Speeding Up Discovery of Auxetic Zeolite Frameworks by Machine Learning
Texte intégral
Figure





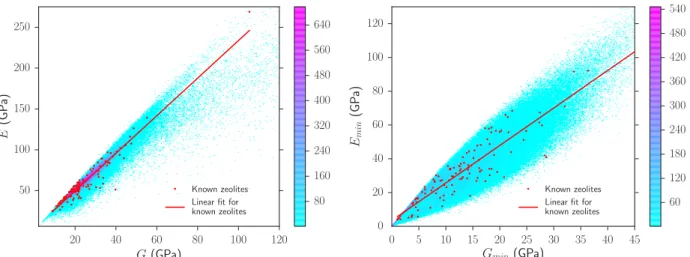


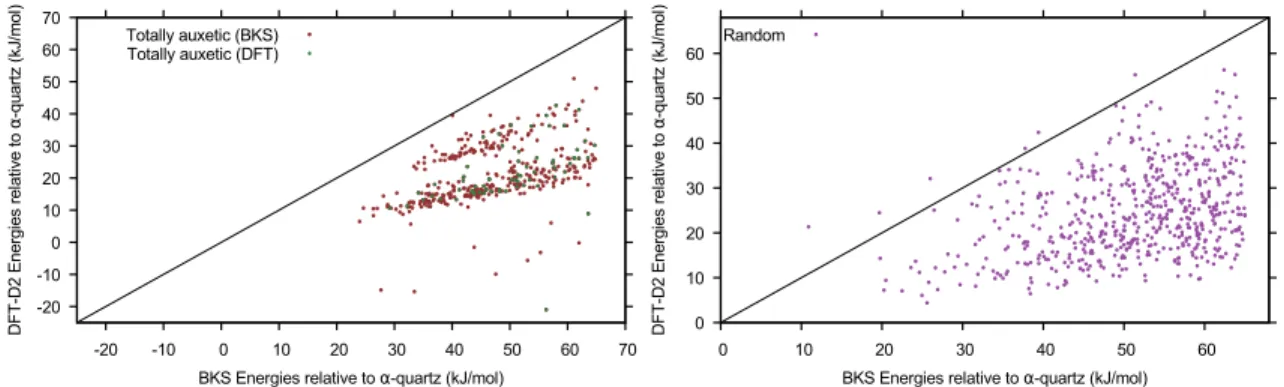
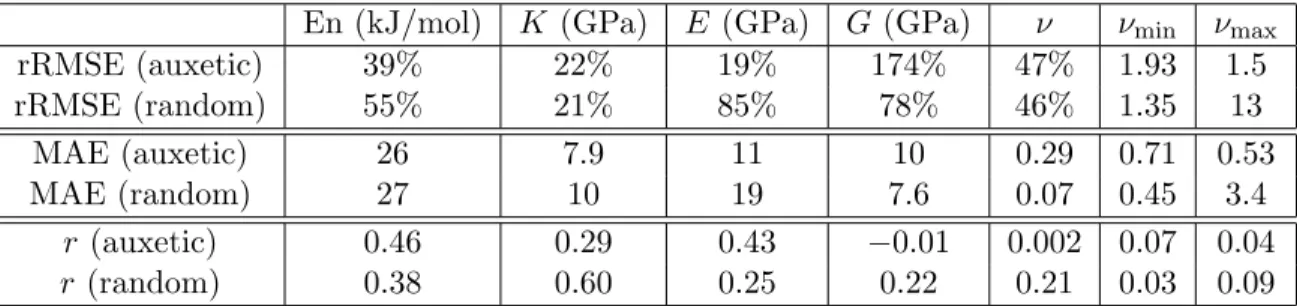
Documents relatifs
Our contribution in this paper is twofold: (i) we learn a joint Pair-Hidden Markov model from a training set composed of (situation,sentence)-pairs; (ii) we study the impact of
Integrated with the data modeling components, the application framework enables an integrated set of services that collaborate to provide a reusable architecture
Figure 6.24 shows the evolution of modulus and hardness (of the titanium carbide sample deposited with 5 SCCM of methane) as a function of the maximum penetration depth..
En effet, en milieu hospitalier, les méningites nosocomiales sont les plus fréquemment rencontrées et leur diagnostic est souvent malaisé du fait d’un tableau
Le premier chapitre traite de la représentation matricielle implicite d’une hypersurface rationnelle dans un espace projectif et propose une nouvelle méthode pour traiter le
Un autre élément à prendre en compte pour évaluer la potentialité des enseignes néon- xénon à remplacer les enseignes au mercure est leur comportement dans le temps.
This report aimed to analyze the LR failure patterns in HNC patients treated with definitive IMRT using a com- prehensive approach for salivary gland-sparing, which integrated in
Cette étude est consacrée à une approche complémentaire à la TVRC (Théorie Variationnelle des Rayons Complexes), basée sur les approximants de Padé, pour la reconstruction rapide
