ERADication of EDEM1 occurs by selective autophagy and requires deglycosylation by cytoplasmic peptide N -glycanase
17
0
0
Texte intégral
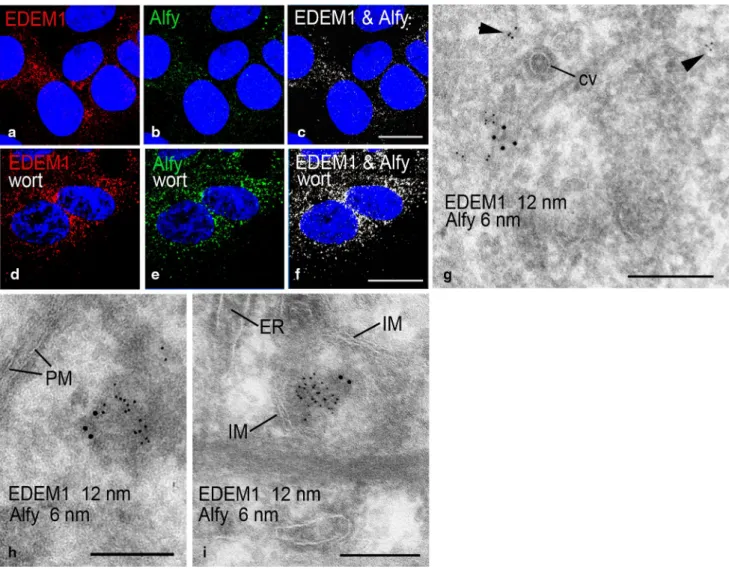
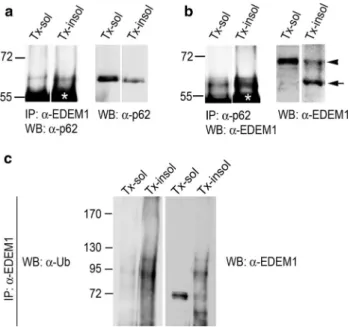
Figure
+5


Documents relatifs