FACULTÉ DES SCIENCES
Rabat
N° d’ordre:2309THÈSE DE DOCTORAT
Présentée par
TALIDI Abdellah
Discipline : Chimie
Spécialité : Electrochimie et Chimie analytique
Etude de l’élimination du Chrome et du bleu de méthylène en milieux
aqueux par adsorption sur la pyrophyllite traitée et non traitée.
Soutenue le 30 juin 2006, Devant le jury :
Président :
K. EL KACEMI : Professeur de la Faculté des Sciences de Rabat
Examinateurs :
A. CHAKIR
: Professeur de la Faculté des Sciences et Techniques
Mohammadia
M. EL RHAZI
: Professeur de la Faculté des Sciences et Techniques
Mohammadia.
M. TAIBI
: Professeur de l’Ecole National Supérieur
A. GUESSOUS
: Professeur de la Faculté des Sciences de Rabat
Z. EL ABBASSI
: Professeur de la Faculté des Sciences de Rabat
Invité :
M. F. HAMMOUDA : Directeur de Pôle Ressources ONEP
Le travail présenté dans ce mémoire a été réalisé dans le cadre d’une action intégrée entre le laboratoire d’Electrochimie et de Chimie Analytique (LECA) de la Faculté des Sciences de Rabat et le Département de Chimie Agricole, Géologie et Etudes des Sols, de l’Université de Murcia.
Je tiens à exprimer toute ma reconnaissance et ma profonde gratitude à Monsieur le Professeur Kacem
EL KACEMI, Responsable du Laboratoire d’Electrochimie et de Chimie Analytique (LECA) à la Faculté
des Sciences de Rabat pour la confiance qu’il m’a témoignée en m’accueillant au sien de son laboratoire et de m’avoir facilité mon intégration au sien de son équipe. Je voudrai lui exprimer mes meilleurs remerciements pour avoir dirigé ce travail avec beaucoup d’intérêt et de patience et pour l’honneur qui m’a fait pour présider ses juges.
Je remercie profondément Madame A. CHAKIR, Professeur à la Faculté des Sciences et Techniques de Mohammadia, pour l’intérêt qu’elle a porté à ce travail en acceptant de co-diriger cette étude, pour sa disponibilité, ses remarques fructueuses. Qu’elle trouve ma profonde gratitude.
Je suis très sensible à l’honneur que m’a fait Madame M. EL RHAZI, Professeur à la Faculté des Sciences et Technique de Mohammadia, pour m’avoir honoré de sa présence en acceptant d’être rapporteur de ce travail et de le juger.
Je prie Monsieur M. F. HAMMOUDA, Professeur, Directeur de pole de l’Office National de l’Eau Potable de croire à mes sincères remerciements d'avoir accepté notre invitation pour juger ce travail.
Je tiens à remercier Madame A. GUESSOUS, Professeur de faculté des Sciences de Rabat, pour avoir très aimablement accepter de faire partie de ce jury, et pour ses précieuses discussions scientifiques. Je remercie vivement madame Z. EL ABBASSI, Professeur à la Faculté des Sciences de Rabat, pour l’aide scientifique qu’elle a bien voulu apporter à ce travail et pour avoir accepter de l’examiner.
collaboration fructueuse à la réalisation de ce travail, je le remercie également d’avoir accepté à le juger.
Mes remerciements s’adressent également à Monsieur A. MOURAD, Chef de Service Physico-chimie à l’Office National de l’Eau Potable d’avoir accepté notre invitation pour juger ce travail.
Je remercie vivement Monsieur A. BENBRAHIM, Professeur Assistant de notre laboratoire (LECA) pour ses conseils et son aide efficace, en particulier, dans l’analyse par absorption atomique.
Je n’oublie pas dans mes remerciements mes collègues du laboratoire d’Electrochimie et de Chimie Analytique qui m’ont aidé directement ou indirectement à réaliser ce travail, en particulier Monsieur A.
EL ALAOUI, pour l’aide qu’il m’a apporté au cours des expériences de traitement.
Enfin, une grande part de mes remerciements va à mes parents, mes frères et ma sœur pour leur soutien précieux au cours de mon travail.
INTRODUCTION GENERALE 1
CHAPITRE I : ETUDE BIBLIGRAPHIQUE PARTIE 1 : GENERALITES SUR LES ARGILES I- GENERALITES SUR LES ARGILES 4
I-1 Introduction 4 I-2. Minéralogie, structure et propriétés chimiques des argiles 4
I-2.1. Minéralogie et classification des argiles 4
I-2.2 Espaces interfoliaires et capacité d’échange cationique (CEC) 7
I-2.2.1 Les cations compensateurs 7
I-2.2.2 Mesures de la capacité d'échange cationique 9
I-3. La surface spécifique des adsorbants 10
I-4. Rétention des métaux par les argiles 11
I-4.1. Affinité des métaux lourds 13
I-4.2 Théorie d’adsorption en phase liquide 14
I-5. Adsorption du bleu de méthylène 15
II. HYDROLYSE DES METAUX 16
III- ARGILES MODIFIEES 17
1II-1. Généralités 17
III-1.1. Famille des Complexes Organo-Argileux 17
III-1.2. Famille des Complexes Inorgano-Organo-Argileux 19
III-1.3. Famille des complexes inorgano-argileux 19
III-2. Argiles modifiées par différents polymères inorganiques 20
III-3. Modification des argiles par Al13 20
III-3.1. Mode d’obtention des argiles modifiées 20
III-3.2. Facteurs influençant la modification des argiles 21
III-4. Polymère après modification des argiles 21
III.4.1. Structure du polymère 21
III.4.2. Stabilité du polymère après traitement d’argile 21
III-5. Stabilité des argiles 22
III-5.1 Influence de la modification et de la calcination sur la texture des argiles 22
IV- Conclusion 23
PARTIE 2 : GENERALITES SUR LE CHROME I. CHROME ET ENVIRONNEMENT 24
I-1.1 Les sources naturelles 24
I-1.2. Les sources anthropiques 24
II. TOXICITE DU CHROME 25
II.-1. historique 25
II.-2 Doses létales 26
II-3 pathologie attribuée au chrome et ces dérivés 26
II-3.1. lésions cutanées 26
III. PROPRIETES CHIMIQUES DU CHROME 27
III-1. Etats d’oxydation 27
III-2. Spéciation du chrome en solution 27
III-2.1. Précipitation du chrome 29
III-2.2. Phénomène d’oxydo-réduction 30
III-2.2.1. Oxydation du chrome (III) 31
IV. L’élimination du chrome trivalent par des matériaux naturels 32
V. CONCLUSION 34
CHAPITRE II : MATERIELS ET METHODES Introduction 35
I. SPECTROSCOPIE D’ABSORPTION ATOMIQUE 36
I-1. Définition 36
1.2- La loi d'absorption en absorption atomique 36
I-3 Appareillage 37
I-3.1. La lampe à cathode creuse 37
I-3.2 CHOPPER 37
I-3.3 Le nébuliseur 38
I-3.4 La flamme – atomisation 38
I-3.5 Monochromateur avec détecteur 38
II. SPECTROPHOTOMETRIE ULTRAVIOLET/VISIBLE 38
III. DIFFRACTION DES RAYONS X 40
III-1 Introduction 40
III-2 Méthode d’analyse 40
IV. ANALYSE THERMOGRAVIMETRIQUE 41
IV-1. Introduction 41
V. GRANULOMETRIE PAR DIFFRACTION LASER 41
V-1. Principe 41
V-2. Appareillage 42
VI. FLUORESCENCE X 42
VII-1. Principe 42
VI-2. Appareillage 42
VII. MESURE DE LA SURFACE SPECIFIQUE (BET) 43
VIII. Spectroscopie RMN 27Al en solution 43
VIII-1. Généralités 43
VIII-2 Conditions de mesure 43
IX. PROTOCOLE EXPERIMENTAL 44
IX-1. Matériels 44
IX-2. Réactifs 44
IX-3. Adsorption 44
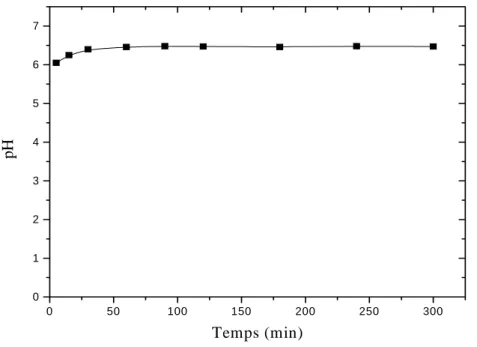
IX-3.1 Cinétique d’adsorption 44
IX-3.2 Influence du pH 44
IX-3.3 Isothermes d’adsorption 45
CHAPITRE III : CARACTERISATION DES ADSORBANTS
Introduction 47
I. Généralités sur la Pyrophyllite 48
I-1. Structure de pyrophyllite 48
I-1.1 Charge de surface de la pyrophyllite : influence du pH sur le potentiel zêta de la pyrophyllite 49
II. Caractérisation de la pyrophyllite 51
II-1 Origine de la pyrophyllite 51
II-2. Analyse chimique quantitative 51
II-3. Analyse par diffraction des rayons X 51
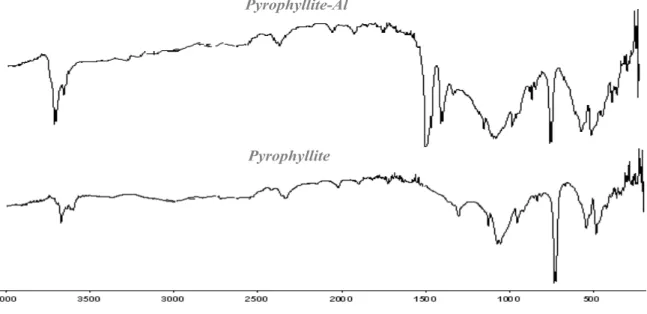
II-4 Analyse par spectroscopie infrarouge 52
II-5 Analyse thermique 53
II-5.1. Analyse thermogravimétrique (ATG) 53
II-5.1. Analyse thermique différentielle (ATD) 54
II-6. Granulométrie 55
II-7. Porosité 56
II-8. Détermination de la capacité d’échanges cationique (CEC 56
II-8.1. Méthode de mesure 56
II-9. Mesure de la surface spécifique 57
II-10. Stabilité de la pyrophyllite en fonction du pH 57
II-11. Stabilité de la pyrophyllite en milieu aqueux 58
III. Le protocole expérimental du traitement de la pyrophyllite 60
III-1. Préparation du polymère d’aluminium 60
III-2 Caractérisation de la solution du polymère 61
III-3. Modification de la pyrophyllite par le polymère d’aluminium 62 IV Caractérisation de la pyrophyllite-Al 63
IV-1 Analyse par diffraction X 63
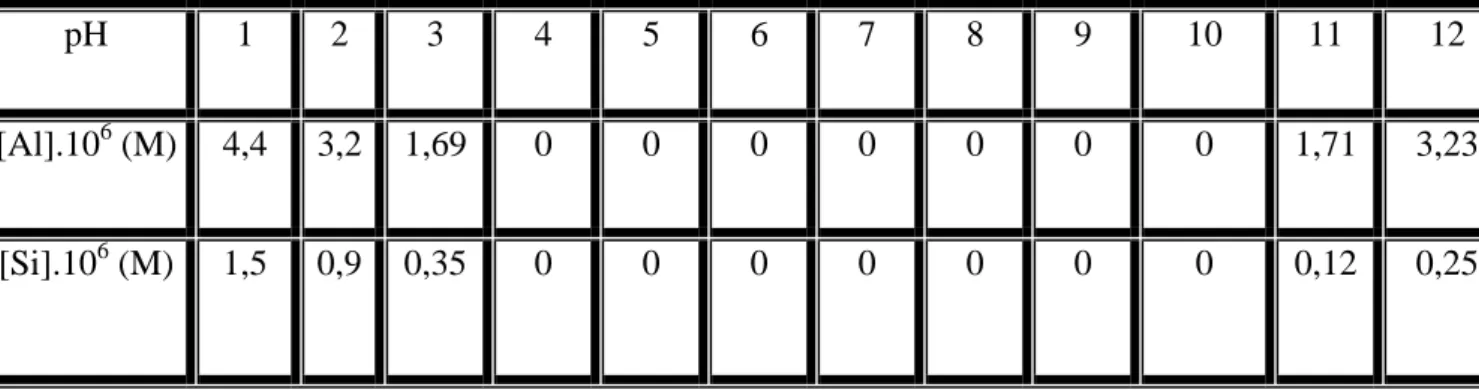
IV-2. Analyse chimique quantitative 65
IV-3. Granulométrie 65
IV-4. Porosité 66
IV-5 Détermination de la capacité d’échanges cationique (CEC 66
IV-6. Surface spécifique 67
IV-7 Analyse par spectroscopie infrarouge de la pyrophyllite-Al 67
IV-8. Comportement de la pyrophyllite en milieu aqueux 68
IV-9. Stabilité de la pyrophyllite en fonction du pH 70
V- Conclusion 71
CHAPITRE IV : ADSORPTION DU Cr(III) SUR LA PYROPHYLLITE ET LA PYROPHYLLITE-AL Introduction 72
I- Etude de l’adsorption du Cr(III) sur la pyrophyllite 73
I-1. Cinétique d’adsorption 73
I-2. Influence du pH 74
II-2 Cinétique d’adsorption 82
II-3. Influence du pH 84
II-4. Influence de la densité de pulpe 86
II-5. Isothermes d’adsorption 87
III- Conclusion 90
CHAPITRE V : ADSORPTION DU BLEU DE METHYLENE SUR LA PYROPHYLLITE ET PYROPHYLLITE-Al Introduction 91
A- ADSORPTION DU BLEU DE METHYLENE SUR LA PYROPHYLLITE ET PYROPHYLLITE-Al A TEMPERATURE AMBIANTE I. L’adsorption du bleu de méthylène sur la pyrophyllite à température ambiante 92
I-1 Introduction 92
I-2 Protocole expérimentale 92
I-3 Influence du temps de la réaction 92
I-4. Influence du pH 94
I-5. L’effet de la concentration initiale Bleu de Méthylène 95
II. L’adsorption du bleu de méthylène sur la pyrophyllite-Al à température ambiante 96
II-1 Introduction 96
II-2 Influence du temps de réaction 96
II-3. Influence du pH 99
II-4. L’effet de la concentration initiale en Bleu de Méthylène 100
II-5. Conclusion 103
B- ADSORPTION DU BLEU DE METHYLENE SUR LA PYROPHYLLITE ET PYROPHYLLITE-Al A DIFFERENTES TEMPERATURES I Adsorption du bleu de méthylène sur la pyrophyllite à différentes températures 104
II. Adsorption du bleu de méthylène sur la pyrophyllite-Al à différentes températures 107
III Calcul des paramètres thermodynamiques liés au processus d’adsorption 109
IV. Conclusion 112
CHAPITRE VI : ELABORATION DE NOUVEAUX SUPPORTS CERAMIQUES A BASE DE PYROPHYLLITE Introduction 113
I. Support céramique 113
I-1. Conditionnement des matières premières 114
I-2. Le solvant 115
I-3. Les additifs organiques 115
I-4. Les procédés de mise en forme 117
I-5. Calandrage 117
II. Traitement thermique 119
III. Le frittage 121
III-1. Le frittage en phase solide 121
III-1.1. construction des ponts 121
IV. Caractérisation des céramiques élaborées 122
IV-1. Evolution de la texture poreuse 122
IV-2 Optimisation de la durée du palier 124
IV-3 Evolution de la résistance mécanique 125
IV-4. Résistance à la corrosion 126
V. Conclusion 127
CONCLUSION GENERALE 129
INTRODUCTION GENERALE
Le secteur de l’eau demeure caractérisé par l’acuité de certains problèmes notamment la dégradation qualitative et quantitative des ressources en eau, auxquels s’ajoutent ceux causés par les conditions climatiques et la prolifération des foyers de pollution. L’eau devient ainsi un vecteur de pollution [1].
Aussi, l’importance de plus en plus grande qu’on attache aujourd’hui à la protection des milieux naturels et à l’amélioration de la qualité des eaux ne cesse de croître et les différentes instances internationales chargées d’inspecter et de surveiller l’environnement sonnent l’alarme à l’occasion de chaque catastrophe et proposent des réglementations de plus en plus strictes.
L’Organisation Mondiale de la Santé et l’Union Européenne quand à elles ne fixent pas de lois en la matière mais donnent des orientations pour la fixation des concentrations maximales admissibles (CMA).
De leur côté, les chercheurs scientifiques de différents horizons (chimie, géologie, agronomie, physiologie végétale, médecine,…) s’intéressent de plus en plus à l’identification et à l’élimination des éléments polluants impliqués directement dans l’apparition de déséquilibres au niveau des écosystèmes ou à l’origine de troubles graves pouvant conduire à la mort, aussi bien chez les animaux que chez l’homme.
Les éléments polluants qui sont introduits de manière importante dans l’environnement sont de nature organique, tels que, les détergents et colorants concentrés en quantité importante dans les eaux résiduaires des industries de textile ou de natures métallique, tels que le cuivre, le zinc, le cobalt et le fer, présents à l’état de traces, sont essentiels pour les organismes vivants. ou enfin, d’autres éléments tels que le mercure, le plomb ou le chrome qui ne peuvent entraîner que des effets néfastes [2].
Le chrome, élément considéré dans ce travail, est, depuis le dix-neuvième siècle, fortement utilisé contre la corrosion dans l’industrie. Les tanneries, à elles seules, constitue à elle seule
effluents liquides et 1,2 à 5,4 g/100g de boues minéralisées) [6], dépassant de loin les normes mondiales et nationales dans les rejets industriels [7]
Pour réduire l’impact de cette pollution plusieurs méthodes ont été utilisées :
La précipitation des métaux lourds est depuis longtemps la technique la plus utilisée [8, 9].
Bien que ce processus soit efficace, il présente des inconvénients : en effet, il produit des quantités importantes des boues dont le temps de tassement est très long.
L’utilisation du charbon dans le processus d’adsorption est également très sollicitée. Le charbon actif présente une forte capacité d’adsorption due essentiellement à sa grande surface spécifique mais ce procédé reste très coûteux.
L’attention a été focalisée par la suite sur l’utilisation de nouveaux adsorbants à base de matériaux naturels abondants. C’est le cas des zéolithes naturelles ou synthétiques, des cendres volcaniques et surtout des argiles [10,11].
De nos jours, une nouvelle famille de solides microporeux à porosité contrôlée semblable aux zéolites et appelé communément argiles modifiés, est très largement étudiée par de nombreux chercheurs. De nombreux travaux sur l’adsorption rapportent des informations sur les différentes méthodes de synthèse et de caractérisation, ainsi, une grande variété d’argiles modifiées par des espèces polymériques cationiques a été mise en oeuvre et utilisée dans plusieurs réactions chimiques.
Par ailleurs, la technique de filtration des effluents par les membranes est utilisée dans les opérations de séparation et de concentration. Elles sont moins coûteuses par rapport aux procédés traditionnels [12, 13], et contribuent à l’amélioration d’autres techniques de séparation par couplage de procédés. Des espèces de ces membranes, à base de matériaux, sont caractérisées par une excellente tenue mécanique, thermique, chimique, mais aussi par une facilité d’utilisation et une grande durée de vie. Toutefois, une membrane céramique est indispensable pour soutenir le support macroporeux qui assure la résistance mécanique. Les supports commercialisés sont relativement chers nous encourageant à l’élaboration des supports à base d’argile marocaine [14, 15].
Le travail présenté dans ce manuscrit, à pour objectifs de valoriser une argile marocaine très abondante : la pyrophyllite, de comprendre les mécanismes de son interaction à l’état naturel ou modifié et d’élaborer de nouveaux supports céramiques à base de la pyrophyllite.
• Le premier chapitre présente une synthèse des différents travaux publiés en matière d’adsorption et de catalyse.
• Le deuxième chapitre présentera les matériels et méthodes, en plus, des caractéristiques des appareillages et produits utilisés, des méthodes de caractérisation des matrices solides, de quantification du soluté et des protocoles expérimentaux utilisés.
• Le troisième chapitre sera consacré à la présentation et au commentaire des différents résultats obtenus.
• Le quatrième chapitre est divisé en deux parties:
Dans la première partie, nous présentons les résultats de l’influence de certains paramètres sur l’adsorption du Cr(III) sur la pyrophyllite naturelle. Dans la deuxième partie, sera réservée à l’impact de ces mêmes paramètres sur la pyrophyllite modifiée.
• Le cinquième chapitre présentera les résultats de l’effet du temps et de la température, sur l’adsorption du pH et du bleu de méthylène sur de ce dernier la pyrophyllite naturelle et traitée.
• Enfin, le dernier chapitre présentera l’élaboration et les caractéristiques de nouveaux supports céramiques à base de pyrophyllite .
CHAPITRE 1
I- GENERALITES SUR LES ARGILES I-1 Introduction
Les matériaux argileux (ou plus simplement "argiles") sont ubiquistes à la surface de la terre. Du fait de leur mode de formation, ce sont en général des matériaux polyphasiques, composés à la fois de phases minérales et organiques [16]. Les phases minérales pures, dites "minéraux argileux", représentent alors des proportions variables du matériau global. Cependant, des conditions hydrothermales ont parfois favorisé la formation de matériaux argileux formés de phases d'une plus grande pureté.
Grâce à leurs propriétés, les argiles sont utilisables pour différentes applications. Outre la fabrication de matériaux de construction, elles sont utilisées, à titre d’exemple, pour l’élaboration de matériaux polymères ou encore le raffinage d'huile alimentaire, la cosmétique ou la médecine. Grâce à leurs propriétés micro et macroscopiques, les argiles, jouent aussi un rôle important dans le stockage des déchets. A cet égard, les argiles ont des propriétés intéressantes pour constituer une barrière imperméable autour de déchets. Au contact des eaux souterraines, la barrière argileuse va se saturer progressivement. Ses propriétés, tant mécaniques qu’hydrauliques et physico-chimiques vont évoluer au cours de cette phase de saturation. En particulier, le spectre de porosité de l’argile va être profondément modifié [17].
Donc, par leur faible perméabilité, leur capacité d'échange de cations permettant le rôle de "piège" face aux pollutions métalliques, les argiles soient sous forme modifiées soient à l’état brut sont d’excellents matériaux utilisés pour centres de stockage de déchets [18, 19].
L’intérêt accordé ces dernières années à l’étude des argiles par de nombreux laboratoires dans le monde se justifie par leur abondance dans la nature, l’importance des surfaces qu’elles développent, la présence des charges électriques sur cette surface et surtout l’échangeabilité des cations interfoliaires. Ces derniers, appelés aussi compensateurs, sont les principaux éléments responsables de l’hydratation, du gonflement, de la plasticité et de la thixotropie et ils confèrent à ces argiles des propriétés hydrophiles.
Dans ce chapitre bibliographique nous parlerons, brièvement, de propriétés des argiles vues sous ce contexte.
I-2. Minéralogie, structure et propriétés chimiques des argiles I-2.1. Minéralogie et classification des argiles
-La couche tétraédrique; -La couche octaédrique.
Les différents groupes de minéraux argileux se différencient par l'arrangement de ces deux couches.
Les travaux de l’AIPEA (Association Internationale Pour l’Etude des Argiles1966-1972) et plus tard, ceux de Pédro (1994) [20], ont permis d’aboutir à une classification qui repose sur l’utilisation des critères suivants:
-Type de feuillets 2:1 ou 1:1; -Charge globale du feuillet; -Nature des cations inferfoliaires. -L'épaisseur et la structure du feuillet. On distingue ainsi 4 groupes [21].
Minéraux à 7 Å: (kaolinite, Halloysite, Dombasite, ….)
Le feuillet est constitué d'une couche tétraédrique et d’une couche octaédrique. Il est qualifié de T:O ou de type 1:1. Son épaisseur est d’environ 7 Å.
Minéraux à 10 Å: (Pyrophyllite, illite, Montmorillionite, Saponite,…)
Le feuillet est constitué de deux couches tétraédriques et d’une couche octaédrique. Il est qualifié de T:O:T ou de type 2:1. Son épaisseur est d’environ 10 Å.
Minéraux à 14 Å: (Chlorites)
Le feuillet est constitué de l'alternance de feuillets T:O:T et de couches octaédriques interfoliaires.
Minéraux interstratifiés: L’épaisseur du feuillet est variable. Ces minéraux résultent du mélange régulier ou irrégulier d’argiles appartenant aux groupes ci-dessus.
Une représentation schématique de la structure des couches tétraédriques et octaédriques, ainsi que de leur empilement, est montrée sur la figure I-1 et la figure I-2, respectivement.
Figure I-1. Représentation des tétraèdres de silicium et des octaèdres d'aluminium ou de magnésium, ainsi que de leur agencement en couches.
Figure I-2. Représentation schématique de l'empilement des feuillets unitaires dans une smectite.
La structure de la couche octaédrique des smectites, ainsi que la localisation des substitutions, ont conduit à une classification de ces minéraux [22]. Ainsi, il existe deux grandes catégories de smectites. Les premières sont dites dioctaédriques du fait de l’occupation de seulement deux sites octaédriques sur trois. Parmi elles, certaines présentent majoritairement des substitutions dans la couche octaédrique (montmorillonite), alors que d’autres sont principalement substituées dans les couches
est remplacé par du magnésium ou du fer, alors que le silicium tétraédrique est remplacé par de l’aluminium.
Il existe un troisième type de smectite dioctaédrique, possédant essentiellement du fer au degré d’oxydation III, dans sa couche octaédrique, remplacé par de l’aluminium ou du magnésium (nontronite). Les autres smectites sont trioctaédriques, car tous les sites octaédriques sont occupés. L’ion en site octaédrique est en général le magnésium. Parmi elles, certaines sont caractérisées par des substitutions du magnésium par le lithium dans la couche octaédrique (hectorite), alors que pour d’autres, les substitutions ont principalement lieu dans la couche tétraédrique, où le silicium est remplacé par de l’aluminium (saponite).
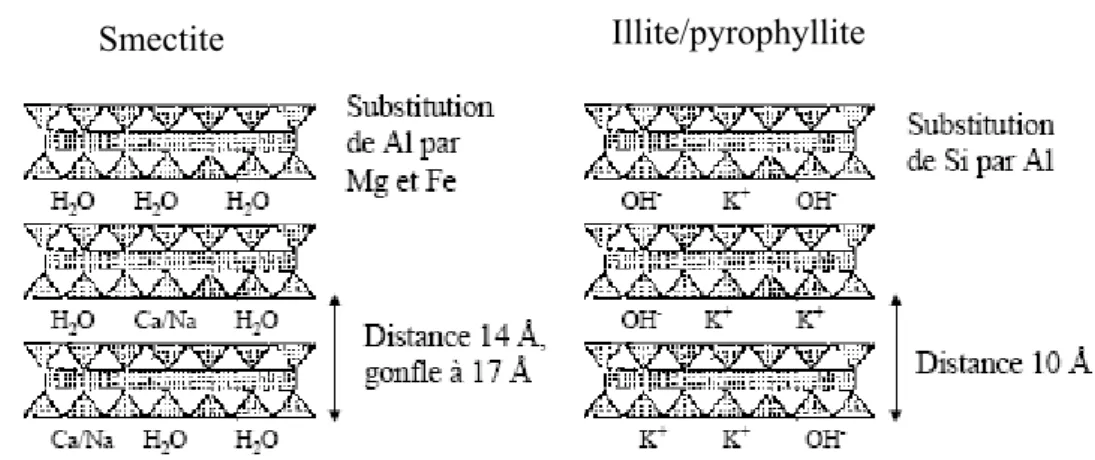
Dans les illites et la pyrophyllite, la charge provient principalement de substitutions dans les couches silicatées, donc plus proches de la surface, ce qui confère alors à ces argiles des propriétés d’adsorption d’ions relativement différentes de celles des smectites, notamment au niveau de la spécificité des sites, une représentation comparative des smectites et de l’illite/pyrophyllite est donnée sur la figure I-3, montrant l’agencement des feuillets ainsi que la localisation des substitutions.
Illite/pyrophyllite Smectite
Figure I-3. Représentation schématique de la structure feuilletée des smectites et des illite/pyrophyllite. [22]
I-2.2. Espaces interfoliaires et capacité d’échange cationique (CEC) I-2.2.1 Les cations compensateurs
Il existe un type d’interaction des ions avec la surface des smectites, intervenant dans la capacité d’échange cationique, sous une forme indépendante du pH. Il s’agit des ions compensant la charge structurale permanente de l’argile. Cette adsorption indépendante du pH
est généralement attribuée à l’échange d’ions dans les interfeuillets, et résulte d’interactions électrostatiques entre les ions et la charge structurale permanente de l’argile. La capacité d’échange associée peut être calculée directement si la composition des feuillets est parfaitement connue.
Les espaces qui se trouvent entre les feuillets peuvent être vides ou remplis :
- Ils sont vides lorsque les différents feuillets sont neutres et liés entre eux par des liaisons hydrogène dans le cas des espèces 1:1, ou par des liaisons de Van der Wals dans le cas des minéraux 2:1 [20].
- Ils sont occupés par des cations dès que les feuillets de l’édifice présentent un déficit de charge à la suite de substitutions isomorphiques. Ces cations rétablissent l’électro-neutralité du système et en même temps assurent la liaison entre les feuillets adjacents, qui est ici de nature ionique [20]. Ces cations peuvent être soit «secs» soit hydratés. Les cations les plus fréquents sont Ca2+, Mg2+, K+, Na+, Li+.
Des substitutions suffisamment nombreuses de Al3+ par Fe2+ ou Mg2+ se produisent dans le feuillet octaédrique, l'excès de charge négative résultant sur une cavité hexagonale proche permet de former des complexes relativement stables avec des cations ou des molécules dipolaires. Si maintenant des substitutions isomorphiques de Si4+ par Al3+ ont lieu au niveau de la couche tétraédrique, l'excès de charge négative est localisé beaucoup plus près des oxygènes de la surface, permettant la formation de complexes cette fois très forts avec des cations ou des molécules d'eau.
a- Phyllosilicates non-expansibles
Les feuillets d’illite et de la pyrophyllite, où la charge est compensée par du potassium, constituent un exemple de ce type d’arrangement. Nous avons affaire à des minéraux à espace interfoliaire anhydre et présentant des espacements constants, voisins de l’épaisseur du feuillet (~10). Les cations interfoliaires ne sont pas en général échangeables par des cations organiques et minéraux, se trouvant dans des solutions mises au contact du phyllosilicate. La présence de potassium dans l’espace interfeuillet, liant très étroitement les feuillets entre eux et empêchant ainsi le minéral de se gonfler en présence d’eau. Le potassium constitue par conséquent un cation difficilement échangeable (figure I-4) [16].
K+
Figure I-4. : Complexe de sphère interne. Il n'y a pas de molécules d'eau entre le cation et la surface. C'est le cas des cations de faible énergie d'hydratation comme potassium
b- Phyllosilicates expansibles
Dans ce cas les cations compensateurs sont hydratés et la présence d’un film d’eau entre les feuillets concourt à leur écartement. On parle alors de minéraux expansibles. La propriété essentielle de ces minéraux est de se disperser au contact de l’eau pour former des suspensions plus ou moins stables. Les cations interfoliaires sont en général échangeables par des cations organiques et minéraux, se trouvant dans des solutions mises au contact du phyllosilicate (figure I-5) [23].
On caractérise alors chaque phyllosilicate par sa «Capacité d’Echange Cationique» (CEC) définie comme étant le nombre de cations monovalents (équivalents chimiques) qu’il est possible de substituer aux cations compensateurs pour compenser la charge électrique de 100 g de minéral calciné.
Figure I-5 Complexe de sphère externe. Le cation reste totalement hydraté. C'est le cas des cations possédant une énergie d'hydratation élevée, comme le sodium.
I-2.2.2 Mesures de la capacité d'échange cationique
Plusieurs méthodes pour déterminer la CEC ont été engendrées. Au début, la détermination du CEC des argiles a été effectué en saturant l'argile par un cation puis en éliminons l'excès du sel utilisé pour le traitement. Ce cation est échangé par plusieurs cycles d'échange/lavage par un autre cation [24]. Les solutions recueillies sont employées pour la détermination de la quantité du cation remplacé. Une autre méthode a été engendrée en saturant l'argile avec les
ions NH4+, la quantité des ions d'ammonium adsorbée est déterminée par la méthode de la distillation de Kjeldahl [25].
D'autres méthodes ont été proposées par l'utilisation de surfactants cationiques [26, 27]. Le but était de déterminer si la quantité adsorbée pouvait être corrélée à la valeur du CEC ou de la surface spécifique. La problématique générale de l'utilisation des surfactants est que l'excès sera adsorbé sur l'argile, se qui nécessite alors la détermination du point d'équilibre où la quantité équivalente est adsorbée. Finalement, les complexes organo-métalliques sont employés comme cations échangeables. L'affinité des minéraux argileux vers ce type de cation est grande, afin que l'échange complet puisse être accompli dans une unique étape de traitement [28]. Un excès du complexe est additionné à la dispersion d'argile et il reste seulement de déterminer la concentration restante après la réaction d'échange. Les méthodes utilisant les ions cétylpyridinium [26], cobalthéxamine [29], silverthiourea [30], cuivré bis-éthylène diamine [31] ou cuivré triéthylène tétramine [32] peuvent être utilisées pour ce but.
I-3. La surface spécifique des adsorbants
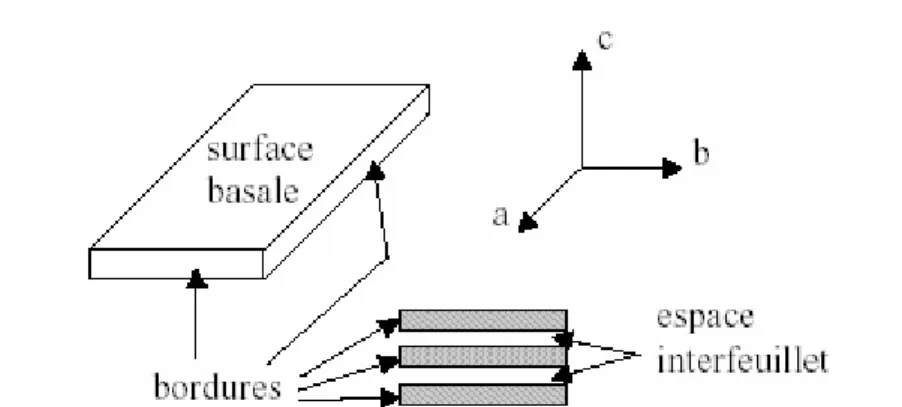
Par définition, la surface spécifique d’un adsorbant est une surface par unité de masse. Elle est généralement exprimée en m2.g-1. Son estimation est conventionnellement fondée sur des mesures de la quantité adsorbée Qads de l’adsorbant en question, correspondant à un adsorbat donné ; la molécule adsorbée doit avoir une surface connue et acceptable. Il suffit à cet effet, de déterminer la valeur de la capacité de la monocouche à partir de l’isotherme d’adsorption. Une discussion compréhensive de la surface spécifique des argiles nécessite la reconnaissance qu'il y a différentes "surfaces spécifiques" pour chaque échantillon d'argile. Différentes méthodes donnent différents résultats. Avant de discuter les méthodes, on doit soigneusement considérer la signification de "la surface spécifique" dans ce type de minéraux. Il est nécessaire de distinguer la surface interne et la surface externe d’un adsorbant. La première est la surface microporeuse Sµ représentée par les parois des micropores ; elle peut atteindre plusieurs mètres carrés par gramme. La deuxième est la surface non-microporeuse ou la surface externe Sext qui comprend les parois des mesopores et des macropores, ainsi que la surface des feuillets aromatiques.
Figure I-6 : Illustration de l'empilement des feuillets d'argile, montrant les surfaces basales, les bordures des particules, ainsi que les espaces interfeuillets.
Dans l'état sec, les minéraux d'argiles s'organisent sous forme de feuillets de silicates dans une orientation face-face. On peut distinguer entre la surface externe, interne et surface totale (figure I-6). Si l'adsorbat ne peut pas entrer dans les espaces interfeuillets, la quantité adsorbée correspond à la surface externe. Toutefois, l'utilité de la surface externe est restreinte, parce que les pores et les trous peuvent aussi contribuer à cette valeur (comme par exemple dans les mesures d'adsorption de gaz). Dans certains cas, les feuillets se collapsent dans le centre alors qu'à proximité des bords les feuillets sont partiellement étendues. Tous ces facteurs compliquent la définition de la surface externe. Certaines molécules polaires peuvent entrer dans les espaces interfeuillets et créent des espacements. Pour déterminer les surfaces spécifiques, il est important de savoir si c'est une monocouche ou une bicouche qui est présente dans l'espace interfoliaire. Si l'espace interfoliaire est étendu par une bicouche d'adsorbat, la surface totale peut être déterminée.
Un point à considérer est la dimension de l'adsorbat: en diminuant la dimension de l'adsorbat la "surface spécifique" augmente [33], à cause de l'augmentation du nombre de sites accessibles, comme le cas des trous ou des pores. Une surface plate expose la même surface pour les petites et grandes molécules. Une surface avec des pores, des bords, des trous et des angles, expose une plus grande surface aux molécules plus petites.
I-4. Rétention des métaux par les argiles
Des mécanismes distincts peuvent conduire à la rétention d'éléments par une surface solide. Ces mécanismes sont:
a- la précipitation :
Engendrant l’apparition d’une nouvelle phase solide sur la surface du minéral. Elle résulte d’une rupture de sursaturation, après nucléation hétérogène;
b- l’absorption :
Correspondant à la migration d’espèces dissoutes vers l’intérieur du solide; c- l’échange d’ion :
Correspondant au remplacement d’un ion initialement présent en position échangeable (ex : interfoliaire), par un ion présent à l’état dissous.
d- l’adsorption :
• Définition de l’adsorption
La sorption est définie comme étant la rétention de substances à la surface des solides : mécanisme d’échange d’ions, complexation de surface et précipitation de surface.
L’adsorption est définie comme étant la fixation des molécules de solutés (contenue dans une phase liquide ou gazeuse) à la surface d’un solide par l’intermédiaire de liaisons de type de Van Der Waals ou chimique. Le processus d’adsorption se produit jusqu’à l’obtention d’un état d’équilibre auquel correspond une concentration bien déterminée du soluté.
• Types d’adsorption
La nature des liaisons formées ainsi que la quantité d’énergie dégagée lors de la rétention d’une molécule à la surface d’un solide permettent de distinguer deux types d’adsorption : adsorption physique et adsorption chimique [34, 35].
• Adsorption physique
Appelée également physisorption, elle est caractérisée par une faible énergie de liaison inférieure à 10 Kcal/mol et elle correspond aux liaisons de nature électrostatique de type Van Der Waals [34, 35]. Dans le cas d’une telle adsorption, le temps de rétention de la substance adsorbée est court et la surface adsorbante peut être recouverte de multiples couches moléculaires de produit adsorbé [34].
• Adsorption chimique
Appelée également chimisorption, elle met en jeu une énergie élevée (supérieure à 10 Kcal/mol) et correspond aux liaisons covalentes plus permanentes entre l’adsorbant et la
première couche liée à la surface adsorbante est chimiquement adsorbée, les autres couches, dans le cas où elles existent, sont retenues par physisorption [34].
• Nature du mode d’adsorption
L’adsorption d’une substance est gouvernée par de multiples types d’interaction. Selon la nature des constituants de l’adsorbant et des molécules adsorbées, différents types de liaisons peuvent exister simultanément. Les liaisons les plus importantes sont [35, 37] :
- Liaison de London-Van Der Waals ; - Liaison ionique ;
- Liaison hydrogène ; - Liaison covalente ;
- Liaison par transfert de charge ;
• Principaux facteurs influençant l’adsorption
Un grand nombre de paramètres et de propriétés peuvent affecter l’adsorption d’une substance sur un support, desquels nous citons [36] :
- La polarité et la polarisabilité des molécules adsorbées ; - La taille de ces molécules ;
- La nature de leurs groupements fonctionnels ; - Leur pKa ;
- Leur solubilité ;
- La composition du milieu adsorbant (teneur en argile, en matière organique, en eau, température, …) ;
- Le pH du milieu [34].
I-4.1. Affinité des métaux lourds Adsorption de sphère externe
Les cations peuvent se fixer en «sphère externe», à la surface des argiles. La tendance de la surface à former des complexes de sphère externe avec un cation fait intervenir deux facteurs : la valence du cation et son rayon hydraté. Plus la valence du cation est élevée et plus l'affinité est forte. A valence égale, un cation à faible rayon hydraté présentera plus d'affinité qu'un cation à fort rayon hydraté. A valence égale se sont donc les cations volumineux qui seront fixés préférentiellement en sphère externe.
Il existe une corrélation entre la tendance d’un cation à former des paires d'ions en solution et sa tendance à former des complexes de sphère externe.
Adsorption de sphère interne
Il existe une corrélation entre la tendance du cation métallique M2+à former des complexes de sphère interne et sa tendance à former des complexes en solution, en particulier du type MOH. Aussi la constante d'équilibre KHde la réaction en solution (réaction 1) détermine-t-elle le comportement à l'adsorption de chaque métal [38].
Reaction(1) : M2+ + H2O MOH+ + H+ La proportion d'adsorption en sphère interne sera d'autant plus grande que pKHest faible. Ainsi, selon Brummer cité par Marcos [39], le plomb (pKH= 7.7) se fixe en sphère interne dans des proportions plus grandes que le zinc (pKH= 9). La complexation en sphère interne suppose en effet la rupture de la sphère d'hydratation du métal et la formation d'une liaison chimique avec un groupement réactionnel de surface. La présomption de fixation du plomb en sphère interne est, par ailleurs, tout à fait confirmée par des considérations spectroscopiques
[40].
I-4.2. Théorie d’adsorption en phase liquide
La modélisation de l'adsorption, d'un soluté en phase liquide sur un matériau solide, emprunte certaines relations utilisées pour l'étude de l'adsorption de gaz sur un oxyde métallique dans des conditions réversibles [41]. La représentation la plus utilisée est l'isotherme d'adsorption qui exprime, à température constante, la relation entre les concentrations de soluté et d'adsorbat.
L'utilisation d'isotherme d'équilibre permettra d'atteindre les valeurs thermodynamiques induites par le phénomène mais sans aucune spéculation quant au chemin, souvent très complexe, suivi par la réaction d'adsorption ou de désorption.
Plusieurs isothermes établies de façon théorique ou empirique sont utilisées au cours de ce travail :
- L'équation de Langmuir basée sur la fixation d'une couche mono-moléculaire d'adsorbat. - La relation de Freundlich, quant à elle, permet souvent une représentation pratique de l'équilibre d'adsorption entre un micropolluant et la surface d'un support solide.
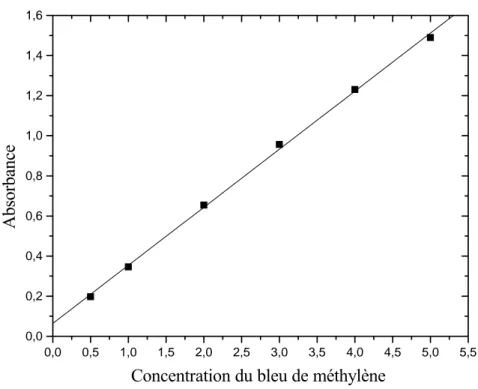
I-5. Adsorption du bleu de méthylène
Les méthodes pour déterminer la surface spécifique sont généralement basées sur l'adsorption des liquides polaires comme l'eau [42, 43] ethylènglycol ou l'éthylène glycol monoethylether. Les colorants sont aussi employés pour la détermination des surfaces, par exemple le bleu de méthylène ou le p-nitrophenol [44]. Helmy et al [45], ont comparé l'adsorption du phenanthroline, bipyridine, quinoline, oxine, le bleu de méthylène, le glycérol, le glycol d'éthylène et le glycol d'éthylène monoethylether pour déterminer les surfaces du kaolin, α-Fe2O3 et l'hydroxy-Al montmorillonite.
L'adsorption du bleu de méthylène (MB) est une méthode commune pour estimer la surface spécifique de certaines smectites. Un grand nombre de publications se rapportent à l'application de cette méthode [45-48]. Accomplir un titrage d'une argile dispersée dans l'eau, Hang et Brindley [45] ont supposé qu'une couverture mono-moléculaire de la surface est atteinte au point spécifique lorsque des traces de bleu de méthylène sont décelées dans la solution. Ils appellent ce point "le point de floculation optimum". A faible concentration en BM, tout le colorant est adsorbé par l'argile. Si "le point de floculation optimum" est en excès, le bleu de méthylène libre peut être déterminé comme un cerne autour de l'argile floculée. En traçant la quantité de BM adsorbée par l'argile en fonction de la quantité introduite en solution, ils ont trouvé une ligne droite pour les petites concentrations. Ils ont considéré "le point de floculation optimum" comme étant le point où la courbe dévie de la linéarité. Si des particules d'argiles non floculées sont présentes dans la solution, la détermination de ce point devient difficile. On doit aussi noter qu'à très basses concentrations en BM un cerne apparaît aussi, indiquant qu'un minimum de concentration en BM est requis avant que l'adsorption n'ait lieu. Toutefois, "le point de floculation optimum" mène à confusion, car ce n'est pas la floculation qui est examinée mais l'adsorption. Donc, ce point sera appelé "le point de cerne".
II. HYDROLYSE DES METAUX
Selon Baes et Mesmer [48], les premières recherches sur l'étude de l'hydrolyse des cations ont débuté dès le début du XXème siècle. En effet, les premiers travaux sur l'hydrolyse des cations et en particulier sur l'atome de chrome ont permis de conclure que les espèces polynucléaires se formaient à partir des monomères simples. A cette même période, déjà, un concept nommé "aquo-acidité" fut proposé dans lequel l'hydrolyse de cations est envisagée
comme étant un déplacement progressif des protons à partir des molécules d'eau [M(H2O)6]z [M(H2O)5(OH)]z-1 [M(H2O)4(OH)2]z-2 ,…
A partir de 1950, Sillen [49] et ses collaborateurs à Stockholm, se sont intéressés à nouveau à l'hydrolyse des cations. Déjà en 1959 [50], ils orientèrent toutes leurs recherches sur les différentes espèces issues des éléments de la troisième période du tableau périodique.
A travers les résultats obtenus, ils ont proposé deux types d'espèces:
- En milieu acide: [Mg(H2O)6]2+, [Al(H2O)6]3+, Si(OH)4, PO(OH)3, SO3(OH)-, ClO4-
- En milieu basique: [Mg(H2O)5(OH)]+, [Al(H2O)2(OH)4]-, [SiO2(OH)2]2-, [PO4]3-, [SO4]2-, [ClO4]-, etc.
Ainsi, selon eux, la variation des espèces est due principalement à la densité de charge du cation central qui, dans le même temps, préserve son nombre de coordination.
Le phénomène d’hydrolyse-polymérisation-précipitation des espèces métalliques se caractérise en général par le rapport molaire d’hydroxylation noté "r" qui s’écrit de la manière suivante :
r = OH/Me (ou H/Ti dans le cas du titane).
De très nombreux auteurs se sont intéressés à la chimie en solution de l’aluminium trivalent et plusieurs modèles ont été proposés pour la structure et la composition des espèces polymères. Selon Baes et Mesmer [48], la chimie de l’aluminium dans l’eau peut être décrite fidèlement en utilisant :
- cinq monomères : Al3+, Al(OH)2+, Al(OH)2+, Al(OH)3, Al(OH)4 -- trois polymères: [Al2(OH)2]4+, [Al3(OH)4]5+, [Al13O4(OH)13]18+ - un précipité Al(OH)3(s)
Les travaux les plus approfondis ont essayé de relier la structure des polymères à celles des hydroxydes d’aluminium cristallisés qui sont principalement la gibbsite et la bayérite [51, 52].
Certaines espèces comme [Al2(OH)2(H2O)8]4+ et la structure de Keggin
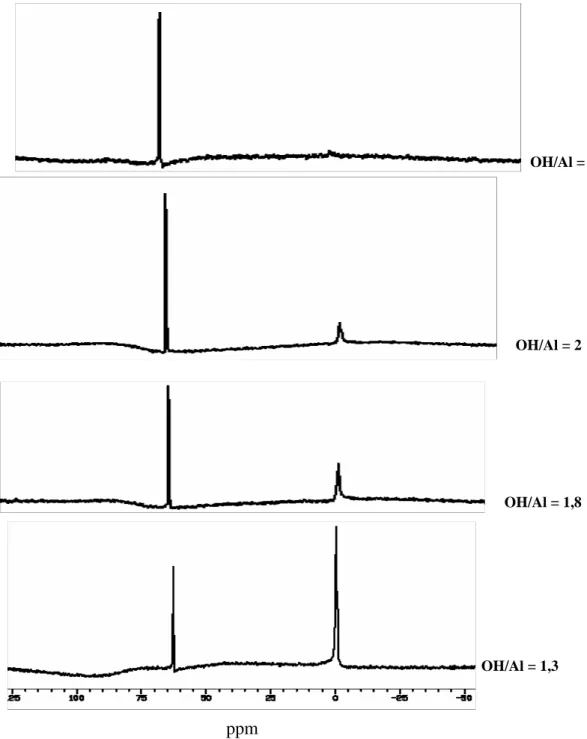
[Al13O4(OH)24(H2O)12]7+ ont été mises en évidence par diffusion des rayons X aux petits angles ou par RMN de 27Al [53, 54].
D’autres espèces telles que [Al8(OH)20]4+ [55] ou [Al6(OH)15]3+ [56] et [Al7(OH)17]4+ et [Al13(OH)32]7+ [49] ne sont que des modèles proposés pour expliciter certains résultats expérimentaux. De nombreuses études sont actuellement en cours pour mettre en évidence la
III- ARGILES MODIFIEES III-1. Généralités
Les premiers travaux sur la synthèse et les propriétés texturales des minéraux argileux modifiés inorganiques et similaires à celles des zéolites ont été réalisés par quelques laboratoires pionniers [57-59].
A cause de leur grande performance et surtout leurs stabilités thermiques et dans une dynamique de recherches scientifiques pluridisciplinaires, de nombreux laboratoires de recherches scientifiques de différents horizons et de différentes spécialités se sont intéressés aux argiles modifiées. De nouvelles équipes et laboratoires auront de nouvelles perspectives sur les différentes méthodes de préparation des argiles modifiées et surtout leur utilisation. La modification des argiles réside dans l’intercalation entre leurs feuillets de gros polycations métalliques simples ou mixtes dans le but d'obtenir des matériaux microporeux, à structure rigide, avec un grand espacement interfoliaire.
De nombreux travaux sur les argiles modifiées rapportent des informations sur les différentes méthodes de synthèse et de caractérisation texturales.
Dans le domaine de l’adsorption et malgré leurs instabilités thermiques, les complexes organoargileux, hydrophobes et organophiles, ont été largement utilisés dans la dépollution des eaux contaminées par certains micropolluants organiques tels que des phénols, des pesticides, des colorants, …
Quelques autres laboratoires ont développé une troisième catégorie de matrices adsorbantes désignées sous l’appellation "complexes organo-inorgano-argileux" ou argiles modifiées mixtes destinées essentiellement au traitement des eaux par adsorption.
Nous nous proposons de présenter en revue et pour chaque famille d’argiles modifiées, une synthèse bibliographique des différents travaux effectués dans ces domaines et publiés par ordre chronologique.
On peut donc classer les argiles modifiées en trois grandes catégories : les complexes organoargileux, les inorgano-argileux et les organo-inorgano-argileux.
III-1.1. Famille des Complexes Organo-Argileux
Les argiles modifiées par des composés organiques décrites initialement par Barrer [60] qui introduisit dans l’espace interfoliaire des ions alkylammoniums et qui ont ensuite été développées par d’autres auteurs à partir du 1,4-diazobicyclo(2,2,2) octane [61]. Leur
utilisation comme catalyseurs a été restreinte à des réactions effectuées à des températures inférieures à 300°C, au-delà de laquelle, les intercalaires organiques sont décomposées, conduisant à l’effondrement de la structure microporeuse.
III-1.2. Famille des Complexes Inorgano-Organo-Argileux
Les argiles modifiées par des composés organométalliques, particulièrement intéressant en catalyse, ont été développées par Pinnavaia et al. selon une méthode qui consiste à immobiliser dans l’argile des espèces cationiques de type catalyseurs de Wilkinson telles que par exemple : Ru3(CO)12 ; Ir(CO)12 ; Cl2PdIINC5H4CH2+[62]. Ils conduisent à des activités et des sélectivités particulières dans les réactions d’hydrogénation et d’isomérisation.
III-1.3. Famille des complexes inorgano-argileux
Pour s’affranchir de l’inconvénient que représente la faible stabilité thermique des organo-argileux, l’idée fut de synthétiser des structures pseudo chlorites qui sont des argiles modifiées par des composés inorganiques. Ceci est le plus aisément réalisé à partir d’hydroxydes de cations facilement hydroxylables tels que l’aluminium [63, 64] et étendu ensuite au Zr [65], Ti [ [66], Fe [67], et Si [ [68], Ti, Ga, Nb, V et n’importe quel oxyde métallique en solution, qui forme une espèce polynucléaire par hydrolyse [59]. Ce type d’argile modifiée a été introduit par Brindley et Sempels [69] en 1977 en utilisant une solution d’hydroxyde d’aluminium. Lahav et al. [70] et Shabatai [71-a] se sont intéressés à la modification des montmorillonites par l’insertion, entre les feuillets, des polycations (Keggin ions ou Al13) [Al13O4(OH)24(H2O)12]7+. Par la suite ce type de traitement ainsi que par les cations [Zr4(OH)8(H2O)16]8+ ont été les plus étudiés.
Actuellement, il est connu que la nature du sel précurseur est primordiale dans la modification des argiles, et le traitement est obtenu généralement par hydrolyse d’un sel métallique par une base forte (ou un acide fort) selon la nature du métal choisi. Après calcination, à différentes températures, les polycations insérés se transforment en grappes d’oxydes métalliques rigides et résistants, confèrent à ces solides une stabilité thermique élevée, et une surface microporeuse développée [71-b].
De leur coté, Lahav et al. [70] ont pu, grâce à l’analyse par DRX, confirmer l’intercalation des polycations d’aluminium avec des espacements basaux de l’ordre de 18 Å à température
III-2. Argiles modifiées par différents polymères inorganiques
La préparation de ces argiles modifiées est basée surtout sur les propriétés de gonflement de ces matériaux. Ces argiles modifiées sont caractérisées par les espaces interfeuillets et interpiliers [72]. Selon leur habilité à échanger les ions, les argiles telles que les smectites se comportent comme de bons accepteurs et elles ont une bonne nature gonflante ce qui leurs confère l’aptitude de changer leurs ions compensateurs [73]. Le nombre d’ions échangeables par le minéral argileux détermine la quantité d’ion (polymère) pouvant être intercalé entre les feuillets de l’argile.
Les pores produits par le pontage dépendent de plusieurs facteurs. On peut citer la nature, la granulométrie et les propriétés physicochimiques de l’argile de départ (Surface spécifique, CEC, espace interfoliaire, cation échangeable, …). D’un autre coté, ils dépendent du type et des conditions du traitement. Les structures de ces argiles modifiées forment des galeries avec des dimensions dépendantes de la nature du polymère introduit et de la distance entre eux. La possibilité de combiner deux types de métaux de certains polycations a aussi permis de synthétiser de nombreux types d’argiles intercalées par des espèces polymériques mixtes: par intercalation d’un doublet d’oxydes métalliques Al-Fe [74], Al-Cu [75], Al-Si [68], Zr-Cu [75], Ti-Cu [75] et Al-Mg [76] ou d’un triplet oxydes métalliques Al-Ce-X [77] avec : X=Co, Ni, Zn, Mg.
Ce type d’intercalation conduit à des argiles modifiées avec des propriétés spécifiques selon l’objectif d’utilisation du matériau traité, tel qu’une augmentation de l’activité catalytique [78, 79] ou une augmentation des propriétés d’adsorption, se présentant par différentes dimensions des pores [80]. Ces argiles modifiées sont parfois utilisées comme adsorbants sélectifs pour la séparation des gaz [81]. Elles permettent une approche rationnelle pour désigner une nouvelle famille de matériaux microporeux car avec le contrôle des conditions dans le processus de traitement, on peut systématiquement contrôler les dimensions et la distribution des pores formés entre les feuillets.
III-3. Modification des argiles par Al13
Dans cette partie, on se réfère essentiellement aux résultas obtenus à partir de la modification des argiles par le polymère aluminium.
III-3.1. Mode d’obtention des argiles modifiées
- La méthode fréquemment utilisée est de rajouter des polymères préalablement formés (soit fraîchement hydrolysés, soit utilisation du chloridrole commercial) à une suspension d’argile (2 à 5 %). L’argile modifiée est alors obtenue par centrifugation et/ou dialyse suivi d’un séchage à l’air ou d’une lyophilisation.
- La méthode opposée consiste à rajouter l’argile à la solution de polymères. D’après cet auteur, des concentrations supérieures à 40% de solide peuvent être utilisées sans provoquer ni gélification ni délamination de l’argile.
Deux autres méthodes que l’on peut appeler in-situ, car elles ne nécessitent pas la fabrication initiale du polymère avant contact avec l’argile sont :
- Hydrolyser le cation compensateur de l’argile (cation initial de l’argile de départ, préalablement échangé par le cation que l’on veut hydrolyser). Cette méthode ne donne pas de bons résultats avec l’aluminium [82].
- Rajouter simultanément le sel et la base directement à la suspension d’argile, à des vitesses contrôlées. La question demeure cependant ouverte de savoir si le polymère se fait directement dans l’espace interlamellaire, ou si il s’intercale après formation à la surface externe des agrégats.
Enfin une dernière méthode consiste à sécher à l’air [83], sur une surface plate, une suspension d’argile de tailles de particules contrôlées, en présence du polymère cationique ce qui permet de synthétiser un film d’argile à pilier.
III-3.2. Facteurs influençant la modification des argiles
Les facteurs influençant le pontage des argiles sont nombreux, nous les citons comme suit : - La nature du cation échangeable initial de l’argile joue un rôle certain, puisque la première étape du traitement est une intercalation par échange cationique. Souvent ce cation est le sodium, ce qui permet d’avoir une argile de départ bien dispersée où l’échange est plus facile à réaliser.
- La nature de l’argile elle-même est très importante, tant du point de vue minéralogique (composition des feuillets, origine de la charge, densité de la charge et sa distribution) que du point de vue textural (dimension et forme des feuillets et ses agrégats).
- La concentration initiale de l’argile dans l’eau joue sur la taille des agrégats. A des concentrations très faibles (< 0.1 %), il peut y avoir disparition complète des tactoides.
- Le rôle du pH qui est lié à la fois aux concentrations initiales et au rapport OH-/Al. - Le rôle de la température de traitement.
- Le temps et la température de maturation de l’argile dans la solution du polymère.
- Le rôle de la dialyse, qui semble mieux organiser les polymères dans l’espace interlamellaire.
- Le mode de séchage. La lyophilisation crée une porosité plus élevée (macroporosité) des argiles trioctaédriques [84].
III-4. Polymère après modification des argiles III.4.1. Structure du polymère
Plee et al. [85] ont montré par R.M.N. du solide de l’27Al que c’est bien le polymère Al13 défini avant pontage qui est inséré dans les argiles trioctaédriques (hectorite, laponite) qui ne contiennent pas d’aluminium dans leur structure. Cependant un léger déplacement dans les fréquences de RMN, par rapport aux fréquences de l’Al13 avant pontage, a été attribué plus tard par Fripiat [86], à une association des polymères Al13.
III.4.2. Stabilité du polymère après traitement d’argile
La question est de savoir ce que deviennent les oxyhydroxydes d’aluminium insérés dans les espaces interlamellaires après calcination. Trois cas de figures peuvent en général se présenter :
- Ils peuvent ressortir de l’espace interlamellaire, l’argile présente alors un espacement de 9.6 Å correspondant à des feuillets fermés.
- Les polymères peuvent parfois se dégrader in situ, en couche d’hydroxyde d’aluminium pour donner un espacement de l’ordre de 14Å correspondant à une pseudo-chlorite.
- Lorsque le pontage est réussi, les polymères commencent à se déshydrater à des températures inférieures à 300°C mais leur structure proposée auparavant de Al13 est conservée. Entre 300 et 400°C, les polymères commencent à se déshydroxyler [87]. A des températures plus élevées, le polymère se transforme en piliers progressivement. Toutefois, l’argile garde l’espacement initiale, pratiquement invariant. D’autre part Suzuki et al87] montrent par RMN que la structure de l’Al13 est pratiquement la même pour une hectorite pontée calcinée à 500°C et qu’au-delà de cette température, le remplacement des OH et des H2O de l’Al13 par des atomes d’oxygène conduit à des piliers formés de colonnes d’oxyde
dont la structure est similaire à celle de l’alumine. Cependant Plee et al. [85] observent la disparition des pics de résonance de l’aluminium tétraédrique (RMN) pour une beidelite pontée calcinée à 500°C.
En résumé, les argiles modifiées en général (sauf la beidelite) ont des piliers de structures très voisine de l’Al13 jusqu’à des températures d’environ 500°C. Au-dessus de cette température, la nature exacte des piliers reste à élucider.
Cependant, cette différence de comportement à l’égard des traitements thermiques pose le problème de savoir si la stabilité est due à la nature même du pilier, à celle du support argileux où à une véritable liaison chimique entre les piliers et l’argile.
Plusieurs paramètres expérimentaux peuvent influencer sur le degré de polymérisation du cation hydroxy-oligomérique en solution aqueuse et par conséquent sur les propriétés physico-chimiques de l’argile modifiée, entre autres nous citons :
• La concentration de l’ion métallique
• L’alcalinité ou le degré d’hydrolyse (exprimé par OH/M) • La température de préparation
• Le temps et la température d’agitation • Le type de contre-ions
• La méthode de préparation III-5. Stabilité des argiles
III-5.1 Influence de la modification et de la calcination sur la texture des argiles
L’intercalation par échange cationique n’affecte pas l’ordre à faible distance à l’intérieur des feuillets argileux. Donc la modification des argiles n’affecte pas la structure du feuillet, ceci est vérifie par les spectres en RMN de 29Si des argiles avant et après traitement qui sont quasiment identiques [85]. Il n’en est pas de même en ce qui concerne la texture des argiles modifiées. L’intercalation d’un gros cation, fortement chargé, a forcement une influence sur l’agrégation des feuillets. Comme déjà signalé, dans les facteurs influençant le traitement, une floculation des montmorillonite ou une destruction des tactoides peuvent se produire selon la
trioctaédriques, dans le plan ab [84]. Cette délamination est encore favorisée par le séchage par lyophilisation (qui favorise en plus la désorientation des tactoides). Mais la cause réelle de la délamination n’est pas en générale connue [88].
IV- Conclusion
Dans ce premier chapitre, nous avons rapporté une étude bibliographique sur les minéraux argileux, ainsi différents traitements réalisés sur les argiles, en l’occurrence le pontage par des cations métalliques, afin d’améliorer les propriétés physicochimiques de ces matériaux, en particulier l’augmentation de la surface spécifique, du volume poreux, la capacité d’adsorption des polluants minéraux et organiques et l’organisation des feuillets.
Le traitement des argiles par la technique de pontage dépend de nombreux paramètres dont on cite la nature de l’argile de départ, du polymère intercalé et de l’objectif attendu de l’utilisation du matériau ponté final.
I. CHROME ET ENVIRONNEMENT
Le chrome occupe le 21ème rang dans le classement des éléments par ordre d’abondance dans la croûte terrestre. Sa concentration moyenne dans les roches est de 100 mg/Kg de roche. Il est largement présent dans les roches (jusqu’à 3400 mg/Kg de roche ignée ) où il est souvent en substitution du fer ( rayons ioniques très proches : Fe (III) = 0.067 nm et Cr(III) = 0.064 nm). Le chrome (III) remplace le fer(III) ou Al(III) dans d’autres minéraux comme les tourmalines, micas et grenats. Les traces de chrome présentes dans ces minéraux sont souvent responsables de leur couleur : le vert de l’émeraude ou le rouge du rubis [89]. Le tableau I.1 présente les concentrations en chrome rencontrées dans des échantillons référencés de roche et de minéraux.
Tableau I.1 : concentrations moyennes en chrome dans différent minéraux [90] .
Minéraux Péridots Basaltes gabbros Argiles Micas Feldspaths Quartz
[Cr]en ppm
3200/2900 400/300 450 200/150 50 25/5 5
I.1. Sources d’émission du chrome
Le chrome présent dans l’environnement a pour origine, d’une part des sources naturelles, et d’autres part des activités industrielles. La source principale étant anthoropogénique.
I-1.1 Les sources naturelles
Les principales sources d’émission naturelles de chrome sont par ordre d’importance [91] :
Altération et érosions des roches :
Environ 50.103 tonnes de chrome/an sont libérés suite à l’altération et l’érosion des roches
Emission volcanique :
Environ 4.103 tonnes de chrome/an sont rejetés lors des émissions volcaniques.
I-1.2. Les sources anthropiques
Le chrome est, le plus souvent, extrait d’un minerai de type oxyde mixte Fe Cr2O4 : la chromite. Les utilisations industrielles de ce métal sont nombreuses et conduisent pour certaines à de graves pollutions environnementales.
chimiques où il sert de catalyseur dans les synthèses organiques [92, 93], dans l’industrie des peintures et colorants [94-96], dans l’industrie du bois du pétrole [97], les industries agroalimentaires [98] et dans la production de films photographiques et de cassettes magnétiques [93, 99]. Une autre source importante de pollution industrielle des eaux naturelles par le chrome est l’industrie de cuir où les sels de chrome trivalent sont largement utilisés comme agent de tannage pour les peaux.
Des stockages inadaptés ou des infrastructures défaillantes sont à l’origine de graves pollutions industrielles. On estime, les rejets anthropiques, dans les différents compartiments de biosphère à [89] :
- 30.103 tonnes par an de chrome émis dans l’atmosphère,
- 140.103 tonnes par an de chrome rejeté dans les eaux de surface, - 900.103 tonnes par an de chrome rejeté dans les sols.
Ce rejet important du chrome d’origine anthropique dans les eaux de surface est assez problématique car ce compartiment de biosphère est très utilisé par l’homme.
Par ailleurs, au cours de ces dernières années, dans de nombreux pays (France, Allemagne…) les quantités de chrome rejeté dans le milieu naturel sont devenues fortement réglementées [100]. Le Maroc est cependant très en retard dans ce domaine, notamment pour le traitement des rejets de tanneries. En effet, dans les effluents hydriques des tanneries, les teneurs en chrome représenté essentiellement par sa forme trivalente sont très importantes et varient de 0,06 à 0,09 g/l. dans les boues, la concentration du chrome est de l’ordre de 1,2 à 5,4 g/100g de boues minéralisées [101].
II. TOXICITE DU CHROME II.-1. historique
Les risques associés à l’emploi des dérivés chromiques sont apparus à la suite des multiples atteintes par des cancers du poumons reportées en Germanie en 1930, chez des ouvriers d’industrie de production de chromates [102].
Au début des années 1970, des travaux épidémiologiques effectués aux états unis, ont montré l’augmentation du risque d’exposition au cancer des poumons chez les employés des usines de production de chromates et d’industrie de fabrication des pigments à base de chrome [103].
En 1978, Adason et Bowden [1104] ont reporté la mort de 12 personnes après l’application d’un onguent dans lequel le soufre était remplacé par un dérivé hexavalent du chrome.
II.-2 Doses létales
La toxicité du chrome dépend non seulement de sa concentration mais aussi de son degré d’oxydation. En effet, il est communément admis que le chrome (VI) est beaucoup plus toxique que le chrome (III). Ce dernier même à très faibles doses, est un élément essentiel aux êtres vivants puisqu’il joue un rôle indispensable dans le métabolisme glucidique comme activateur de l’insuline [89, 105, 106].
L’intoxication au chrome peut être accidentelle par manque d’hygiène, surtout dans les lieux de travail, ou volontaire dans une tentative de suicide. L’ingestion d’un sel de chrome (VI) ou chrome (III) cause une nécrose sélective des cellules des tubes proximaux [107]. Généralement la mort peut survenir pour des doses comprises entre 100 et 300 mg. Après ingestion, les premiers signes d’une gastro-entrite hémorragique apparaissent, suivi d’une insuffisance hépatocellulaire avec ictère et syndrome de coagulation intercellulaire disséminée.
Les tests de toxicités effectués sur différents organismes, ont montré que des concentrations supérieures à 100 mg de Cr (VI)/Kg de poids peuvent devenir létales pour l’homme [92]. La CL50 (concentration de toxicité provoquant la mortalité de 50 % de la population testée en expérience, à la fin d’un temps donné) chez les souris est de 32 mg/Kg et est de 11 mg/Kg chez le lapin [97].
II-3 pathologie attribuée au chrome et ses dérivés II-3.1. lésions cutanées
Un contact avec du chrome contenu dans l’eau, des poussières ou des particules de sol provoquent des alliages cutanés [106-108].
Les dérivés du chrome peuvent conduire à des lésions caractéristiques appelées « pigeonneau » ou « rossignol ». Elles débutent par des fissures douloureuses qui s’accroissent progressivement et deviennent des ulcérations ; outre ces lésions typiques, on
peut observer des dermatoses eczématiformes, et dans certains cas, il y a développement d’une dermatose de contact d’origine allergique, et plus rarement de l’acné [109].
II-3.2. lésions pulmonaires :
Des inhalations prolongées induisent des cancers brancho-pulmonaires chez les personnes en contact dans leur vie professionnelle, principalement dans les industries de production de dichromate et pigments [97, 105, 108].
Une étude sur les effets cancérigène du chrome conduit à un principe probable d’action au niveau des cellules. Contrairement au Cr(III), le Cr(VI) traverse plus rapidement la peau et les membranes cellulaires à l’intérieur desquelles il est rapidement réduit en Cr(III). La substance agissant au niveau des sites actifs est très probablement le Cr(III). Ensuite, ce serait l’intéraction directe Cr(III) et matériel génétique ou processus génétiques qui serait à la base de l’effet cancérigène [105, 108]. Cet effet a lieu aussi bien sur les humains que sur les animaux de laboratoire et des études in vitro montrent qu’il est également responsable de toxicité génétique sur des bactéries ( E. Coli et S. Thyphymirium), des champignons et des cellules animales [97, 105, 108].
III. PROPRIETES CHIMIQUES DU CHROME III-1. Etats d’oxydation
L’isotope du chrome le plus abondant est le 52Cr. Comme les autres métaux de transition, il peut exister à différents états d’oxydation : de la forme métallique Cr(0) à Cr(VI) la forme la plus oxydée. Cependant seuls les états d’oxydation (III) et (VI) sont présents dans l’environnement.
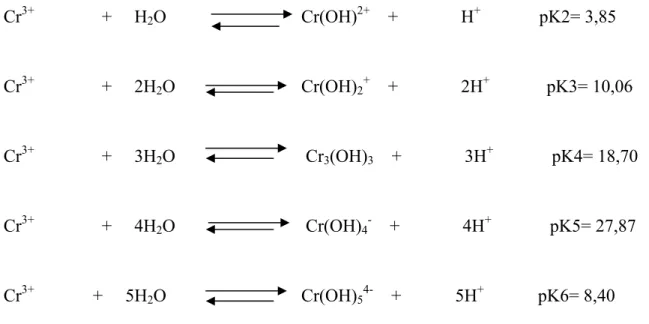
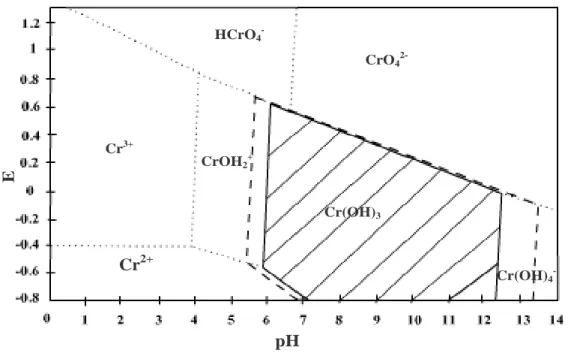
III-2. Spéciation du chrome en solution
L’ion chromique Cr3+ s’hydrolyse très facilement pour produire principalement les espèces suivantes [110, 111] : Cr(OH)2+, Cr(OH)2+, Cr3(OH)54+, Cr(OH)3 et Cr(OH)4-.
Les équilibres correspondant à ces différentes formes peuvent être schématisés comme suit :
Cr3+ + H2O Cr(OH)2+ + H+ pK2= 3,85
Cr3+ + 2H2O Cr(OH)2+ + 2H+ pK3= 10,06
Cr3+ + 3H2O Cr3(OH)3 + 3H+ pK4= 18,70
Cr3+ + 4H2O Cr(OH)4- + 4H+ pK5= 27,87
Cr3+ + 5H2O Cr(OH)54- + 5H+ pK6= 8,40
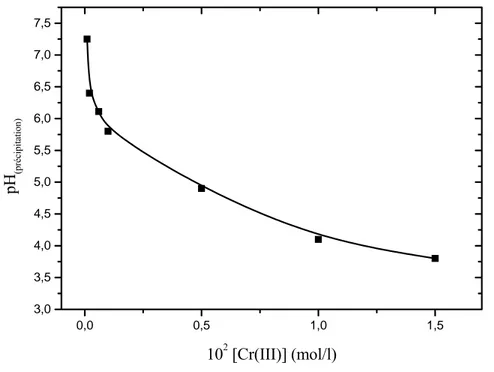
a partir de ces équilibres et des valeurs des constantes correspondantes Lyva-Ramos et al [112] ont établi, pour une concentration 5.10-4 M, le diagramme de distribution des espèces du Cr(III), en solution aqueuse, en fonction du pH (figure I-7)
pH Distribution molair e (% )