I
Les leucémies aiguës (LA) constituent un groupe hétérogène de pathologies et sont dues à la prolifération clonale de précurseurs hématopoïétiques immatures. 1
D’un point de vue moléculaire, la leucémie humaine résulte de multiples mutations qui mènent aux anomalies dans l'expression ou la fonction des produits de gènes qui affectent l’équilibre entre la prolifération, la différentiation et l’apoptose. Les leucémies sont caractérisées par l'acquisition des aberrations génétiques récurrentes principalement des translocations chromosomiques.2
La principale conséquence de cette prolifération est l’installation d’un tableau d’insuffisance médullaire associant une neutropénie fébrile, un syndrome anémique et un syndrome hémorragique.1 On distingue, selon l’origine du précurseur impliqué, les leucémies aiguës lymphoblastiques de la lignée B ou T, des leucémies aiguës myéloïdes 3
Selon la classification internationale du cancer de l’Enfant, la leucémie représente 32.6 % de tous les cancers d'enfant, dont la leucémie aiguë lymphoblastique représente approximativement 72% des cas de leucémie chez les enfants (age 0-19 ans). 4
Le traitement de référence de première intention d’une LA inclut une chimiothérapie.5 Et comme la chimiothérapie classique entraîne beaucoup d’effets secondaires, du fait qu’elle ne fait pas de distinction entre les cellules saines et les cellules cancéreuses détruisant ainsi les deux types de cellules. C’est la raison pour laquelle les traitements ciblés semblent si attirants. Puisqu’ils tâchent de détruire uniquement les cellules cancéreuses et d’épargner les cellules saines, ce qui devrait entraîner moins d’effets secondaires.
Les objectifs de notre thèse consistent à :
- Rapporter les mécanismes moléculaires impliqués dans la physiopathologie des leucémie aigues.
- Répertorier les principaux médicaments ciblés utilisés dans les leucémies aigues de l’enfant en insistant sur leurs mécanismes d’action.
P
P
a
a
r
r
t
t
i
i
e
e
I
I
G
G
é
é
n
n
é
é
r
r
a
a
l
l
i
i
t
t
é
é
s
s
s
s
u
u
r
r
l
l
e
e
s
s
l
l
e
e
u
u
c
c
é
é
m
m
i
i
e
e
s
s
a
a
i
i
g
g
u
u
e
e
s
s
d
d
e
e
l
l
’
’
e
e
n
n
f
f
a
a
n
n
t
t
1- Définition de leucémie aiguë :
Les leucémies aiguës sont des maladies malignes liées à la prolifération dans la moelle osseuse d’un précurseur hématopoïétique bloqué à un stade donné de maturation.6 On distingue, selon l’origine du précurseur impliqué, les leucémies aiguës lymphoblastiques de la lignée B ou T, des leucémies aiguës myéloïdes.3
Les leucémies aiguës sont les plus fréquentes des maladies malignes de l’enfant : elles représentent environ un tiers de ces affections au cours de 15 premières années de la vie. Trois quarts des leucémies de l’enfant sont aiguës et lymphoblastiques .6
La leucémie lymphoblastique aiguë (LLA) est la maladie maligne la plus fréquente avant l’âge de 14 ans ; elle représente environ 25 % de l’ensemble des cancers pédiatriques. Cette maladie résulte de la prolifération incontrôlée des lymphoblastes (cellules progénitrices des lymphocytes T et B) qui envahissent la moelle osseuse, le sang et d’autres organes. La LLA de type pré-B est celle qui affecte le plus fréquemment l’enfant (80 % des patients) comparativement au type pré-T (15 %).7
La leucémie aiguë myéloïde (LAM) est une expansion clonale de blastes myéloïdes. C’est une pathologie, tous âges confondus, plus fréquente que la LAL (70 % des cas de LA sont myéloïdes) et qui touche surtout les adultes. Elle représente de 75 à 80 % des cas de LA chez l’adulte et de 15 à 20 % chez l’enfant.8
2- L’hématopoïèse en cas normal :
L'hématopoïèse physiologique, qui a lieu dans la moelle osseuse chez l'adulte, assure le remplacement continu des cellules sanguines matures à capacité de prolifération nulle ou très faible et à durée de vie limitée, grâce à un pool de cellules particulières, les cellules souches. Celles-ci sont capables de multiplication sans différenciation (auto-renouvellement) et de prolifération avec différenciation en l'une ou l'autre des cellules hématopoïétiques spécialisées : érythrocytes, polynucléaires neutrophiles, éosinophiles et basophiles, plaquettes, monocytes/macrophages, lymphocytes T et B.9
2-1 Les différents compartiments de cellules hématopoïétiques :
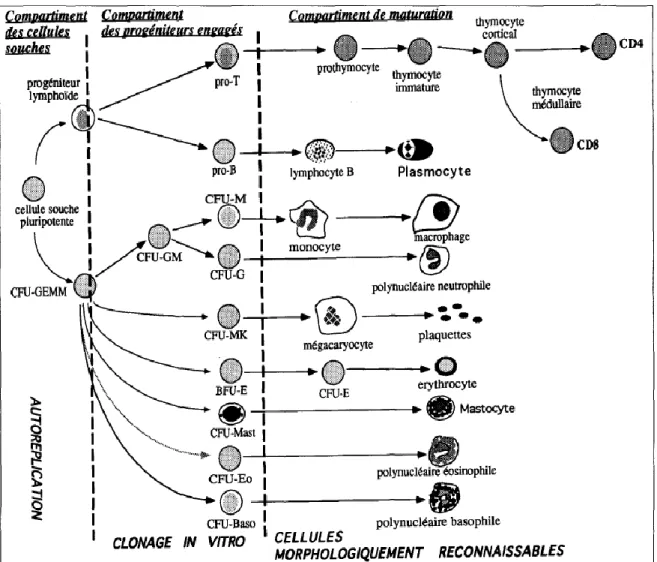
Hématopoïèse est un phénomène complexe présentant une hiérarchie cellulaire, et comprenant schématiquement trois compartiments (Figure 1):
- Un compartiment de cellules très primitives et multipotentes ou cellules souches hématopoïétiques : de durée de vie longue, capables de se différencier vers toutes les lignées hématopoïétiques y compris les lignées lymphoïdes T et B.10
- Un compartiment de progéniteurs clonogéniques, cellules capables de proliférer in vitro en se différenciant sous l'effet de facteurs de croissance. Ces cellules sont habituellement déterminées vers un seul lignage cellulaire, mais certaines peuvent avoir encore plusieurs potentialités. Il est habituel de différencier les progéniteurs matures déterminés vers une seule lignée, qui répondent à un seul facteur de croissance, des progéniteurs plus primitifs pluripotents, qui répondent à des combinaisons de facteurs de croissance ou à
des molécules d'origine stromale.10 Parmi les é1éments du compartiment des progéniteurs, on distingue les cellules souches lymphoïdes B et T, la CFU-GEMM (Colony Forming Unit-granulo-erythro-mono-megakaryocyte), la BFU-E (Burst forming unit-erythroblast, progéniteur des érythroblastes), la CFU-GM (Colony Forming Unit-Granulocyte/Monocyte, progéniteur des cellules des lignées granulocytaires et macrophagiques), la CFU-Eo (progéniteur de l'éosinophile), la CFU-Baso (progéniteur du basophile), la CFU-Mast (progéniteur du mastocyte) et la BFU-Mk (Burst Forming Unit-Megakaryocyte).9
Les frontières entre progéniteurs primitifs et les cellules souches qui reconstituent l'hématopoïèse sont actuellement floues, ce qui entraîne une confusion dans le terme de cellules souches hématopoïétiques.
Ces deux compartiments de cellules (cellules souches et progéniteurs) correspondent à des cellules non morphologiquement reconnaissables et rares parmi les cellules hématopoïétiques (autour de 0,3 % des cellules médullaires pour les progéniteurs matures, de 0,03 % pour les progéniteurs primitifs et vraisemblablement dix fois moins pour les cellules pluripotentes lymphoïdes/myéloïdes). Cette rareté explique les efforts menés pour purifier, essentiellement sur des antigènes de différenciation, les cellules souches hématopoïétiques et les progéniteurs.10
- Un compartiment de maturation, celui des précurseurs hématopoïétiques, où les cellules sont morphologiquement reconnaissables. A la fin de leur processus de maturation, les cellules hématopoïétiques passent dans la circulation.9
Figure 1 : Schéma général de l’affiliation des é1éments des différents compartiments de l'hématopoïèse normale chez l'homme 9
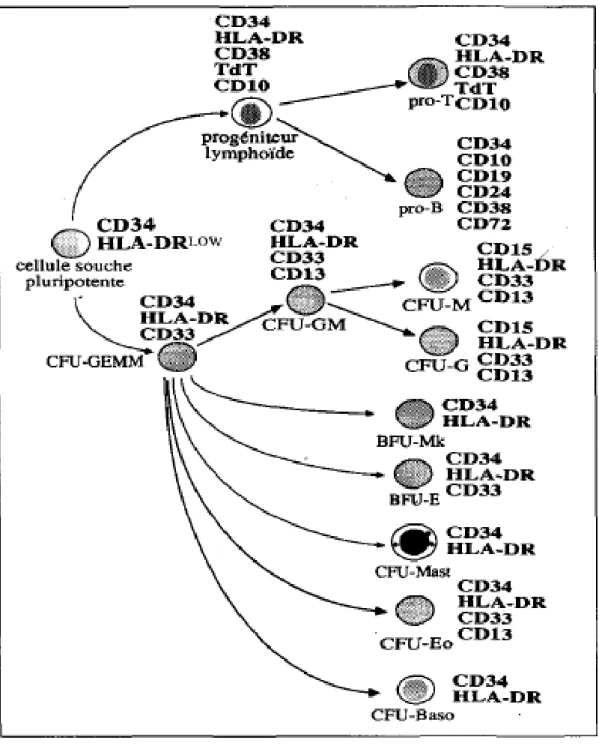
L'ensemble des données connues concernant l’affiliation des différentes cellules hématopoïétiques s'est fortement enrichi avec la mise en évidence à la surface de ces différents é1éments de l'expression transitoire ou constante de divers antigènes membranaires ou « CD » (« cluster of différentiation ») dont la révélation permet, à l'aide d'anticorps spécifiques, de disséquer très précisément les multiples stades de maturation intervenant au cours de l'hématopoïèse, et de purifier les cellules appartenant à chacun de ces stades.
2-2 Les marqueurs antigéniques des cellules hématopoïétiques :
Les marqueurs immunologiques sont des Ag de différenciation. Ils ne sont pas toujours spécifiques d'une étape, mais sont différemment exprimés dans des étapes distinctes de maturation.
L’antigène CD 34 :
CD34 est un antigène de surface des cellules souches hématopoïétiques immatures mais également des cellules déjà engagées dans une lignée. L’antigène CD34 est aussi présent sur les cellules endothéliales des petits vaisseaux et sur les fibroblastes embryonnaires.11, 12
La population exprimant fortement CD34 contient la plus grande part des cellules progénitrices hématopoïétiques immatures alors que les cellules déjà engagées dans une lignée expriment moins intensément l’antigène CD34 qui est une protéine transmembranaire de type I de 115 kDa fortement glycosylée,12 dont le rôle exact est mal connu (intervient dans l’adhérence et la migration).13 En ce qui concerne le rôle de CD34 dans la différenciation, les choses sont encore peu claires : il semble toutefois que la diminution de l’expression de CD34 soit nécessaire pour que la différenciation ait lieu.12
Récemment, on a mis en évidence de façon indiscutable que la population CD34– (caractérisée en plus par une absence de marqueurs de lignée) contient également des cellules à activité CSH, capables de générer une hématopoïèse à long terme. La question reste ouverte de la nature exacte de ces CSH (population plus primitive que les CSH CD34+ ou état transitoire et réversible) .13
L’antigène de surface CD38 : n’est pas exprimé par les cellules souches
hématopoïétiques.12
L’antigène de surface HLA-DR: dans le sang de cordon fœtal, dans le foie
fœtal et dans la moelle osseuse fœtale, les cellules coexprimant HLA-DR avec CD34 sont enrichies en cellules souches.12,14 En revanche, dans la moelle adulte, les progéniteurs primitifs n’expriment pas HLA-DR.12
Le marqueur c-kit ou CD117 : Il s’agit d’un récepteur membranaire
possédant une activité tyrosine kinase intrinsèque codée par le proto-oncogène c-kit. Il est exprimé sur les cellules souches de la moelle osseuse 12,15 ,16 mais il semble que les progéniteurs les plus primitifs n’expriment CD117 que faiblement à leur surface 12,17 La population qui montre une forte expression de l’antigène c-kit est majoritairement celle des cellules progénitrices engagées dans la lignée granulomonocytaire ; puis, au fil de la maturation, l’expression de c-kit diminue et disparaît. Le ligand de c-kit est connu : il s’agit du SCF (stem cell factor), facteur de croissance des cellules souches. Il semble que la transduction du signal consécutive à l’interaction de CD117 avec son ligand joue un rôle important dans les stades précoces de l’hématopoïèse.
Une série d’antigènes de surface appartenant à la même famille de récepteurs tyrosine kinase que CD117 ont été identifiés : il s’agit de
FLK-2/FLT-3, du produit du proto-oncogène c-fms, de TIE qui semblent tous
jouer un rôle important dans la prolifération cellulaire. Tous ces antigènes paraissent exprimés sur les cellules souches hématopoïétiques.12
Le facteur de croissance hématopoïétique FLT-3 joue un rôle clé dans la croissance des cellules souches hématopoïétiques. Le récepteur de FLT-3 est un
récepteur tyrosine kinase qui est exprimé sur les cellules hématopoïétiques normales et malignes. CD135 (= le récepteur de FLT-3)12,18 est exprimé sur les cellules souches hématopoïétiques précoces et aussi sur les cellules engagées dans la lignée monocytaire mais le niveau d’expression reste toujours relativement faible. 12
Le récepteur tyrosine kinase, CD115 ou produit du proto-oncogène c-fms est également exprimé sur une sous-population de cellules souches hématopoïétiques : il semble toutefois que son expression soit plus restreinte aux cellules ayant un devenir dans la lignée monocyte/macrophage.12, 19
Un certain nombre d’antigènes sont utilisés pour déterminer l’engagement d’une cellule dans une voie de différenciation hématopoïétique. CD33 permet ainsi d’identifier les cellules engagées dans la voie myéloïde, CD64 est un marqueur de l’engagement granulomonocytaire,12,20
CD7 et CD19 permettent, respectivement, d’identifier les cellules engagées vers les lymphocytes T ou B.12
Les relations hiérarchiques entre les différentes cellules hématopoïétiques primitives et les principaux CD utilisés pour les distinguer sont analysées dans la figure 2.
Figure 2 : Principaux antigènes de différenciation exprimés par les cellules souches hématopoïétiques et les progéniteurs engagés chez l'homme.9
3- Physiopathologie de leucémie aigue :
L’hématopoïèse leucémique conserve certaines caractéristiques comparables à celles de l’hématopoïèse normale. En effet, le clone leucémique est organisé de façon hiérarchique en trois compartiments distincts :
1) Un compartiment minoritaire de cellules souches leucémiques de phénotype immature (CD34+ CD38- CD123+), pour la plupart quiescentes mais capables d’auto-renouvellement ;
2) un compartiment plus mature de progéniteurs leucémiques ayant perdu des capacités d’auto-renouvellement mais ayant des propriétés clonogènes et de différenciation limitée ;
3) Un compartiment majoritaire de cellules leucémiques bloquées à un stade donné de maturation.21
3-1 Leucémie aigue myéloïde:
Dans le processus de la leucémogénése ; les cellules progénitrices perdent non seulement la capacité de différenciation, mais échappent également de la régulation de prolifération et de survie.22
D’un point de vue moléculaire, il est considéré que le phénotype leucémique est conféré à une cellule souche normale ou à un progéniteur hématopoïétique engagé dans un processus de maturation par l’acquisition d’au moins deux événements mutationnels.21 Des mutations dites de classe I qui affectent des récepteurs à activité tyrosine kinase (récepteur FLT-3 dans 25-30 % des cas ou c-Kit dans 3-5 % des cas) et/ou des protéines clés de la signalisation (N-Ras et
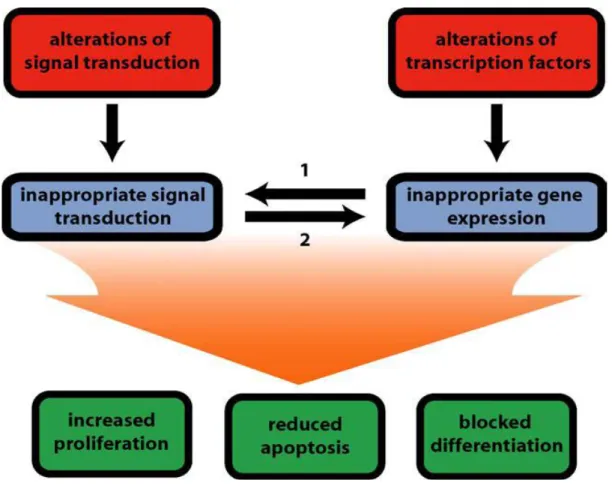
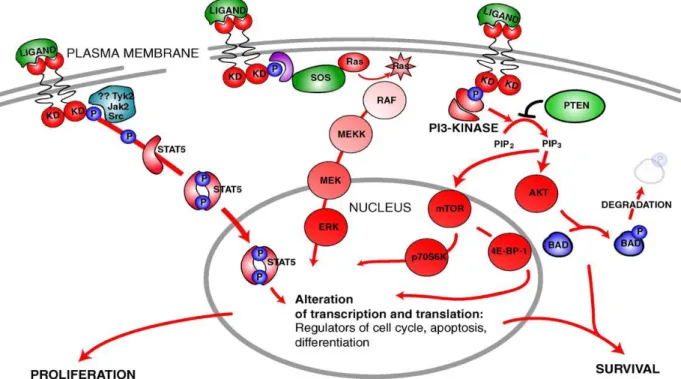
K-Ras, dans 20 s cas) conduisent à des signaux prolifératifs et de survie cellulaire.23,24 Elles sont classiquement associées à des mutations de classe II impliquant des facteurs de transcription qui interfèrent sur les processus de différenciation. Ainsi, les cellules leucémiques présentent certaines voies de signalisation intracellulaire constitutivement actives conduisant à une survie et à une prolifération accrue.24 Les mécanismes de transduction du signal impliquant les mitogen-activated protein kinases (MAPK), les signal transducer and activator of transcription (STAT3 et 5), ou, les phospho-inositides 3-kinases (PI3K) et Akt (PKB) sont souvent mis en jeux dans les LAM .23, 24(Figure 3).
Figure 3 : Intéraction des différentes altérations moléculaires dans le développement d'AML.22
Altérations des médiateurs du signal de transduction mènent au signal de transduction inapproprié, alors que les altérations des facteurs de transcription donnent lieu à l'expression inappropriée des gènes. En conséquence, l'altération de l'expression de signalisation intermédiaire influence le signal de transduction dans les cellules (1). Vice versa, les voies de transduction du signal inapproprié influencent l'expression des gènes par modification post-traductionnelle et influence indirectement sur l'expression des facteurs de transcription impliqués dans la différenciation myéloïde (2). Ces deux mécanismes contribuent conjointement à la pathogenèse de l'AML par l'induction d’une prolifération accrue, réduction de l'apoptose et blocage de la différenciation.
3-1-1 Altérations de signal de transduction :
De nombreux sous types d’AML sont caractérisés par des mutations qui confèrent un avantage de prolifération et / ou de survie aux progéniteurs hématopoïétiques sans beaucoup d’impact sur la différenciation.25
On pense que l’augmentation de la prolifération cellulaire et de survie des blastes leucémiques (mutations de classe I) sont causés par altération de la voie de signalisation RTK. Dans ce contexte ; il a été démontré que les mutations de FMS-like tyrosine kinase 3 (Flt3), c-Kit et Ras sont fréquent dans AML.22 ; 26, 27 Le statut d’activation des voies de croissance et de survie y comprise phosphoinositide 3- kinase (PI3K)/Akt a été également augmenté dans les blastes d’AML .27, 28
Les Récepteurs Tyrosine kinase (RTKs) sont des éléments centraux des réseaux de signalisation cellulaire et jouent un rôle crucial dans le processus physiologique central tel que l’embryogenèse et le développement. L’ensemble des RTK contrôle des activités cellulaires fondamentales ; en incluant la prolifération cellulaire et de la survie, le contrôle du cycle cellulaire, le métabolisme aussi bien que la forme et le mouvement des cellules.27
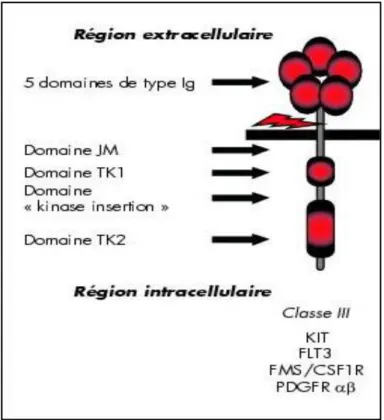
Les RTK sont dans leur quasi- totalité composés d’une seule chaîne polypeptidique qui comporte un domaine extracellulaire responsable de la fixation du ligand et une partie cytoplasmique portant l’activité enzymatique (l’activité kinase).
La région extracellulaire composée de 5 domaines de type immunoglobuline (Ig) possédant de nombreux sites potentiels de N- glycosylation. La région
cytoplasmique se divise en cinq sous domaines structuraux\ fonctionnels : la région juxtamembranaire (JM), la séquence d’insertion hydrophile (KI interkinase) séparant le domaine tyrosine kinase (TK) en deux parties : TK1 correspondant au site de fixation de l’ATP et TK2 présentant l’activité phosphotransférase, et enfin les 50 acides aminés de la partie C- terminale (Figure 4).29 La classe 3 des RTK regroupe les récepteurs majeurs de l’hématopoïèse. Elle se compose des récepteurs c-KIT, c-FMS, FLT3 et des 2 récepteurs α et β du PDGF .29, 30
Trois RTKs de classe 3, c-FMS, c-KIT et FLT3, sont essentiels pour le développement d’une hématopoïèse normale. Les récepteurs du PDGF n’ont pas quant à eux, de fonction hématopoïétique majeure, en dehors du chimiotactisme des neutrophiles et des macrophages.29, 31
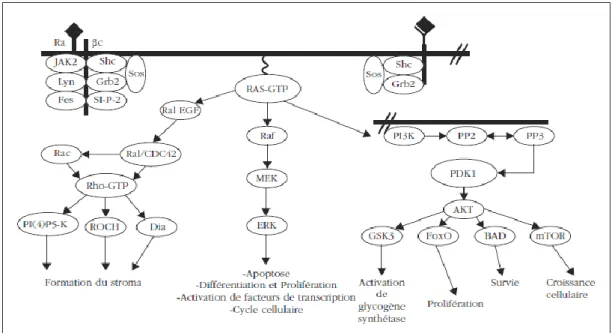
La fixation du ligand conduit à la dimérisation du récepteur suivie de son activation par modification conformationnelle et par transphosphorylation sur résidu tyrosine. Ces deux modifications vont permettre le gain ou la perte d’interaction avec des protéines cytoplasmiques qui conduiront à la transduction d’un signal mitogénique ou de différenciation. Trois voies principales sont alors stimulées, la voie de prolifération\ différenciation liée à l’activation de la voie Ras\MAP- kinase, la voie Jak \STAT, importante dans la réponse proliférative ; enfin, la survie cellulaire assurée par l’activation de la voie PI3-Kinase\AKT.29 (Figure 5).
La meilleure compréhension de la pathogenèse de l’AML découvre plusieurs anomalies génétiques nécessaires pour le développement de la maladie. Les modifications structurelles peuvent conduire à une activation
constitutive de RTK, une subversion des mécanismes de contrôle moléculaire et des altérations de signal de la transduction. Les délétions dans le domaine extracellulaire de fixation de ligand altèrent la sensibilité au ligand, ou éliminent les mécanismes de contrôle négatifs que cette structure pourrait exercer sur le domaine kinase. Même les mutations ponctuelles peuvent induire des altérations conformationnelles en général ligand- indépendant et donc l’activation des RTK. En plus des altérations génétiques ; surexpression du récepteur sauvage et / ou activation du récepteur autocrine sont connues jouer un rôle important dans la transduction du signal aberrant .27
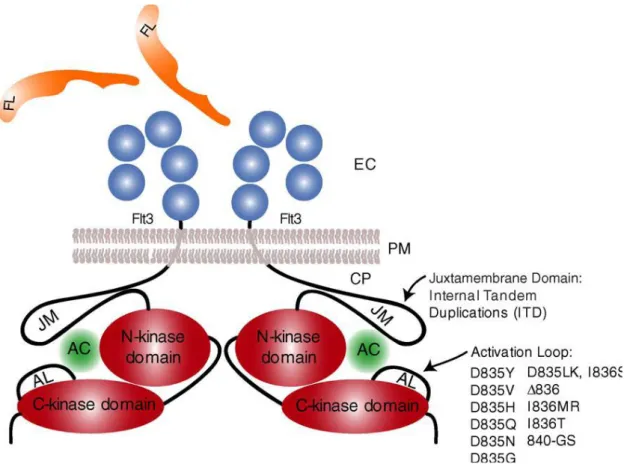
Fms-like tyrosine kinase 3 (FLT3): (Figure 6)
FLT3 (Fms-like tyrosine kinase 3), également connu sous le nom de FLK-2 (fetal liver kinase-2) et STK-1 (human stem cell kinase- 1) ,32 est un récepteur tyrosine kinase impliqué dans la différenciation et la prolifération des cellules souches hématopoïétiques et aussi exprimé sur les blastes d’AML.33
Le ligand de Flt3 (FL) est une protéine transmembranaire27 et aussi exprimé dans la plupart des cellules leucémiques suggérant l'existence de boucles autocrines. Le récepteur des cellules leucémiques est fonctionnel. En effet, dans les cellules de LAM, l'activation du récepteur flt3 par son ligand entraîne la prolifération des cellules leucémiques. Il agit non seulement en stimulant la prolifération mais également en inhibant l'apoptose. Ainsi, l'activation constitutive du récepteur pourrait conduire à une prolifération incontrôlée, et favoriser la leucémogénése.34 Deux types de mutations du récepteur Flt3 aboutis à une activation constitutive du récepteur. Une duplication interne en tandem du Flt3 (Flt3-ITD) est une duplication interne en tandem (ITD) dans le domaine juxta
membranaire du récepteur Flt3.35 ,36 Elle allonge le domaine juxta membranaire.34 Des Mutations ponctuelles dans le domaine kinase du récepteur Flt3 peuvent aussi conférer à une activation du récepteur Flt3. l’activation des mutations ponctuelles la plus commune est la substitution de tyrosine par l’acide aspartique en position 835 dans le boucle d’activation du domaine kinase.36,37
Figure 4 : Représentation schématique de la structure des RTKs hématopoïétiques.29
Figure 5: Représentation schématique de certaines voies de signalisation intracellulaire avec la pertinence de la pathogenèse de l'AML.22
Figure 6 : Décrits des sites d’activation des mutations de Flt3. 22
Le diagramme représente le récepteur dans l’état d’auto-inhibition, avant la fixation du ligand à la partie extracellulaire (EC) du récepteur qui consiste en cinq domaines semblables d’immunoglobilines. Dans l’état d’auto-inhibition, le centre actif (AC) de la kinase est obstrué par des séquences du domaine juxta membranaire (JM) et de la boucle d’activation (AL). Le ligand induit l’activation du récepteur résultant de phosphorylation des résidus critiques de tyrosine dans les régions JM et AL du récepteur et la libération de l’AC. Les mutations activatrices se produisent comme duplication interne en tandem dans le domaine JM ainsi que les mutations ponctuelles dans l’AL et ont pensé de perturber leurs fonctions. Les mutations ponctuelles décrites sont indiquées.
CP: cytoplasme ; PM: membrane plasmique; FL: Flt3-ligand
c-Kit (stem cell factor receptor/CD117):
Le kit est un récepteur de tyrosine kinase codé par le gène c-kit. 38, 39, 40 Les différentes types de cellules y comprise les cellules précurseurs hématopoïétiques expriment c-Kit.40, 41 Dans des situations normales, l'activité
de kinase du c-Kit est étroitement régulé par son ligand, facteur de cellule souche (SCF ; également appelé facteur de Steel, facteur de croissance de mastocyte, et ligand de c-Kit). La fixation du Ligand induit l'oligomérisation du récepteur et par la suite auto/transphosphorylation, suivi par le recrutement et l'activation des protéines cytoplasmiques cibles, comme des membres des voies de MAPK et de PI3K, de tyrosine kinase Src 40,42
La mutation du gène C-KIT est présente dans 3,3 à 11 % des leucémies aiguës myéloblastiques (LAM) de l’enfant.43,44,45
Les mutations de classe I de c-Kit qui provoquent une prolifération excessive des cellules dans le clone malin impliquent l'exon 8 qui code pour le domaine extracellulaire de c-Kit et les mutations de classe II de c-kit qui provoquent le blocage de la différenciation des cellules malignes impliquent l'exon 17(ou des mutations dans la boucle d'activation au codon 816 46), confirmant leur implication dans la transformation maligne.47
Dans AML, les mutations de classe I de c-Kit sont présentes dans 20% de cas avec l'inv (16) et seulement 6% de cas avec t (8 ; 21) tandis que les mutations de classe II de c-Kit sont présentes dans 11-40% de cas avec t (8 ; 21) reflétant la nature hétérogénique de cette maladie maligne.47, 48, 49
Dans AML, le c-Kit est principalement exprimé dans les cellules souches CD34-positive capables de former des colonies de leucémie et de maintenir la maladie.47,50,51 Aussi, l'expression de c-Kit est significativement plus élevée dans des blastes d'AML par rapport à la moelle osseux normale.47,52
RAS (rat sarcoma) :
Les oncogènes de RAS (rat sarcoma) codent une famille des protéines associées à la membrane qui régulent la transduction du signal par liaison à une variété de récepteurs membranaires. Il y a trois gènes fonctionnels de RAS : Le N (de lignée de cellule de neuroblastome), le K (Kirsten), et le H (Harvey) RAS.53
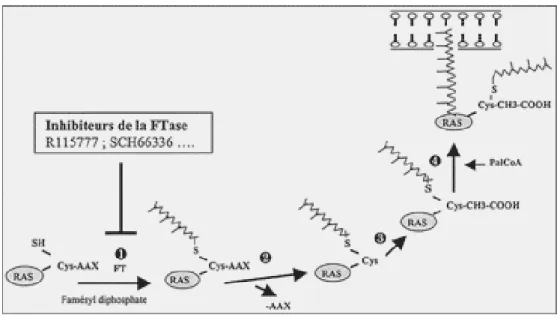
Ras doit se lier à la membrane cellulaire pour exercer son activité.54, 55 Lors de la liaison à la membrane cellulaire, Ras fluctue entre une forme active liée à GTP (deoxyguanosine triphosphate) et une forme inactive liée à GDP (deoxyguanosine diphosphate).54 La possibilité de passage de la forme inactive liée au GDP, à la forme active liée au GTP, est dépendante de la prénylation de la protéine, c’est-à-dire de l’adjonction à la séquence peptidique d’un composé lipidique. La première étape de cette modification post traductionnelle correspond à une farnésylation, catalysée par la farnésyl-transférase (FTase), les étapes ultérieures, à un clivage du peptide -AAX, une carboxylation du résidu farnésylcystéine C-terminal et enfin une palmitoylation à proximité de ce résidu.56 ( Figure 7). À l’état basal, RasGDP est rapidement et transitoirement converti en RasGTP en réponse à divers stimuli extracellulaires. Le signal peut être initié par des facteurs de croissance ou d’autres transmetteurs se liant à des récepteurs membranaires de type tyrosine kinase, ou par d’autres récepteurs tels que la famille Scr. La liaison à ces récepteurs entraîne une dimérisation du récepteur suivie d’une autophosphorylation. En cas de mutation, Ras peut ne pas interagir proprement avec ses régulateurs négatifs et ainsi conduire à son activation sous forme liée à GTP.54,57 Des mutations dans la famille de gènes de RAS ont été décrites dans environ un quart des cas d'AML, principalement dans
les N-Ras, mais aussi dans K-Ras et, bien que très rarement, dans H-Ras.27,58,59 Les mutations de Ras réduisent l’hydrolyse de GTP, entraînant une signalisation Ras constitutive. Cette augmentation du signal peut aussi résulter d’une amplification ou de mutations touchant le récepteur des facteurs de croissance. Ras est le point de départ de plusieurs voies de signalisations telles que Raf/MEK/ERK, et PI3K (phosphatidyl inositol-3 kinase)/AKT.54, 60, 61 (Figure 8)
* La voie Raf/MEK/ERK :
La voie Raf- MAPK (mitogen-associated protein kinase) résulte de l’activation de facteurs de transcription de la famille Ets. Par cette voie, Ras agit sur des gènes impliqués dans les processus de prolifération cellulaire, d’envahissement cellulaire et de formation du stroma. Raf est une sérine/thréonine (S/T) kinase normalement activée par une série d’événements complexes : une interaction avec Ras, une dimérisation de la protéine, une phosphorylation de différents domaines. Raf peut alors initier une suite de phosphorylations se terminant par la phosphorylation de la protéine kinase MAPK.54, 62, 63 Dans le noyau, MAPK active des facteurs de la transcription intervenant dans la gestion de la prolifération cellulaire et de l’apoptose.54, 64, 65
Comme Ras, MAPK peut être surexprimée ou activée dans les hémopathies malignes.54
* La voie PI3K/AKT :
La voie PI3K/AKT joue un rôle dans les signaux de transduction débutant avec les facteurs de croissance et se terminant par la prolifération cellulaire (figure 8). 54, 66, 67 L’activation de Ras représente un des déclencheurs de cette voie en agissant directement avec PI3K dans la phosphorylation et l’activation
de la protéine kinase B-Akt. Akt contribue à activer m- TOR de manière directe en le phosphorylant.24 Akt est activée dans les cellules soumises à l’action de différents stimuli tels que des hormones, des facteurs de croissance, des composants de la matrice extracellulaire. À travers la régulation du cycle cellulaire, AKT module la fonction de nombreuses protéines cellulaires impliquées dans le métabolisme, l’apoptose, la prolifération cellulaire.54, 68
Une des actions de AKT peut conduire à la production de VEGF, connu comme un facteur de croissance et de survie dans certaines leucémies (Cela peut expliquer les relations dans l’oncogenèse entre Ras et l’expression de VEGF, et suggère une action des FTI sur l’apoptose en supprimant l’activité VEGF dépendant de la voie RasPI3K/AKT).54, 69
Figure 7 : La farnésylation de Ras. 56
1 = Farnésylation d'un résidu cystéine de la séquence CAAX ; 2 = Clivage protéolytique du peptide -AAX ; 3 = Carboxyméthylation de la cystéine farnésylée ; 4 = Pamitoylation à proximité de la cystéine farnésylée. FT= Farnésyltransférase.
Figure 8 : Activation de Ras (d’après Morgan et al. 54,70). 54
Les protéines Ras sont activées par des récepteurs de type tyrosine kinase et des récepteurs de cytokines. Après activation, Ras lié à GTP active des molécules telles que Raf, et PI3K. À travers l’activation de Raf, Ras active MEK et ERK, qui sont impliquées dans les processus cellulaires de prolifération et de différenciation, ainsi que le cycle cellulaire. L’activation de la voie PI3K-PDK1 conduit à l’activation de AKT, qui phosphoryle des substrats impliqués dans la prolifération et la survie cellulaire.
3-1-2 Altérations impliquant des facteurs de transcription :
Les mutations de classe II conduisent à une altération de la différenciation hématopoïétique. On pense que l’altération de la différenciation des blastes d'AML se produit par altérations des facteurs de transcription exigés pour la différentiation des cellules myéloïdes normales. Les facteurs de transcription sont fréquemment ciblés par les translocations chromosomiques équilibrées dans AML. Ceux-ci incluent le core binding factor (CBF), le récepteur d’acide retinoique (RAR) et les membres de la famille de HOX des facteurs de transcription. 22, 26, 27, 71 et la protéine MLL.72
Core binding factor (CBF) :
CBF est un facteur de transcription hétérodimérique composé d’un élément de fixation à l'ADN, AML1, et la sous unité CBFß qui fonctionne comme un activateur transcriptionnel d'AML1. CBF contrôle l'expression des gènes qui sont essentiels pour le développement hématopoïétique normal. Les trois translocations les plus communes impliquant CBF sont le t [8 ; 21], inv [16], et t [12 ; 21] qu'ont pour résultat l'expression des protéines de fusion AML1/ETO, CBFß/SMMHC, et TEL/AML1, respectivement.27
Le gène AML1 est remanié par plusieurs translocations dont la plupart conduisent à l’expression de protéines de fusion, qui toutes comprennent le domaine de liaison à l’ADN d’AML1. Les gènes partenaires TEL et ETO codent normalement pour des protéines qui sont des répresseurs transcriptionnels. Les protéines de fusion TEL–AML1 et AML1–ETO ont des propriétés répressives de la transcription et exercent un effet dominant négatif sur l’activité du CBF normal. Au moins pour AML1–ETO et pour TEL–AML1, l’activité de répression passe par le recrutement des complexes corépresseurs et des activités histone déacétylase. Le gène codant pour le CBFb est fusionné avec le gène “Smooth Muscle Myosin Heavy Chain” (SMMHC) a aussi des propriétés de répression transcriptionnelle.73
Le récepteur d’acide rétinoique (RAR) :
RARa est exprimée dans la cellule souche myéloïde et permet en présence de concentrations physiologiques d’AR, de participer à la différenciation granulocytaire et spécifiquement au passage du stade myéloblaste au promyélocyte. Toutes les anomalies du gène codant pour RARa aboutissent à un
blocage au stade de promyélocyte.74 Les translocations les plus communes dans APL sont la fusion de la chaîne α du récepteur de l’acide rétinoique au gène PML (PML- RAR α) ou à la protéine promyelocytic leukemia zinc Finger (PLZF/ RAR α),36,75
Les deux protéines PML/RARa et PLZF/RARa interagissent fortement avec les corépresseurs N-CoR et SMRT et recrutent les protéines HDAC en l’absence de ligand.74 Les HDAC bloquent l’acétylation des histones et donc la transcription de l’ADN.76
( Figure 9)
MLL :
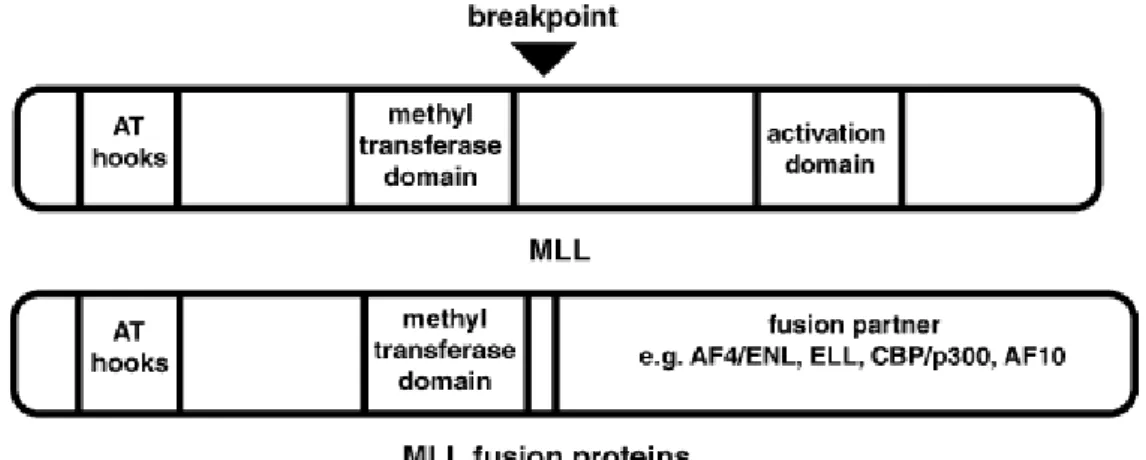
Le gène MLL (Mixed Lineage Leukemia ou myeloid/lymphoid leukemia gène ou encore appelé HTRX, ou ALL1 77), situé sur le bras long du chromosome 11,8 MLL a une activité histone méthyltransférase .22 ,78 Il est impliqué, dans un nombre élevé de translocations avec des partenaires différents, les plus fréquentes étant les translocations (9; 11), (11; 19) ou (6; 11).8
Le domaine de fixation à l’ADN de MLL contient trois « crochet AT » qui change la conformation d'ADN et permet ainsi la régulation de la fixation des facteurs de transcription à l’ADN.22, 79, 80
Les autres domaines de MLL sont homologues au domaine régulateur d'un methyltransferase22, 81, 82 et contrôle l’activation de l'activité transcriptionnelle .22, 83, 84
Les translocations de MLL se produisent non seulement dans AML, mais également dans leucémie aigue lymphoblastique (ALL). Toutes les protéines de fusion contiennent la partie N-terminale comprenant le domaine de liaison à l'ADN et le domaine de répression de MLL fusionné à la partie C- terminale du partenaire de translocation. 22(figure 10).
Certaines données expérimentales indiquent que seul le gène de fusion transcrit à partir du chromosome 11 dérivatif joue un rôle dans la leucémogénése. Il faut également signaler l'existence d'hémopathies myéloïdes aiguës sans réarrangement de 11q23 mais avec duplication partielle du locus MLL.85,86 Cette duplication concerne la région 5' du gène où sont localisés les motifs «crochet AT » et le segment d'homologie avec l'ADN méthyl-transférase. Cela suggère l'importance de cette région et de ces deux motifs dans la physiopathologie de ces hémopathies.85
HOX :
Les protéines homeodomaines HOX font partie d'une grande famille des protéines codées par les gènes homeobox. Leurs gènes sont organisés dans quatre groupes (A-D) sur les chromosomes 7, 17, 12 et 2. Les protéines HOX ont montré d’être des régulateurs importants pour le développement et l’hématopoïèse d’adulte.22, 87, 88
Les produits du gène HOX influencent la transcription par leur liaison à l’ADN comme des monomères ou des hétéromères avec les produits de gènes homeobox non-HOX comme les gènes de Pbx ou Meis1 (les membres de la famille TALE).22, 89, 90 Comme décrit pour beaucoup d’autres facteurs de transcription, aussi les protéines HOX fonctionnent au moins partiellement par des modifications épigénétiques d'ADN. 22,91
Plusieurs membres de la famille du gène HOX, en particulier du groupe de HOXA, sont fréquemment inactivés par hyperméthylation des îlots de CpG dans la leucémie. Pour deux de ces gènes, HOXA4 et HOXA5, hyperméthylation a
été vu dans tous les types de leucémies étudiées, y compris les deux malignités myéloïdes et lymphoïdes.92
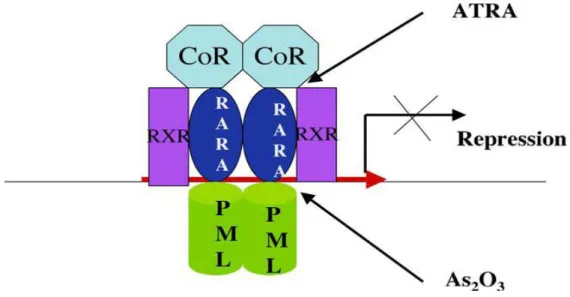
Figure 9 : Répression transcriptionnelle par PML-RARα dans APL et les effets d'ATRA et de Trioxyde Arsenic. 36
En l'absence de l'acide Rétinoique (RA), les hétérodimeres RAR α et RXR recrutent le corépresseur (Cor), qui sert un médiateur de la répression transcriptionnelle. En présence de RA, le Cor est relâché amenant à l'activation transcriptionnelle. Dans APL, la protéine de fusion PML-RARα se lie aux gènes cibles de RARα et recrute CoR, en causant la répression transcriptionnelle et l’inhibition de la différentiation myéloïde. ATRA agit sur RARα et libère le Cor ce qui induit l'activation transcriptionnelle et l'induction de la différentiation. Trioxyde arsenic agit sur PML et provoque une différentiation partielle des cellules d'APL et une apoptose.
Figure 10 : Représentation schématique des domaines de la protéine sauvage MLL et des protéines de fusion MLL.22
3-2 Leucémie aigue lymphoblastique :
On pense que la leucémie lymphoblastique aiguë provient de diverses lésions génétiques importantes dans les cellules progénétrices sanguines qui sont engagées dans la voie de différenciation à la cellule T ou cellule B, y compris les mutations qui donnent la capacité d’auto renouvellement illimité et ceux qui mènent à l’arrêt de développement au stade spécifique précis.93, 94, 95
Les cellules impliquées dans la leucémie aiguë lymphoblastique ont des réarrangements clonaux dans leur immunoglobuline ou des gènes du récepteur de cellule T, expriment des molécules antigéniques du récepteur et d’autres liées à la différenciation glycoprotéines de surface cellulaire qui récapitulent largement ceux des cellules progénitrices lymphoïdes immatures dans les premières étapes de développement des lymphocytes normaux T et B.93,94,95 ; 96
Les translocations chromosomiques qui activent des gènes spécifiques sont une caractéristique définissante de leucémies humaines et en particulier de
leucémie lymphoblastique aiguë.93, 94, 95 Habituellement, les translocations activent les gènes des facteurs de transcription, qui peuvent dans beaucoup de cas contrôler la différenciation cellulaire (plutôt que sa division cellulaire), régulent le développement, et encodent fréquemment des protéines au sommet des cascades transcriptionel important.93,95
Environ 25% des cas de leucémie aiguë lymphoblastique à précurseur lymphocyte B, la forme la plus fréquente de leucémie aiguë chez les enfants, portent le gène de fusion TEL-AML1 généré par la translocation chromosomique t (12 ; 21) (p13 ; q22).93, 94 La protéine produite par le gène AML1 est un facteur de régulation de transcription dont l’action physiologique est de modifier la structure chromatinienne en agissant sur l’acétylation des histones, ce qui permet à l’ADN d’être mieux traduit. Le transcrit de fusion TEL-AML1 produit une protéine hybride dont l’action sur l’acétylation des histones est pathologique.6
La translocation (9;22) ou chromosome Philadelphie, 93 résulte de la fusion du gène BCR sur le bras long du chromosome 22 et du gène ABL sur le bras long du chromosome 9. Dans la moitié des cas, le transcrit de fusion (BCR-ABL) est une protéine (p210)(ou M-BCR pour Major BCR 97) identique à celle de la leucémie myéloïde chronique. Dans les autres cas, le transcrit de fusion BCR-ABL) est une protéine (p190) (ou m-BCR pour minor BCR 97) de plus bas poids moléculaire.8 Les deux types de protéines (P190 et P210) ont une activité tyrosine kinase très augmentée par rapport à celle du proto-oncogène ABL.97
Plus de 50% de cas de leucémie aiguë lymphoblastique à cellule T ont des mutations activatrices qui impliquent NOTCH1, 93, 98 un gène codant pour un
récepteur transmembranaire qui règle le développement du lymphocyte T normal. 93,99
Les récepteurs membranaires Notch (il en existe 4, mais c’est surtout de Notch1 qu’il s’agit ici), après interaction avec des ligands présents sur d’autres cellules, subissent une cascade de clivages protéolytiques, notamment par la γ-sécrétase, qui libère leur partie intracellulaire qui agit ensuite comme cofacteur transcriptionel. Chez les patients atteints de LAL-T, l’activation de Notch est souvent associée à des mutations du gène Notch1 qui augmentent sa sensibilité au clivage par la γ-sécrétase (mutations du domaine d’hétérodimérisation), ou qui entraînent des troncations dans la partie carboxyterminale augmentant l’activité de cette protéine.98 ,100
4- Facteurs favorisants :
Dans la très grande majorité des cas, les LA surviennent sans élément étiologique identifiable. Cependant, on connaît certains facteurs favorisants qui, bien que rarement retrouvés, sont considérés comme indiscutables : le benzène, les radiations ionisantes, certaines chimiothérapies et quelques désordre génétiques ou familiaux (tableau I).5
4-1 Facteurs génétiques constitutionnels :
La LAL est particulièrement fréquente chez l’enfant atteint de trisomie 21 (le risque est multiplié par un facteur 20 environ) ainsi que dans d’autres maladies génétiques : ataxie télangiectasie, syndrome de Bloom, maladie de Shwachman. Dans la maladie de Fanconi, la neurofibromatose de type 1 et la
maladie de Blackfan-Diamond, l’augmentation de fréquence des hémopathies malignes porte essentiellement sur les hémopathies myéloïdes.6
Le risque de développer une LAL dans l’année est estimé à 20-25 % pour le jumeau monozygote d’un patient atteint de LAL. Pour la fratrie dizygote, le risque est quatre fois supérieur à celui observé dans la population générale.101
4-2 Exposition aux radiations et aux toxiques :
L’exposition à des radiations ionisantes augmente le risque de leucémie aiguë lymphoïde et myéloïde. Ce rôle a été analysé pour trois types différents d’exposition : avant la conception, pendant la vie intra-utérine et après la naissance. Certains auteurs ont mis en évidence une augmentation du risque chez les enfants nés d’un père ayant vécu à proximité d’une centrale nucléaire mais ceci n’a pas été confirmé dans d’autres études. Le risque lié à l’exposition pendant la vie intra-utérine ou après la naissance est bien documenté mais n’est actuellement en cause que dans un très faible pourcentage de cas de LAL de l’enfant. Les leucémies secondaires aux médicaments cytostatiques et agents chimiques cancérigènes sont dans la majorité des cas des leucémies aiguës myéloïdes ou des myélodysplasies plutôt que des LAL.6, 102
Les solvants, certains produits chimiques et les pesticides ont également été reliés aux LA.5,103,104
4-3 Facteurs infectieux :
Le rôle d’agents infectieux qui agiraient comme cofacteurs de la leucémogenèse a été suggéré de longue date (hypothèse de Greaves) mais n’est
démontré que dans le cas du virus Epstein-Barr (EBV), qui est impliqué dans la leucémie de Burkitt (LAL3).6
Tableau I : Facteurs favorisants classiques des leucémies aiguës.5
5- Signes cliniques :
L’expression clinique de la maladie peut se traduire par deux types de signes : ceux qui sont liés à l’insuffisance médullaire et ceux qui sont liés à la
prolifération tumorale. En dehors des formes découvertes à un stade très avancé, le tableau clinique est généralement incomplet, pouvant regrouper des combinaisons très variables de symptômes cliniques.
5-1 Signes en rapport avec l’insuffisance médullaire :
5-1-1 Signes d’anémie :
L’asthénie est très fréquente, bien que totalement non spécifique. L’entourage ou le médecin peuvent avoir remarqué une pâleur anormale. Dans d’autres cas, l’enfant se plaint d’une fatigabilité inhabituelle à l’effort, de vertiges, de lipothymies. Il est rare de découvrir l’anémie à la phase de défaillance cardiorespiratoire : dyspnée de repos et tachycardie, voire hypotension et troubles de la conscience.6
5-1-2 Signes liés à la neutropénie :
L’angine ulcéronécrotique est aussi classique que rare. La fièvre est très fréquente au moment de la découverte de la maladie, souvent plus liée à la prolifération de lymphoblastes qu’à une infection ORL ou bronchique. L’extrême gravité potentielle des infections bactériennes chez les patients profondément neutropéniques doit cependant conduire à considérer que toute fièvre chez un patient en agranulocytose est liée à une septicémie bactérienne jusqu’à preuve du contraire. En conséquence, hémocultures et antibiothérapie intraveineuse sont indispensables si le taux de polynucléaires neutrophiles est inférieur à 0,5 giga/l.6
5-1-3 Signes liés à la thrombocytopénie :
Le purpura est typiquement de type thrombocytopénique : pétéchial et ecchymotique, sans prédominance topographique particulière. Si le taux plaquettaire est inférieur à 20 giga/l, on peut observer des bulles hémorragiques sur la muqueuse buccale. Lorsque des hémorragies surviennent, elles sont généralement limitées à des épistaxis, mais des hémorragies plus sévères sont possibles, la plus grave étant l’hémorragie cérébrale. 6
5-2 Signes liés à la prolifération tumorale :
5-2-1 Hypertrophie des organes hématopoïétiques :
Les adénopathies superficielles sont davantage observées dans les LAL, particulièrement chez l’enfant (80 % des cas). Les adénopathies médiastinales sont très évocatrices de LAL de type T et peuvent occasionner un syndrome compressif. Les LAL3 s’accompagnent fréquemment d’une masse ganglionnaire abdominale de croissance rapide.La splénomégalie est un élément commun au cours des LAL (75 % des cas), surtout de l’enfant, et des LAM (50 %) dans les variétés monocytaires (une hépatomégalie associée peut se rencontrer jusque dans 50 % des LAL et un peu moins souvent dans les types M4 et M5). 5
5-2-2 Syndrome de leucostase :
Dans les formes hyperleucocytaires des LAM (en pratique pour des chiffres excédant 100 109/L), on peut rencontrer des phénomènes de leucostase s’exprimant principalement dans la circulation cérébrale (céphalées, torpeur pouvant aller jusqu’au coma, ataxie, troubles visuels avec signes au fond d’œil) et pulmonaire (hypoxémie, dyspnée, anomalies radiologiques : opacités diffuses
bilatérales). Ces signes sont la traduction de phénomènes thrombotiques (occlusion des artérioles cérébrales et pulmonaires par les agrégats blastiques) ou hémorragiques (en particulier intracérébraux). Le syndrome de leucostase concerne environ 10 % des patients et est très rapidement fatal en l’absence de cytoréduction rapide (chimiothérapie associée aux leucaphérèses). La rareté du phénomène de leucostase dans les LAL, même à des taux de lymphoblastes circulants très élevés, s’explique par la plus petite taille, la plus grande déformabilité de ces cellules et l’absence de phénomène d’adhésion entre elles, contrairement à ce qui est observé dans les LAM.5
5-2-3 Localisations extra-hématologiques :
5-2-3-1 Localisation neuro-méningée :
L’atteinte du liquide céphalorachidien (LCR) s’observe plus spécialement dans tous les types de LAL (et à des fréquences extrêmes dans la LAL3), les LAM à composante monocytaire (LAM4, LAM4 à éosinophiles, LAM5) et de façon générale en cas d’hyperleucocytose ou d’élévation importante des lacticodéshydrogénases (LDH). 5,105 L’expression clinique est variable : signes d’hypertension intracrânienne (céphalées, nausées, vomissements, oedème papillaire au fond d’œil), atteinte des nerfs crâniens, syndrome méningé, troubles des fonctions supérieures, troubles du comportement alimentaire (boulimie), signe de la houppe du menton (anesthésie de la région mentonnière témoignant d’une atteinte de la base).5,106,107,108
Néanmoins, la majorité des patients avec atteinte du LCR sont asymptomatiques.5,108
5-2-3-2 Atteinte osseuse :
C’est un élément relativement fréquent dans les LAL de l’enfant (environ un cas sur cinq) et beaucoup plus rare dans les LAM. L’atteinte osseuse se traduit par des douleurs localisées aux os longs ou plus diffuses, spontanées ou provoquées (pression du sternum). Lorsqu’elles constituent la manifestation inaugurale, ces douleurs sont parfois faussement étiquetées douleurs de croissance, rhumatisme inflammatoire, ostéomyélite... 5, 106, 109 Le mécanisme causal inclut une expansion de l’espace intramédullaire ou un envahissement direct du périoste par les cellules leucémiques.5, 108
5-2-3-3 Syndrome médiastinal : 6
L’atteinte du médiastin antérieur et supérieur est pratiquement spécifique des formes lymphoblastiques T. Il peut s’agir soit d’une atteinte thymique, soit d’adénopathies, soit d’une association des deux atteintes. Un épanchement pleural uni- ou bilatéral, contenant des cellules leucémiques, est fréquemment associé. L’atteinte médiastinale peut être asymptomatique. Elle peut également être révélée par des signes de compression de la trachée ou de la veine cave supérieure. C’est alors une urgence médicale à prendre en charge dans un centre spécialisé de cancérologie pédiatrique. L’atteinte médiastinale doit rendre très prudent dans les indications d’anesthésie générale.
5-2-3-4 Atteintes cutanéomuqueuses :
La présentation prédominante consiste en des nodules ou des placards violacés multiples, non prurigineux, durs et indolores. Ils correspondent histologiquement à une infiltration blastique du derme. Ce tableau atteint
jusqu’à 10 % des malades porteurs d’une LAM, en général de type M4 ou M5. Il est en relation avec une modification des propriétés d’adhésion des cellules leucémiques et s’accompagne volontiers d’autres atteintes extramédullaires, en particulier méningées.5, 107
5-2-3-5 Atteintes gonadiques :
Elles sont classiquement décrites au cours des LAL de l’enfant. L’atteinte du testicule (hypertrophie indolore) est beaucoup plus fréquente que celle de l’ovaire. Il s’agit d’un tableau clinique davantage observé en situation de rechute qu’au diagnostic initial .5, 106, 109
6- Diagnostic biologique :
6-1 Hémogramme 6 :
Quatre signes peuvent être diversement associés : l’anémie, la thrombocytopénie, la neutropénie et la présence de lymphoblastes dans le sang. L’anémie est normochrome, normocytaire ou légèrement macrocytaire et arégénérative. La présence de lymphoblastes malins dans le sang est responsable d’une hyperleucocytose dont l’intensité est très variable. Environ 20 % des enfants ont des leucocytes supérieurs à 50 giga/l mais le taux peut être bien supérieur, notamment dans les formes T et chez les nourrissons âgés de moins de 1 an.
Un petit nombre de patients n’ont pas de lymphoblastes cytologiquement détectables dans le sang, le tableau biologique se résumant à une ou plus généralement plusieurs cytopénies. Le myélogramme est alors l’examen clé du diagnostic. En cas de LAL, la moelle est envahie par des lymphoblastes.
6-2 Myélogramme :
En dehors des formes sans lymphoblaste circulant, il confirme généralement un diagnostic déjà fait dans le sang .6
Le myélogramme est un examen clé, qui met en évidence une infiltration blastique supérieure à 30 %, définissant le diagnostic de LA. Plus récemment, la classification de l’OMS a établi ce seuil à 20 % incluant l’entité « anémie réfractaire avec excès de blastes en transformation ». La moelle est en général hypercellulaire, mais des aspects hypoplasiques, voires aplasiques, ne sont pas exceptionnels. L’aspiration du suc médullaire est parfois impossible (dry tap) en cas de fibrose. Ce cas de figure est fréquent lors de la LAM de type mégacaryoblastique (LAM7). La réalisation d’une biopsie médullaire est alors impérative.5
6-3 Cytochimie:
Les études cytochimiques complètent l’interprétation purement cytologique. Les colorations de routine concernent essentiellement deux types d’activités enzymatiques : les myéloperoxydases, caractéristiques des LAM, et les estérases qui sont positives sur les cellules granuleuses et monocytaires.5
6-4 Bilan d’hémostase :
La CIVD est quasi constamment observée dans les LAM3, mais tous les types, en particulier les LAL et LAM4 et 5, peuvent en comporter.5, 107 Elle se caractérise par une baisse du fibrinogène, des plaquettes, de certains facteurs de la coagulation et la présence de complexes solubles. L’acide tout-trans rétinoïque permet en règle un contrôle rapide de ce problème au cours des
LAM3. Dans les autres types cytologiques, le clinicien peut utiliser, selon les cas, les transfusions de plaquettes, le plasma frais, les faibles doses d’héparine ou les concentrés d’antithrombine III. 5
6-5 Dosages biochimiques :
Ils permettent de déceler des anomalies métaboliques : hyperkaliémie (en cas d’acidose et dans les formes hyperleucocytaires) ou hypokaliémie; acidose lactique; 5 hyperuricémie.5, 107. Le taux de LDH (lacticodéshydrogénase) constitue un élément pronostique dans les LAM et dans les LAL.5 De fausses hypoglycémies sont possibles en cas de forte blastose. 5,108
7- Classification :
7-1 Leucémie aiguë lymphoblastique (LAL) :
7-1-1 Classification cytologique (Figure : 11):
La classification morphologique FAB, 8 initialement décrite par les auteurs français, américains et britanniques, est utilisée très largement. 6 On distingue les formes L1, L2, L3 en fonction de l’aspect du noyau et des nucléoles, du rapport nucléocytoplasmique, de la basophilie du cytoplasme, de la présence de vacuoles cytoplasmiques.
Les lymphoblastes L1 (90 % des patients) sont de petites cellules à très haut rapport nucléocytoplasmique ; les nucléoles ne sont généralement pas visibles ; le cytoplasme dépourvu de granulations est réduit à un petit liseré périphérique (Figure 11.A).
Les lymphoblastes L2 sont des cellules plus grandes, leur cytoplasme est plus abondant, les nucléoles sont visibles (Figure 11.B). La distinction L1/L2 n’est pas corrélée à des profils phénotypiques ou cytogénétiques particuliers.
Les lymphoblastes L3 sont en revanche très spécifiques de la leucémie de Burkitt, c’est-à-dire de la leucémie à cellule B mature qui porte des immunoglobulines de membrane. Le lymphoblaste L3 est facilement reconnaissable par l’intense basophilie du cytoplasme et la présence de nombreuses vacuoles cytoplasmiques (Figure 11.C).6
Figure 11: Classification cytologique des leucémies aiguës lymphoblastiques. 6 Figure 11.A : le rapport nucléocytoplasmique très élevé de
L1 et l’absence de nucléole.
Figure 11.B : les nucléoles de L2 sont bien visibles (flèche) et le cytoplasme
plus abondant.
Figure 11.C : Noter l’intense basophilie du cytoplasme et les vacuoles des lymphoblastes L3
7-1-2 Classification immunophénotypique :
La mise en évidence par cytométrie en flux de marqueurs cellulaires (membranaires et intracytoplasmiques) a permis d’affirmer la nature lymphoïde des blastes et d’individualiser différents sous-groupes. Ces constatations constituent la base de la classification immunophénotypique de l’EGIL (European Group for the Immunological Characterization of Leukaemias), largement utilisée aujourd’hui et qui reconnaît quatre sous-groupes au sein des LAL B et quatre sous-groupes au sein des LAL T ; au sein de ces groupes sont individualisés les cas dont les blastes coexpriment un marqueur myéloïde (LAL My +). 8
LAL-B 110 :
Pour la lignée B (Tableau II), les marqueurs pan-B sont CD19, cCD22, cCD79a. Les marqueurs de stade de la lignée B étant CD10, cμ (chaîne μ d’immunoglobuline [Ig] cytoplasmique) et sIgM (IgM de surface), le CD20 et le marqueur de maturité FMC7. La molécule HLA-DR et l’Ag nucléaire TdT (désoxynucléotidyl transférase terminale), non spécifiques de la lignée B sont présents dans presque tous les cas de LAL-B .110, 111
Les précurseurs B très immatures sont caractérisés par l’absence d’Ig, que ce soit en surface (sIgM) ou en intracytoplasmique (cμ) et se divisent en deux catégories, les lymphoblastes pro-B (LAL pro-B ou de type I) n’exprimant pas l’Ag CD10 et les blastes pré-pré B (LAL pré-pré B ou de type II) qui l’expriment. Les lymphoblastes pré-B (LAL pré B ou de type III) restent CD10 positifs et acquièrent l’expression de la chaîne lourde μ intracytoplasmique. Enfin, les cellules B matures (LAL-B matures ou de type IV) sont définies par
l’expression en surface d’une Ig complète. La LAL-3 de type Burkitt (classification FAB) 110 (ou lymphome / leucémie de Burkitt selon la classification OMS 110, 112) est une entité particulière où le phénotypage ne correspond à aucun stade physiologique de la maturation d’une cellule B, co-exprimant le CD10 et l’IgM de surface.110
LAL-T 110 :
Pour les LAL-T (Tableau III), les marqueurs pan-T utilisés sont les CD2, CD5, CD7 et le CD3 intra cytoplasmique. Les molécules rigoureusement spécifiques de la lignée T, le récepteur T (TCR) et le CD3, dont l’apparition à la surface des cellules est assez tardive dans la différenciation, ne sont le plus souvent pas retrouvées à la surface des cellules leucémiques. L’Ag cortical thymique, CD1a, et les Ag des sous-populations T, CD4 et CD8 peuvent être utilisés pour classer les différents précurseurs des LAL-T : les cellules blastiques T très immatures CD34+, CD1a–, sCD3–, CD4– et CD8– (LAL pro-T ou de type I) ; les cellules immatures de même phénotype mais CD34– (LAL pré-T ou de type II) ; les thymocytes communs CD1a+, sCD3–, CD4+ et CD8+ (LAL-T commune ou de type III) ; et les cellules T matures CD1a–, CD3+, CD4+ ou CD8+ (LAL-T mature ou de type IV).
Tableau II : Caractéristiques phénotypiques des leucémies aiguës lymphoïdes B .110
cCD79a : CD79a intracytoplasmique ; μ : chaîne μ intracytoplasmique ; sIgM : immunoglobuline M exprimée en surface.
Tableau III : Caractéristiques phénotypiques des leucémies aiguës lymphoïdes T.110
7-1-3 Classification cytogénétique :
La nouvelle classification des leucémies aiguës (LA) proposée par l’Organisation mondiale de la santé (OMS) 8,112,113
intègre des données génétiques et cliniques aux données morphologiques et immunophénotypiques déjà utilisées dans les précédentes classifications du groupe French-American-British (FAB) et du European Group for the Immunological Characterization of Leukemias (EGIL). Ainsi sont prises en considération la valeur pronostique de certaines anomalies génétiques, qui est désormais bien établie, et l’existence d’un traitement cytotoxique antérieur ou d’un antécédent de myélodysplasie qui conditionne le devenir de l’affection. Des entités ayant des caractéristiques morphologiques, immunophénotypiques, génétiques et cliniques particulières ont ainsi été définies. Pour les cas qui ne répondent à ces critères, le recours aux classifications actuellement en cours (FAB et EGIL) est nécessaire .8
Les leucémies aiguës lymphoblastiques (LAL) comprennent deux grandes catégories :
- Leucémies aiguës lymphoïdes avec anomalies génétiques récurrentes. - Leucémies aiguës lymphoïdes sans anomalies génétiques significatives.
Classification des leucémies aiguës lymphoïdes sur la présence d’anomalies génétiques récurrentes :
Leucémies aiguës lymphoïdes hyperdiploïdes à plus de 50 chromosomes (entre 51 et 64) :
Leur fréquence est estimée à 20-25 % des LAL chez l’enfant (surtout entre 2 et 10 ans) et à 4-9 % chez l’adulte. Il s’agit le plus souvent de LAL communes (BII) non hyperleucocytaires, dont le bon pronostic chez l’enfant est maintenant établi.8
Leucémies aiguës lymphoïdes hypodiploïdes à moins de 45 chromosomes :
Leur fréquence est estimée à 5 % environ, aussi bien chez l’enfant que chez l’adulte. Il s’agit habituellement de LAL B communes (BII) mais parfois aussi de LAL T dans 20 % des cas.8
Leucémies aiguës lymphoïdes B avec t (12; 21) (p13; q22) :
Cette forme est fréquente chez l’enfant. Elle concerne en effet 25 % des cas de LAL B de l’enfant, alors que les cas rapportés chez l’adulte sont très rares. Il s’agit le plus souvent de LAL communes (BII) avec un marqueur myéloïde CD13 ou CD33 fréquemment associé. 8(figure 12)
Leucémies aiguës lymphoïdes B avec t (1; 19) (q23; p13.3) :
Elle est mise en évidence dans 25 % des LAL pré B (BIII) ; elle représente 6 % des cas de LAL B chez l’enfant et environ 3 % chez l’adulte. 8
Leucémies aiguës lymphoïdes B avec t (9 ; 22) (q34;q11.2) :
Cette forme est fréquente chez l’adulte, chez qui elle représente environ 30 % des cas de LAL, alors qu’elle est rare chez l’enfant (de 3 à 4 %). Il s’agit le plus souvent de LAL communes (BII), parfois pré-B (BIII) ou plus rarement
pro-B (BI) dont les blastes coexpriment, dans plus de la moitié des cas, un ou plusieurs marqueurs myéloïdes (LAL B My +).8
Leucémies aiguës lymphoïdes B avec anomalies du gène MLL (11q23) :
Sa fréquence chez l’enfant est estimée à 2-3 %, mais elle constitue 60 % environ des LAL B de l’enfant de moins de 1 an. Ces patients sont souvent hyperleucocytaires et le profil immunophénotypique est de type pro-B (BI) avec un marqueur myéloïde, CD15 et/ou CD65. La survie des patients est significativement plus courte que dans les LAL B sans anomalie du gène MLL.8
Leucémies aiguës lymphoïdes B avec t (8 ; 14) (q24 ; q32) :
La translocation (8;14)(q24;q32) et ses rares formes « variantes », t(2;8)(p13;q24) et t(8;22)(q24;q11) sont retrouvées dans le lymphome de Burkitt et les LAL type Burkitt c’est à- dire les LAL3 de la classification FAB qui sont des LAL « matures » exprimant les immunoglobulines de surface ; elle représente 5 % des LAL de l’adulte ou de l’enfant .97
Leucémies aiguës lymphoïdes T avec t (5; 14) (q35; q32) :
Cette translocation est associée à l’expression anormale du gène Hox11L2. Cette anomalie est présente dans 25 % des LAL T de l’enfant et semble associée, dans les formes hyperleucocytaires, à un plus mauvais pronostic que celui des LAL T Hox11L2 négatifs. Cette catégorie, décrite récemment, ne figure pas dans la classification OMS.8, 114, 115
Leucémies aiguës lymphoïdes sans anomalies génétiques significatives :
D’autres anomalies cytogénétiques sont mises en évidence dans les LAL, mais elles ne sont pas associées à des entités particulières. Il existe en outre des LAL B ou T pour lesquelles aucune anomalie génétique n’est actuellement détectée. 8 (figure 13)
Figure 12 : Leucémie aiguë lymphoïde B (BII) avec t (12;21) (analyse moléculaire effectuée par le Dr E Delabesse, laboratoire d’hématologie, hôpital Necker-Enfants
Malades, 75743 Paris cedex 15). 8
Frottis médullaire : blastes de petite taille, faible extension cytoplasmique, contour nucléaire régulier ou encoché, chromatine fine
Figure 13 : Leucémie aiguë lymphoïde T (IV, TCR ab+), pas d’anomalies décelées sur le caryotype (caryotype effectué par I Radford-Weiss, laboratoire de cytogénétique,
hôpital Necker-Enfants Malades, 75743 Paris cedex 15). 8
Frottis médullaire : blastes de taille petite à moyenne, cytoplasme basophile, contour nucléaire régulier, chromatine fine, nucléoles visibles ;
7-2 Leucémie aiguë myéloïde :
7-2-1 Classification cytologique :
La leucémie est classée selon l’une des catégories cytologiques décrites par le groupe FAB (tableau IV), en fonction du degré de différenciation, de l’implication d’un contingent monoblastique ou monocytaire, de l’atteinte des lignées érythroblastiques ou mégacaryocytaires. La présence de granulations spécifiques sous forme d’un corps d’Auer (figure 14) et/ou la positivité de la réaction cytochimique identifiant une activité peroxydasique dans les myéloblastes permettent le diagnostic .116
Tableau IV : Classification cytologique des leucémies aiguës myéloïdes 116
Figure 14 : Myéloblastes présentant un bâtonnet d’Auer, spécifique de la leucémie aiguë myéloïde. 116
7-2-2 Classification immunophénotypique :
Les caractéristiques immunophénotypiques de LAM ont permis de mieux caractériser les leucémies d’aspect indifférencié et de « reconnaître » les leucémies biphénotypiques ou biclonales.8
Les anticorps fluorescents permettent parfois de mieux caractériser certaines formes de myéloblastes indifférenciés, par la présence d’une positivité de la myéloperoxydase, par l’expression de certains antigènes membranaires des lignées granuleuses, monocytaires, mégacaryocytaires, des marqueurs d’immaturité tels que le CD34, antigène porté par la cellule souche hématopoïétique.116 (Tableau V)
Leucémie aiguë myéloïde peu différenciée (FAB : LAM-0) : 110
La caractérisation immunologique des blastes prend un intérêt majeur, puisqu’il s’agit de cellules indifférenciées, myéloperoxydases (MPO) négatives en intra- cytoplasmique, et pour lesquelles l’étude cytologique et cytochimique