in Physics 2010 - Advanced Information". Nobelprize.org. Nobel Media AB 2013. Web. 14 Mar 2014. <http://www.nobelprize.org/ nobel_prizes/physics/laureates/2010/advanced.html>
3 Owens, F. J. J. Chem. Phys. 2008, 128, 194701.
4 Banhart, F.; Kotakoski, J.; Krasheninnikov, A. V. ACS Nano 2011, 5, 26.
5 Zhu, Y.; James, D. K.; Tour, J. M. Adv. Mater. 2012, 24, 4924.
6 Song, X.; Hu, J.; Zeng, H. J. Mater. Chem. C 2013, 1, 2952.
7 Tour, J. M. Chem. Mater. 2014, 26, 163.
8 Morin, J.-F. Synlett 2013, 24, 2032.
9 Rondeau-Gagne, S.; Morin, J.-F. Chem. Soc. Rev. 2013, 43, 85.
10 Bai, J.; Duan, X.; Huang, Y. Nano Lett. 2009, 9, 2083.
11 Kosynkin, D. V.; Higginbotham, A. L.; Sinitskii, A.; Lomeda, J. R.; Dimiev, A.; Price, B. K.; Tour, J. M. Nature 2009, 458, 872.
12 Schwab, M. G.; Narita, A.; Hernandez, Y.; Balandina, T.; Mali, K. S.; De Feyter, S.; Feng, X.; Müllen, K. J. Am. Chem. Soc. 2012, 134, 18169.
13 Wu, J.; Gherghel, L.; Watson, M. D.; Li, J.; Wang, Z.; Simpson, C. D.; Kolb, U.; Müllen, K. Macromolecules 2003, 36, 7082.
14 Yang, X.; Dou, X.; Rouhanipour, A.; Zhi, L.; Räder, H. J.; Müllen, K. J. Am.
Chem. Soc. 2008, 130, 4216.
15 Fogel, Y.; Zhi, L.; Rouhanipour, A.; Andrienko, D.; Räder, H. J.; Müllen, K.
Macromolecules 2009, 42, 6878.
16 Dössel, L.; Gherghel, L.; Feng, X.; Müllen, K. Angew. Chem. Int. Ed. 2011, 50, 2540.
17 Xu, Y.; Smith, M. D.; Geer, M. F.; Pellechia, P. J.; Brown, J. C.; Wibowo, A. C.; Shimizu, L. S. J. Am. Chem. Soc. 2010, 132, 5334.
18 Néabo, J. R.; Rondeau-Gagné, S.; Vigier-Carrière, C.; Morin, J.-F. Langmuir
23
19 Neabo, J. R.; Vigier-Carriere, C.; Rondeau-Gagne, S.; Morin, J.-F. Chem.
Commun. 2012, 48, 10144.
20 Rondeau-Gagné, S.; Néabo, J. R.; Desroches, M.; Larouche, J.; Brisson, J.; Morin, J.-F. J. Am. Chem. Soc. 2013, 135, 110.
21 Wegner, G. Z. Naturforsch. B, 1969, 824.
22 Wegner, G. Makromol. Chem. 1972, 154, 35.
23 Wenzel, M.; Atkinson, G. H. J. Am. Chem. Soc. 1989, 111, 6123.
24 Chance, R. R.; Baughman, R. H.; Müller, H.; Eckhardt, C. J. The Journal of
Chemical Physics 1977, 67, 3616.
25 Lauher, J. W.; Fowler, F. W.; Goroff, N. S. Acc. Chem. Res. 2008, 41, 1215.
26 Néabo, J. R.; Tohoundjona, K. I. S.; Morin, J.-F. Org. Lett. 2011, 13, 1358.
27 Schulz, K.; Hofmann, J.; Zimmermann, G. Eur. J. Org. Chem. 1999, 1999, 3407.
28 Zimmermann, G.; Nuechter, U.; Hagen, S.; Nuechter, M. Tetrahedron Lett. 1994,
35, 4747.
29 Kaafarani, B. R.; Neckers, D. C. Tetrahedron Lett. 2001, 42, 4099.
30 Tinnemans, A. H. A.; Laarhoven, W. H. J. Chem. Soc., Perkin Trans. 2 1976, 1104.
25
Chapitre 2
Matériau Graphitique Multicouche à partir
d’un Précurseur Moléculaire
Layered Graphitic Materials From a Molecular Precursor
Isabelle Levesque, Jules Roméo Néabo, Simon Rondeau-Gagné, Cécile Vigier-carrière,
Maxime Daigle, and Jean-François Morin
Département de Chimie and Centre de recherche sur les matériaux avancés (CERMA), 1045 Ave. de la médecine, Université Laval, Québec, Canada G1V 0A6.
26
2.1. Résumé
Un matériau graphitique multicouche a été préparé à partir d’un réactif moléculaire contenant des alcynes à basse température et sans l’utilisation de catalyseur. Le nanomatériau obtenu est fait de quelques nanofeuillets partiellement graphitisés et est soluble dans les solvants organiques usuels. De plus, lorsque mis en solution, il présente une fluorescence verte.
2.2. Abstract
A layered graphitic material was prepared from an alkyne-containing, reactive molecular precursor at low temperature without catalyst. The resulting nanomaterial is made of stacks of a few partially graphitized nanosheets and is soluble in common organic solvents in which it exhibits green fluorescence.
27
2.3. Introduction
In the past five years, a wealth of efforts has been devoted to the preparation and characterization of two-dimensional materials, including single-layer graphene, 1 graphene oxide,2 few-layer graphene3 and other organic polymers. 4,5 The broad interest for these materials, especially graphene, comes from their two-dimensional electrons delocalization that leads to exceptional semi-conducting (finite ribbons) or conducting (“infinite” sheets) properties that, in many cases, surpass the best silicon-based materials known to date.6 Thus, many perceive two-dimensional materials as the next generation of active materials in electronic devices such as field-effect transistors,7 supercapacitors,8 solar cells9 and sensors.10 Despite the success stories behind graphene-based materials, an efficient and reliable method to prepare high-quality two-dimensional semiconducting materials is still lacking.11 Micromechanical cleavage,12 liquid-phase exfoliation,13 unzipping of carbon nanotubes,14 chemical vapor deposition15 and chemical reduction of graphene oxide2 are all “physical” methods for the production of graphene materials that possess evident advantages, but also important drawbacks that slow the industrial production of these materials.
One promising way to prepare well-defined two-dimensional materials with finite structural parameters is to use the so-called “all-organic” method.16 The Müllen group pioneered this field of research by preparing nanographenes and graphene nanoribbons from macromolecular carbon-rich precursors using a combination of C-C cross-coupling reactions and cyclodehydrogenation (Scholl) reactions.17 Although very useful and versatile, this innovative strategy possesses some intrinsic limitations regarding the size of graphene nanosheets that can be prepared. Thus, different strategies leading to graphenic nanomaterials with sizes of 50 nm and higher are desirable in order to further expand the scope of organic synthesis for the preparation of such materials.
Recently, others and us used a combination of both organic and physical methods, called the “hybrid” method, in which reactive molecular precursors obtained in few synthetic steps are transformed using a physical stimulus to provide carbon nanomaterials. Using the topochemical polymerization of butadiynes- 18 or oligoynes-containing
28
molecules, various types of carbon nanomaterials such as microbeads,19 nanoparticles,20 nanotubes,21 nanorods,22 nanofibers23 and other nanoarchitectures24 have been prepared. However, two-dimensional nanomaterials such as nanosheets and nanoribbons have never been obtained using this versatile strategy.
Herein, we report the synthesis of a new graphitic materials from a reactive molecular precursor consisting of a linear oligomer with three butadiyne units embedded within the oligomer backbone and separated by a phenyl group (BD3, Scheme 2.1). The creation of a covalent, two-dimensional network made of a polydiacétylene (PDA) skeleton
(PDA-BD3) was achieved through the topochemical polymerization of a xerogel of BD3.
Graphene-like nanosheets, thereafter called G-BD3, were obtained after partial graphitization of PDA-BD3 at low temperature and without metal catalyst. The key structural feature for such a graphitization is the presence of the arylenyne motif known to cycloaromatize upon heating or irradiation (Scheme 2.1).25 The resulting graphenic material was characterized using electron microscopy, Raman, infrared and optical spectroscopy.
29
Schéma 2.1 Synthesis of the reactive molecular precursor BD3 and the general
strategy for the preparation of graphitic materials from assembled butadiynes-containing molecular precursors. The green region shows the arylenyne motif. Side groups on PDA-BD3 have been omitted for clarity.
30
2.4. Résultats et discussion
The reactive molecular precursor (BD3) was carefully designed to self-assemble through gelation process. First, both ends of the molecule are made of a phenyl group possessing a long alkyl chain, an amide function and a short hydrophilic chain. This particular unit is very efficient to drive the gelation of butadiyne-containing molecules in different solvents, especially aromatic ones.26 Second, two methyl groups were added on the central phenyl groups in order to increase the solubility and to facilitate the gelation process. We thus assumed that further graphitization reactions would not be complete since the sp2 carbon atoms bearing these methyl groups cannot undergo a cycloaromatization reaction.
The synthesis of the reactive oligomer BD3 is presented in Scheme 2.1. Starting from compound 1,20b the butadiyne unit was deprotected in basic conditions and the resulting intermediate was coupled to compound 2 27 through a standard Castro-Stephen-Sonogashira coupling to give compound 3 in moderate yield. Then, the terminal alkyne was deprotected and a homocoupling reaction in Glaser-Hay conditions was performed to yield the hydroxyl-protected linear oligomer 4. Finally, the two primary alcohols were deprotected using a tetrabutylammonium fluoride to give the final oligomer BD3.
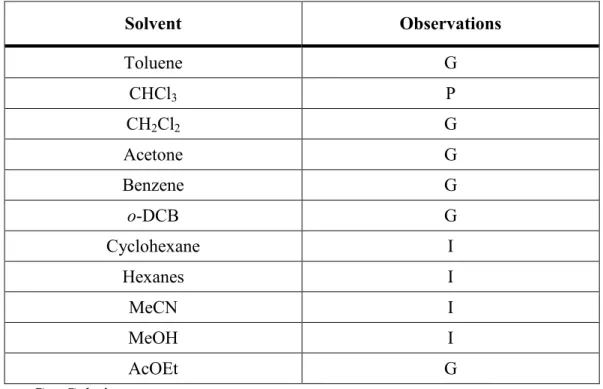
Self-assembly of BD3 was achieved by gelation process in ethyl acetate at a concentration of 10 mg/mL. Using conventional heating/cooling process, 170 mg of the organogel were spread onto a large, clean glass substrate and dried under ambient conditions to yield a thin film of xerogel. Scanning electron microscopy (SEM) images (Figure 2.6) revealed wrinkled sheets rather than the expected fibrillar structure, although presence of nanofibrils with diameter too small to be observed using SEM cannot be discarded. More precise data regarding the molecular organization have been obtained from powder X-ray diffraction (PXRD). Indeed, PXRD pattern of the xerogel of BD3 (Figure 2.8) suggests a rather loosely packed lamellar arrangement in which each molecule are facing each other in a one-dimensional fashion owing to the presence of intermolecular hydrogen bonding. Then, the xerogel was subjected to irradiation under a 254 nm, 100 watt UV lamp for 72 hours. The deep purple solid thus produced, associated
31 to the formation of polydiacétylene (PDA), was purified in a Soxhlet apparatus using acetone to remove short oligomers and unreacted BD3. After 48 hours, 33 mg (19% yield) of a pure cross-linked PDA material, thereafter called PDA-BD3, was dried thoroughly. Recovering of the unreacted BD3 from the Soxhlet apparatus and further iteration of the gelation/irradiation process can be achieved to increase the yield.
PDA-BD3 is readily soluble in THF, but barely soluble in chloroform and aromatic
solvents and it absorbs in the orange-red region of the visible spectrum with a peak at 644 nm and a shoulder at 586 nm (Figure 2.7), which are typical of diaryl-substituted PDA.26a Expectedly, the calculated band gap of PDA-BD3 (1.80 eV) is slightly smaller than its linear analogues (1.90 eV) due to its extended conjugation length in the aryl-aryl axis.
Raman spectroscopy analysis was performed in order to assess whether all the butadiyne units have reacted to form enyne (PDA) moieties (Figure 2.9). As expected, the peak at 2201 cm-1 associated to the butadiyne units in BD3 completely disappeared and a new set of peaks at 2113 cm-1 and 1455 cm-1, attributed to the stretching vibration modes of newly formed alkene and alkyne, respectively, appeared.
Based on PXRD result and other studies on PDA formation from one-dimensional aggregates,26a,28 we expected PDA-BD3 to consist of 2 nm-wide nanoribbons. Thus, we performed transmission electron microscopy (TEM) to confirm the presence of such polymeric material. Surprisingly, no nanoribbon was observed (Figure 2.10). Instead, large domains of amorphous carbon-rich materials with no precise shape appeared, suggesting that the topochemical polymerization did not proceed through a one-dimensional pathway as expected, but rather through a two- or three-one-dimensional pathway. Considering the strict structural parameters required for the topochemical polymerization of butadiynes to take place,18,29 this result is quite surprising and suggests that several assembly modes took place during organogel formation. Thus, we have decided to pursue with the partial graphitization of PDA-BD3 to determine if this two-dimensional morphology can be retained to create graphitic materials.
32
The transformation of PDA-BD3 into a partially graphitized material, thereafter called
G-BD3, was attempted in different conditions. Remarkably, the PDA-BD3 underwent a
modification of its chemical nature at a temperature as low as 55°C when heated in inhibitor-free THF, as indicated by the significant decrease of the absorption bands of
PDA-BD3 at 586 nm (Figure 2.1a and 2.15). A complete disappearance of this absorption
band was observed when a diluted THF solution of PDA-BD3 was refluxed for only few minutes. Moreover, the resulting solution became fluorescent upon heating with an emission band peaking at 533 nm (Figure 2.1b), indicative of the creation of a new chemical species, most likely graphitic materials (vide infra).26b The fluorescence properties of G-BD3 of in methanol has also been measured on its aggregated form using confocal fluorescence microscopy and the results are shown in Figure 2.16. G-BD3 is soluble in common organic solvents and can be dried and resolubilized many times without deterioration of its physical properties. Interestingly, the same chemical transformation occurred when the solution was stored under ambient light for several hours, also producing a yellow solution exhibiting green fluorescence. Although we cannot prove it yet, this result strongly suggests that the chemical transformation went through a radical reaction path. All further characterizations on G-BD3 were performed on a sample that was filtered on a 0.45 μm filter in order to coarse contamination.
33
Figure 2.1 UV/visible (a) and photoluminescence (b) of PDA-BD3 in dilute THF solution as a function of temperature. The photoluminescence peak at 533 nm is attributed to the formation G-BD3. The green curve represents the photoluminescence spectrum of G-BD3 once it is has been heated to reflux in THF for few minutes. Inset: color change in THF solution from
PDA-BD3 (left) to G-BD3 (right).
In order to study the chemical nature of this newly formed materials, Raman spectroscopy analysis was performed and the result is shown on Figure 2.2. The pattern of the spectrum, especially the presence of two bands at 1360 cm-1 and 1580 cm-1, associated with the D and G band, respectively, is clearly indicatives of the presence of
(a)
34
graphitic materials. The relatively high intensity of the D band compared to the G band indicates that the graphitic materials formed upon gentle heat treatment contains defects, mostly coming from edges but also from inside the graphenic nanosheets. In fact, the presence of four methyl groups per BD3 molecule prevents the corresponding sp2 carbon atoms to undergo a cycloaromatization reaction, providing materials with intrinsic defects. It is noteworthy that the presence of defects can be beneficial to give graphitic materials an electronic band gap, opening the way to their use as semi-conductors in different applications.30
Figure 2.2 Raman spectra of G-BD3 films.
To obtain stronger evidences of the presence of a graphitic material, electron energy lost spectroscopy (EELS) was recorded on G-BD3 and the result is shown in Figure 2.11. Peaks at 285 and 295 eV, associated to the 1s-π* and 1s-σ* transitions, respectively, are in agreement with those usually found for graphitic materials.31
G-BD3 was imaged using SEM and high-resolution TEM (HRTEM) and
representative examples of images obtained are shown in Figure 2.3. Images on Figures 2.3a and 2.3b were obtained after a THF solution of G-BD3 was slowly evaporated on top of a SEM substrate. G-BD3 appears as very thin, semi-transparent flakes, which is