Changes in readthrough acetylcholinesterase expression modulate amyloid-beta pathology
Texte intégral
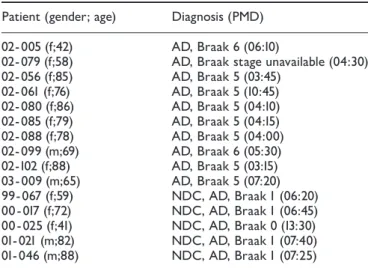
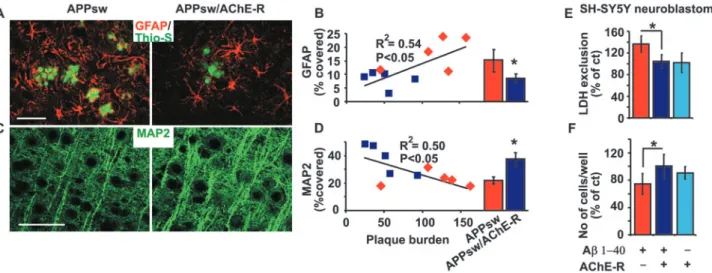
Figure



Documents relatifs
Diets with ideal nutrient composition may be most easily composed in habitats with high resource diversity, where animals can mix resources with variable nutrient contents obtained
However, current implementations of interval arithmetic on SIMD architectures either rely on IEEE-754 rounding modes not available on GPUs [ 12 ], or just ignore rounding
such models are defined in terms of spin degrees of free- dom interacting via infinite-ranged interactions, the deep connections between them and the mode-coupling the- ory of the
We decided to synthesized the 2:1 [Aza/α]-tripeptide sequence aVal-aAla-Val-CONH 2 (Molecule 5.2) (Figure 5.2), composed by two aza-amino acids aVal and aAla and one natural amino
To explore the relationship between the peptide sequence and its ability to form multiple polymorphic forms, we built the peptide sequences GNNQQNY, SSTSAA, and GGVVIA into
In order to probe the origins of the diversity in fibrillar twist we use the weighted-histogram analysis method of atomistic molecular dynamics (WHAM) to measure the free energy
Kinetics of nanophase separation sintering. The above obser- vations suggest a possible explanation for the rapid sintering in this W–Cr alloy: if a second phase precipitates,
Pour le volume total aérien, par exemple, faute d’un nombre suffi - sant de données dans certains sous- groupes, il a fallu se contenter de modèles gris feuillus ou résineux,
