Aneuploidy causes proteotoxic stress in
Saccharomyces cerevisiae.
By
Ana Belen Oromendia
B.S. Biochemistry
University of Minnesota- Twin Cities
ARtCHNEU
MASSACHUSCE ETTYftg
TJUN
3
0 2014
LIBRA RIES
SUBMITTED TO THE DEPARTMENT OF BIOLOGY IN PARTIAL FULLFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF
DOCTOR OF PHILOSOPHY IN BIOLOGY AT THE
MASSACHUSSETTS INSTITUTE OF TECHNOLOGY JUNE 2014
( Ana B. Oromendia. All rights reserved.
The author hereby grants to MIT permission to reproduce and to distribute publically paper and electronic copies of this thesis document in whole or in part in
any medium now know or hereafter created
Signature of author:
Signature redacted
Certified by: Accepted by:
I
Signature redacted
"' I A1Signature redact
Department of Biology June, 2014 Angelika Amon Professor of Biology Thesis Supervisord
Michael Laub Professor of Biology Chair, Committee for Graduate Students, Microbiology Graduate ProgramAneuploidy causes proteotoxic stress in
Saccharomyces cerevisiae.
By
Ana Belen Oromendia
Submitted to the Department of Biology on May 1", 2014 in Partial Fulfillment of the
Requirements for the Degree of Doctor of Philosophy in Biology
ABSTRACT
Gains or losses of entire chromosomes lead to aneuploidy, a condition tolerated poorly in all eukaryotes analyzed to date. How aneuploidy affects organismal and cellular physiology is only beginning to be understood. Aneuploidy also has a profound impact on human health; it is the leading cause of mental retardation and spontaneous abortions and a key characteristic of cancer, as more than 90% of all solid human tumors have aneuploid genomes. Systematic analyses of aneuploid yeast and mouse cells suggested that aneuploidy causes chromosome-specific effects elicited by the amplification of specific genes and general aneuploidy-associated phenotypes Here I describe a phenotype that is shared by most if not all aneuploid yeast cells- I find that aneuploid budding yeast cells are under proteotoxic stress. I show that aneuploid strains are prone to aggregation of endogenous proteins as well as of ectopically expressed hard to fold proteins such as polyQ stretch-containing proteins. Prion conversion rates are also increased in most aneuploid yeast strains. Protein aggregate formation in aneuploid yeast strains is likely due to limiting protein quality control systems, since I present data showing that at least one chaperone family, Hsp90, is compromised in many aneuploid strains. The link between aneuploidy and the formation and persistence of protein aggregates has important implications for diseases such as cancer and neurodegeneration.
Thesis Supervisor: Angelika Amon Title: Professor of Biology
This thesis is dedicated with much love and admiration to Ana Maria Vigliocco.
"If you are not lost you are at a place that someone else has aheady found..."
Acknowledgements
During the course of this thesis, many people have been invaluable in their support, advice and encouragement. First and foremost, I would like to thank my advisor Angelika Amon. Angelika: it has been a privilege to learn how to think about science from you- I couldn't have asked for a better scientific role model. The Amon Lab was an amazing place to learn how to be a scientist and I will take away with me many memories forged in the old CCR and the KI building. Luke, Elcin, Matt, Leon, Folkert and Stefano were an invaluable source of knowledge and technical expertise and always willing to discuss data when I needed a sounding board. I would especially like to thank Jeremy and Michelle for teaching me, and Stacie, Megan and Juliann for teaching me how to teach. Sarah was ever so patient in helping me learn how to work with mammalian cells and scientific discussions with her are some of my greatest memories. I'm thankful for the many fun times and wonderful friendships I forged with Michelle, Sarah, Kristin, Stacie, Megan, Elcin, Luke Matt and Jeremy.
A huge thank you goes to my committee members: Frank Solomon and Susan Lindquist. Your input and support was greatly appreciated. I would also like to thank Randy King for participating in my defense.
I am incredibly grateful to David Schauer and Alan Grossman for starting the Interdepartmental Microbiology Graduate Program
@
MIT and for including me in the founding class. Their dedication to the program, and to my success while at MIT had no bounds. I would especially like to thank Frank Solomon and Alan Grossman for their continuous encouragement- thank you for being straightforward and honest and for believing in me every step of the way; I cannot explain how much it has meant to me.A very special thank you goes to the Massachusetts General Hospital and the wonderful doctors and nurses there, in particular Dr. Christopher Oglivy and Dr. Patricia Musolino. I truly could not have done this without you.
The support I have received from many friends during the last years both in the form of lengthy conversations and late night drinks has been invaluable. I am better for having had you in my life. Heather, Marina, Caro, Meche, Jordan and Cristian- thank you for being my family in Boston. I would especially like to thank the MIT Micro dudes:Ben and Tyler, who have been here with me from the very beginning. Saydi, there are no words for how much your love and support has buoyed me through, thank you for always being my #1 cheerleader!
Finally, I would like to thank my family. You, and your unrelenting support and love that has no bounds means more than I can ever explain. I thank you for encouraging the curiosity and creativity that has led me to pursue science. To my siblings Mercedes, Clara, Milagros and Manuel- you are my best friends, and your unwavering encouragement has kept me going all these years. To my mom and dad: I am so incredibly grateful for all the sacrifices you have made to give us choices; thank you for always being on my side and encouraging my dreams. To Ana V: you inspire me as a scientist and as a person- I hope that when I grow up I can be half the person you are.
Table of contents
ABSTRACT 2
ACKNOWLEDGEMENTS 4
TABLE OF CONTENTS 5
CHAPTER 1: 7
ANEUPLOIDY DISRUPTS CELLULAR BALANCE 8
Genome maintenance 8
Comparison between aneuploidy and polyploidy 9
Origins of whole-chromosome aneuploidy 9
Saccharomyces cerevisiae models of aneuploidy 14
Cellular consequences of aneuploidy 19
Aneuploidy results in reduced proliferation 21
Transcriptional response to aneuploidy 22
Aneuploidy results in proteome alterations 23
PROTEIN QUALITY CONTROL MAINTAINS THE PROTEOME 29
Protein Folding 29
Controlling Protein Aggregation 35
Protein Degradation 36
Cellular responses to acute proteotoxic stressors 37
ANEUPLOIDY, PROTEIN QUALITY CONTROL AND DISEASE 38
Aneuploidy in Cancer 39
Whole-organism aneuploidy 40
Aneuploidy and Neurodegeneration 43
Aneuploidy and aging 43
Concluding Remarks 44
References 46
CHAPTER 2: 50
Introduction 51
Results 53
Disomic yeast strains harbor a higher load of endogenous protein aggregates. 53
Adaptation to proteotoxic stress is delayed in disomic yeast strains. 57
Meiotic and mitotic chromosome mis-segregation leads to protein aggregate formation. 68 Aneuploid strains fail to efficiently fold the protein quality control sensor VHL. 71
Hsp90 folding capacity is reduced in many disomic yeast strains. 76
Aneuploid strains are more susceptible to protein aggregates associated with human
disease. 80
Discussion 87
Why are aneuploid cells aggregate-prone? 88
Aneuploidy in cancer and neurodegenerative diseases. 90
Materials and Methods 91
Strains used in this study. All straisn are of the W303 background 97
References 107
CHAPTER 3: 111
Summary of key conclusions 112
Aneuploidy exhausts the cell's protein quality control capacity 114 Why are aneuploid cells aggregate-prone? 114 The folding capacity of chaperones is altered by genomic imbalances 118
Aneuploidy is a chronic stress, distinct form environmental proteotoxic stressors 119
The composition of protein aggregates in aneuploid yeast 122
Aneuploidy in mammalian cells alters protein quality control 127
Interface between aneuploidy, aging and neurodegeneration 129
Chapter 1:
Introduction
Sections of this introduction have been reproduced with permission from DMM
Oromendia, A and Amon, A 'A neuploidy: implications for protein
homeostasis
and disease' DMM, in press 2013Homeostasis is at the crux of biology. Cells must maintain their karyotipic integrity and, at the same time, ensure the maintenance of their proteome even when faced by stressful growth conditions. I have found that the disruption of a balanced karyotype, i.e. aneuploidy results in a disruption in protein homeostasis. This Introduction will expand first on the consequences of aneuploidy, then on the cellular mechanisms that maintain protein homeostasis and finally explore the interactions they share in the context of human disease.
ANEUPLOIDY DISRUPTS CELLULAR BALANCE
Genome maintenance
The maintenance of stable karyotype, i.e. number and identity of chromosomes, is essential to the success of all species. Species exist with varying chromosomal copies, from haploid (1 copy of each chromosome) to the most common diploid (2 copies) but some plant species can have up to 12 copies of each chromosome. Regardless of ploidy, all organisms carry an equal number of each chromosome ensuring a balanced genome in which genes encoded on different chromosomes are present in the same number of copies. It is this balance that gets disrupted in aneuploid cells.
Aneuploidy, defined as a karyotype that is not a whole multiple of the genomic complement results in an 'unbalanced' genome in which chromosomes(s), or pieces of chromosome(s) are missing or supernumerary and thus genes present on different chromosomes are present in varying copy numbers. Several studies have now shown that gene copy number is well correlated with gene expression and, for the most part, well correlated with protein abundance- an imbalance in copy number results in an imbalance of gene products that aneuploid cells are burdened with. Aneuploidy is generally not well
tolerated in nature, giving rise to developmental abnormalities of aneuploid organisms and the impaired fitness of aneuploid cells in all species studied to date (reviewed in (Williams and Amon 2009, Torres, 2008).
Comparison between aneuploidy and polyploidy
Whereas aneuploidy results in an unbalanced, abnormal number of chromosomes and is poorly tolerated in nature, polyploidy does not. Polyploidy is a condition in which cells contain a non-cognate, but balanced number of chromosomes- i.e. cells that of a species that normally maintains a 2n karyotype being tetraploid (4n). Since the relative ratio between gene products is maintained, there is no imbalance for the cell to contend with. Polyploidy, to a degree, is well-tolerated and there are many well documented cases of cells intentionally becoming polyploid to perform their function, such as human megakaryocytes and Drosophila
melanogaster
salivary gland cells (Lacroix and Maddox 2012). It is clear thatwhile there is an optimal karyotype that each species has evolved to have, modifications that alter chromosome number but maintain genomic balance are far less detrimental than those that generate genomic imbalanceby altering the copy number of only a subset of chromosomes.
Origins of whole-chromosome aneuploidy
During the course of cell division cells must replicate their DNA and then segregate it equally so that each daughter cell maintains the same chromosomal content as the mother cell. The cell employs a number of mechanisms to ensure that chromosome segregation has occurred before cell division concludes. The process of chromosome segregation begins when the replicated sister chromatids are linked via cohesin molecules. During prophase,
each pair of sister chromatids forms attachments to the mitotic spindle so that each chromatid's kinetochore is attached to opposing spindle poles via microtubules.
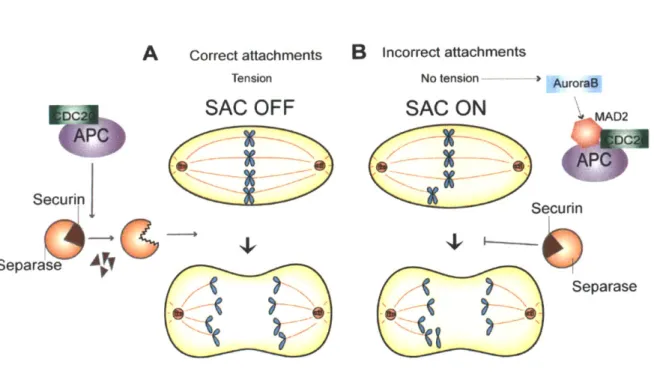
In metaphase, sister chromatids are attached to opposing spindle poles and under tension from pulling forces of rnicrotubules and cohesin molecules holding them together; they are said to be bi-oriented. For accurate chromosome segregation, it is essential to prevent cell cycle progression until all sister chromatid pairs are bi-oriented. The Spindle Assembly Checkpoint (SAC) monitors chromatid attachment and tension and halts the cell cycle until all sister chromatids are properly attached to the mitotic spindle. Once all of the chromatids are appropriately attached, Separase cleaves the cohesin molecules and allows the pulling microtubules to segregate individual chromatids to opposing poles (Figure 1).
Figure 1: The Spindle Assembly Checkpoint (SAC) ensures accurate chromosome segregation
(a) Cohesion between sister chromatids is retained through metaphase until all attachments to the spindle have been properly made. At the metaphase to anaphase transition, APC-CDC20 stimulates the degradation of the inhibitory protein Securin, the degradation of Securin frees Separase to cleave Cohesin. As the chromatids are attached to opposite spindle poles and under tension, they move away from the metaphase plate as the spindle elongates. (b) When chromosomes are not attached, or improperly attached to the spindle, there is a lack of tension. It is this lack of tension, detected, in part, by the kinase Aurora B that activates the Spindle Assembly Checkpoint. MAD2, along with other players, prevents the ubiquitination of Securin by the APC-CDC20. Securin maintains Separase inactive, pausing cell cycle progression. Bypass of the SAC can lead to progression through the cell cycle with improper chromosome attachments resulting in aneuploidy.
Figure
1
A
Securi Separase r Correct attachments TensionSAC OFF
...
_...
B
incorrect attachments No tension - -' AuroraBSAC
ON
MAD2 CDXSecurin SeparaseCompromised SAC function or mis-regulated Separase activity invariably leads to whole-chromosome aneuploidy because the cell cycle is not arrested in cells with unattached or mis-attached chromosomes (Figure 2a). Defects in chromatid cohesion also result in aneuploidy- each chromatid can segregate as it attaches to a microtubule, resulting in almost random chromosome segregation (Figure 2b). Chromatids can also form aberrant kinetochore attachments that are difficult for the SAC to detect. Merotely, when a single sister chromatid kinetochore is attached to microtubules from both spindle poles, is especially difficult to detect as there is still ongoing tension. Often, these resolve by anaphase and do not result in aneuploidy (Thompson and Compton 2008) (Thompson and Compton 2011) but when unequal merotelic attachments occur (kinetochore attached to more microtubules emanating from one pole than from the other), aneuploidy is thought to ensue
Errors in chromosome segregation in meiosis result in the creation of aneuploid
gametes, which can then lead to whole-organism aneuploidy. In mejosis, DNA replication is followed by two rounds of chromosome segregation: first, in Meiosis I homologous chromosomes segregate away from each other, and in Meiosis II sister chromatids segregate. In order to accomplish these orchestrated segregation events, cells have altered the canonical, mitotic chromosome segregation program. To properly segregate homologues, chromosomes undergo crossover events that physically link homologous chromosomes and allow them to align at the Meiosis I metaphase plate, both sister kinetochores must also coorient and attach to the same pole. Additionally, cohesion is lost in a stepwise manner, with arm cohesion being lost first, to allow for homologue segregation in anaphase I and centromere cohesion lost at a later stage to allow for sister chromatid segregation at anaphase II. In metaphase II, sister kinetochores must bi-orient and attach to opposing poles for sister chromatids to segregate to either pole (reviewed in (Miller et al. 2013)). Failure in any of several meiotic chromosome segregation events can lead to mis-segregation, including premature sister chromatid separation, failure to establish crossovers between homologous chromosomes in Meiosis I and various chromosome attachment defects in either Meiosis I
or Meiosis II (Figure 2d).
Errors in chromosome segregation can arise via many different means, and understanding the consequences of these events on cellular physiology is of critical importance. Aneuploidy has been shown to have severe consequences and to be detrimental in most cases studied to date.
FIGURE 2: Whole chromosome aneuploidy arises through errors in mitosis or meiosis (adapted from
J.
Siegel and Amon 2011)Cells missegregate chromosomes in mitosis by: (a) mutations in the Spindle Assembly Checkpoint (SAC) in which mis-attached kinetochores do not trigger a cell-cycle arrest, (b) premature loss of sister chromatid cohesion where sister chromatids attach to spindle poles and segregate randomly, and (c) merotelic attachments in which a single kinetochore attaches to microtubules emanating from both poles. (e) Aneuploidy can also arise from errors in chromosome segregation in either Meiosis I or Meiosis II.
Figure 2
A
Spindle AssemblyB
Pre-mature Loss of C Aberrant KinetochoreCheckpoint Mutations Chromatid Cohesion Attachments
[ED
E
IIC
-cD
* 1
.
Lurin eiosis~ Meiotic Segregation d 0 Errors mis-segregation durin meiosis i'
EN
[NxSaccharomyces cerevisiae models of aneuploidy
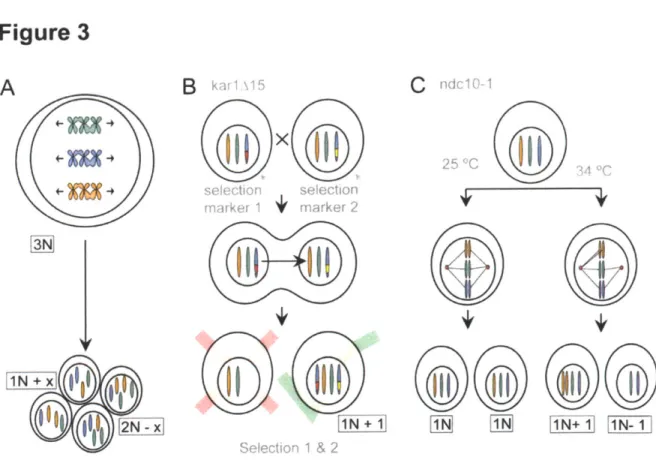
In this thesis I have used aneuploid Saccharomyces cerevisiae strains of various karyotypes generated via three different methods (Figure 3). I generated highly aneuploid, highly genomically unstable strains via triploid meiosis and using mutants that readily mis-segregate chromosomes during mitosis. Additionally, I used a set of stably aneuploid strains that carry one extra chromosome that were generated by direct chromosome transfer. Using this wide panel of aneuploid strains, I was able to ensure that the phenotypes observed are not due to any particular karyotype nor to the method via which they were constructed; I am confident that the phenotypes observed in the majority of the strains are consequences of being aneuploid.
Saccharomyces cerevisiae strains that carry large, random, genomic imbalances were created by inducing missegregation of chromosomes either in meiosis or in mitosis. I created a triploid strain (3n, genotype a/a/a), induced it to undergo meiosis via starvation and recovered the meiotic products (Figure 3a). Triploid cells induced to undergo meiosis produce highly aneuploid progeny, with karyotypes ranging from diploid to highly aneuploid (St Charles et al. 2010). The majority of the aneuploid progeny is inviable (Parry and Cox 1970), but some genetically unstable aneuploid strains can be obtained (Pavelka et al. 2010b) (Sheltzer et al. 2011) (Zhu et al. 2012). As colony formation is a prerequisite for the recovery of these strains, the aneuploidies that cause severe growth defects and do not form colonies will not be analyzed. This approach to generating aneuploid strains is beneficial in that it rapidly allows one to generate a pool of strains with high karyotype variability, as these cells are highly unstable, one is limited to colony formation or single cell assays and must take into account that the analysis will be biased towards 'healthier' aneuploidies that do not impinge greatly on colony formation or growth.
Figure 3: Generating aneuploid Saccharomyces cerevisiae strains (Adapted from (Siegel and Amon 2012))
Triploid strains induced to undergo meiosis produce highly aneuploid progeny (a). Using the abortive matings of the karyogamy defective karl,15 strain, aneuploid strains can be generated by single chromosome transfer and selection using markers placed at the same locus on both chromosomes (b). Mitotic chromosome mis-seggregation can be induced by
shifting strains carrying temperature sensitive alleles of Iptl or Ndc1O (c)
Figure 3
N{
_B
k; 15 selection~ selecion marker 1 marker 2 Seieotionr 1 & 2C
1
CN0i N[A
25 "COne can also generate random aneuploidies by inducing chromosome missegregation during mitosis (Figure 3c). Strains harboring temperature-sensitive alleles of genes encoding the kinetochore component Ndc10 or the SAC component Aurora B kinase, Ipli can be arrested in G1 under permissive growth conditions, and induced to mis-segregate chromosomes by shifting them to semi-permissive growth conditions. This treatment results in dramatic chromosome mis-segregation, with 29-35% of cells being unable to correctly segregate a chromosome that is marked by integrating a tandem array of tetO sequences. As these strains also carry a TetR-green fluorescent protein (GFP) fusion, one can visualize the tetR arrays and by extension, track chromosome segregation (GFP-dots) (Oromendia et al. 2012). As with aneuploid strains generated by meiotic chromosome mis-segregation, these strains are highly unstable and are best employed for single cell assays or genetic synthetic interaction analysis with other mutant strains.
In order to more carefully characterize aneuploidy and perform population based assays, our lab developed a set of haploid yeast that carry an extra copy of one additional chromosome ((Torres et al. 2007), Figure 3b); these strains have an n+1 karyotype and will be referred to as disomes in this thesis. These disomic yeast strains with defined karyotypes were generated via chromosome transfer from a donor cell to a recipient cell (Figure 4). Disomic strains are low-complexity aneuploidies (only carrying one supernumerary chromosome) but, by adding selectable markers at the same locus in both copies of the disomic chromosome, one can use double selection methods to ensure a stably propagating, pure population of an aneuploid strain with a defined karyotype. These strains have proven to be invaluable in understanding the effects of aneuploidy on cellular physiology, but due to the method in which they are generated one can only create low-complexity (one or two extra chromosomes) aneuploidies.
To comprehensively study the effects of aneuploidy on cellular physiology, I have generated aneuploid strains in various different manners. I used strains that carry stable, low-complexity aneuploidies and unstable high-low-complexity aneuploid strains, strains resulting from mitotic or mitotic chromosome mis-segregation and strains that can be maintained as aneuploid via selection. Using this wide panel of aneuploidies I hope to elucidate the general consequences that aneuploidy has on a cell.
Figure 4: Generating aneuploid strains via failed karyogamy matings (Adapted from Torres, et al 2007)
Strains carrying extra chromosome were generated by a chromosome transfer strategy described by Hugerat et al. (Hugerat and Simchen 1993) A HIS3 cassette is integrated at a particular location on each chromosome using the PCR-based method described by Longetine et al. (Longtine et al. 1998) The strain is then mated to a strain carrying the karlA15 allele, which renders the strain defective in karyogamy (STEP 1).b In addition the strain carries the cyb2-Q37E allele, which confers resistance to cycloheximide in a recessive manner. The mating mixture was then plated on medium lacking histidine and containing 3pg/ml cycloheximide to select for the marked chromosome and to select against diploids and heterokaryons. karz1l5 cells carrying the HIS3 marked chromosome were then mated to cells that carried the kanMX6 cassette at the same genomic locus where the HIS3 was integrated (STEP 2). This strain also carries the cani-100 allele, which confers resistance to canavinine in a recessive manner. Matings were performed and the mating mixture was plated on medium containing G418 and lacking histidine to select for the presence of the disome. To select against mating events the medium also contained canavanine.
Figure 4
Step I
Mata, xxx::HIS3, LYS2, CYCH2, can 1-100 Mato, karlA15, lys2-801, cyh2-Q37E
xxx-,HIS3
xxx:HIS3 Select for: CycR and -His
Step 2
Mata, xxx::HIS3, LYS2, CYCH2, can 1-100 Mata, kar1A15, lys2-801, cyh2-Q37E
xxx..HIS3 xxx::.kanMX6
xxx HIS3 xxx katpMX6 Select for: CanR, -His and KanR
Cellular consequences of aneuploidy
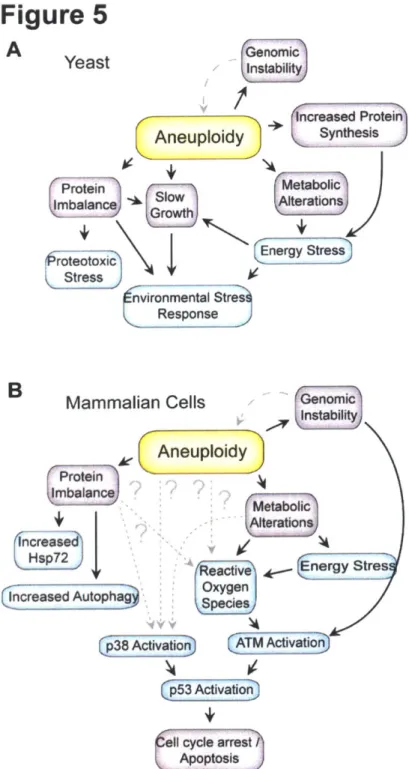
Systematic analyses of aneuploid yeast, mouse and human cells and studies on cancer cell lines suggest that aneuploidy causes chromosome-specific effects that are elicited by the increased (or decreased) number of copies of individual genes and/or combinations of a small number of genes present on the aneuploid chromosome (Tang and Amon 2013). Changes in the gene copy number of regulators of gene expression lead to further disruption of cellular function. Surprisingly, recent studies have shown that aneuploidy also causes chromosome-independent effects, which are a not a consequence of any specific gene imbalance but general consequences of harboring an unbalanced karyotype. These phenotypes include a cell cycle delay in G1 (Torres et al. 2007; Stingele et al. 2012b; Thorburn et al. 2013), metabolic alterations (Williams et al. 2008; Pavelka et al. 2010a), genomic instability (Sheltzer et al. 2011; Zhu et al. 2012) and proteotoxicity (Torres et al. 2007; Tang et al. 2011; Oromendia et al. 2012; Stingele et al. 2012b) (Figure 5). Understanding the origins of these phenotypes is important as this could provide insights into how chromosome mis-segregation and the resulting imbalanced karyotype impacts normal cell physiology and disease states. I describe a subset of these phenotypes in more detail below.
Figure 5: Observed characteristics of aneuploid cells in yeast (a) and mammalian cells (b). Adapted from (Siegel and Amon 2012). Blue boxes show observed physiological stresses and pink boxes show conditional changes resulting from aneuploidy.
Figure 5
A
Yeast
ncreased ProteinAneuploidy
Synthesis(ProteinMetabolic
CProtein rImbalance GSow Alterations
roteotoxic Energy Stress
C Stress nvironmental Stres Response R
Mammalian CE
An
increased Hsp72j
----se dAutophag Lp38 Activatio)uploidy
Metabolic Alterations,Reactive E nergy Stres Oxygin S pe cies ATM Acvatio ~iva~on
+
Insabilenomic
A neuploidy results in reduced
prolferation
Among the most prevalent and key phenotypes of aneuploid cells is their slower proliferation relative to that of euploid cells. First described in fibroblasts derived from individuals with Down's Syndrome (Segal and McCoy 1974), we now know that this phenotype is a general consequence of aneuploidy. Thorough studies in aneuploid Saccharomyces cerevisiae harboring an extra copy of one or two chromosomes (Torres et al. 2007) or derived from triploid meiosis (Pavelka et al. 2010b) and in SchiZosaccharomycespombe aneuploid cells derived from triploid meiosis (Niwa et al. 2006) showed that, irrespective of which chromosomes are present in excess, aneuploidy results in impaired proliferation. Similarly, aneuploid MEFs containing an extra copy of chromosomes 1, 13, 16 or 19
(Williams et al. 2008) exhibit proliferation defects, and MEFs derived from mice carrying a hypomorphic allele of the SAC component BUBR1 show slower proliferation than euploid MEFs at later passages when aneuploidies are allowed to accumulate (Baker et al. 2004). Aneuploid cells obtained by inducing meiotic non-disjunction, MEFs harboring mutations in SAC component Bubi, or mutations that render the checkpoint component Cdc20 non functional also exhibit proliferation defects and are outcompeted by euploid cells in growth assays (Thompson and Compton 2008; Li et al. 2009).
The slow growth phenotype of aneuploid cells has been most extensively studied in
S. cerevisiae, in which a recent study has found that aneuploidy results in an extended G1
phase and a delay into entry of the cell cycle that correlates well with the size of the supernumerary chromosome (Thorburn et al. 2013). This phenotype is dependent on the proteornic consequences of aneuploidy, as strains carrying chromosome sized human DNA
fragments that can be replicated but do not produce any protein do not display a G1 delay. Both cell growth (cell volume accumulation) and entry into the cell cycle appear to be
affected. Although most disomic yeast strains show a cell growth defect, there appear to be no gross defects in global protein synthesis as measured by polysome profiling or ['S]methionine incorporation(Thorburn et al. 2013),although the effects of aneuploidy on these processes may be too subtle to detect by these methods. The growth defect does not appear to be due to diminished amino acid pools or reduced translational efficiency. 10 out of 14 disomic strains analyzed showed a delay in cell cycle entry observed as an increase in critical size (the size at which 50% of cells in a population have budded). All of the strains
analyzed show delayed accumulation of the G1 cyclin CLN2 mRNA, and it was shown that high levels of CLN2 suppress the increase in critical size. Accumulation of Cln3, another G1 cyclin was also delayed in all disomes analyzed
(Thorburn
et al. 2013). It is yet unclear how aneuploidy interferes with the accumulation of Cln3 and whether this is a gene specific effect or a general response to aneuploidy. As it has been observed in almost all aneuploid strains, I favor the idea that the G1 delay is a general consequence of aneuploidy. Interestingly, many environmental stresses (including heat stress) have been shown to cause a transient G1 delay- it is possible that proteotoxic stress in aneuploid yeast is contributing to the G1 delay observed.Transcriptional response to aneuploidy
Several lines of evidence suggest that cells respond to the aneuploid state. Most aneuploid cells studied to date exhibit a transcriptional signature associated with slow growth and stress (Torres et al. 2007; Sheltzer et al. 2012; Stingele et al. 2012b; Foijer et al. 2013). Recent studies have shown that aneuploidy elicits a transcriptional response reminiscent of the environmental stress response (ESR) in species as divergent as budding and fission yeast, Arabidopsis thaliana, and human and mouse cell lines. The ESR consists of -300 genes that
are upregulated and ~600 genes that are downregulated by various exogenous stresses,
including heat shock or oxidative stress (Gasch et al. 2000). Most of these genes also vary in expression in response to growth rate; inducing slow proliferation by nutrient limitation mimics the ESR (Regenberg et al. 2006; Brauer et al. 2008). The high correlation between the ESR-like response seen in aneuploid cells and the transcriptional response observed in slow-growing S cerevisiae strains suggests that the transcriptional response observed in aneuploid cells is, for the most part, due to the slow proliferation observed in aneuploidy (Sheltzer et al. 2012).
A neuploidy results in proteome alterations
The unbalanced genome caused by aneuploidy has been shown to translate into an unbalanced proteome - that is to say that the changes in gene dosage for the most part result in equivalent changes in protein levels (twice as much DNA results in twice as much protein, Figure 5). Studies of Saccharomyces cerevisiae aneuploid strains show that the abundance of approximately 80 percent of proteins changes in proportion to gene copy number (Pavelka et al. 2010b; Torres et al. 2010). Interestingly, many of the proteins for which this is not true are subunits of multimeric complexes (Torres et al. 2007). Indeed, often times, subunits that are endogenously expressed in excess because of aneuploidy retain stoichiometric numbers within multimeric complexes (Torres et al. 2007). Stingele and colleagues showed that this is also true in human aneuploid cells {Stingele, 2012 #1121; Torres et al. 2010). Analysis of the transcriptome and proteome of aneuploid human cells generated by chromosome transfer showed that most genes are expressed according to their copy number, and proteins are translated in strong correlation with the abundance of mRNA, resulting in a dramatic change in cellular protein composition (Stingele, 2012 a).
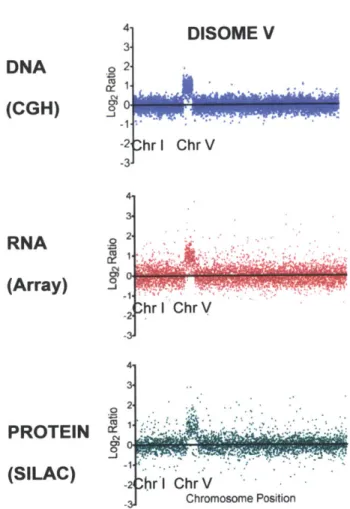
Figure 6: DNA, mRNA and protein levels in yeast disomic for chromosome V (Data from Torres et al 2007).
Disomic S. cerevisiae strains carry an active, replicating chromosome in one additional copy as evidenced by comparative genome hybridization (CGH, top panel). The extra chromosome is transcribed, as seen by the two-fold increase in mRNA present form that chromosome (microarray, middle panel). The majority of the proteins encoded by the chromosome are also expressed and can be found at close to 2 fold higher levels than those encoded by other chromosomes (SILAC, bottom panel)
Figure 6
4-DISOME V
3-DNA
o 2 .(CGH)
(Aray)
-1 -2 !hr i Chr V -3 4-3(RNA
-2 shr I Chr V 4.PROTEIN 20
(SILAC)
1t
-2 br l ChrV SChromosome PositionHowever, as in aneuploid yeast, human aneuploid cells were also found to maintain a subset of proteins (enriched for complex subunits) at stoichiometric levels even if gene copy number was altered. The regulatory mechanisms responsible for this correcting process have not been elucidated. Overall, these data suggest that, although some proteins are maintained at stoichiometric levels, there is no general whole-chromosome 'gene dosage compensation' mechanism for autosomes in yeast and mammals, as has been observed for sex chromosomes. This might not be the case in all organisms, however. Aneuploid
Drosophila S2 cells have been reported to experience dosage compensation at the
transcriptional level by means of the male-specific lethal (MSL) complex and general compensation mechanisms that compensate for differences in non-autosomal chromosome copy number (Zhang et al. 2010). Further studies in Drosophila aneuploid cells are needed to determine the status of their proteome.
A key question resulting from the profound effects of aneuploidy on cellular protein
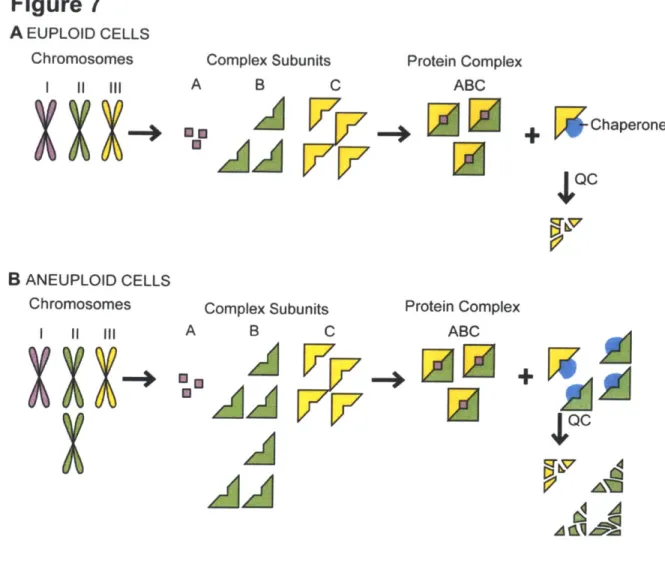
composition is whether the simultaneous changes in the relative ratios of many proteins impacts upon the protein quality-control pathways of the cell. Chaperones and the degradation machinery, the 26S proteasome, proteases and autophagy, ensure that all proteins acquire their native conformation and prevent cellular toxicity by reducing the number of aberrant interactions between proteins. In aneuploid cells, these protein quality-control systems must not only attend to the excess proteins produced from additional chromosomes, they must also support all excess subunits of complexes that are not in stoichiometric ratios with their binding partners (Figure 7).
Figure 7. Aneuploidy causes proteotoxic stress. (a) Cells use protein quality-control and feedback mechanisms to maintain subunit stoichiometries of complexes whose subunits are encoded by different chromosomes. The protein quality-control (QC) machinery ensures accurate folding and maintains complex subunits that lack a binding partner in a soluble state. Eventually, excess and misfolded subunits must be degraded, as illustrated here by the yellow
subunit that has been produced in relative excess. (b) Changes in chromosome number in aneuploid cells (shown here as disomy of the green chromosome) lead to a genomic imbalance that results in stoichiometric protein imbalances. Every subunit encoded by an unbalanced chromosome that functions in a protein complex lacks its binding partner(s) and must rely on cellular chaperones to maintain solubility and, if no binding partner is found, on the cellular proteases for its eventual degradation. This can lead to an increased burden on the protein quality-control systems and the exhaustion of the cellular protein quality-control machinery.
Figure 7
A EUPLOID CELLS Chromosomes I lAX
B ANEUPLOID CELLS Chromosomes 1 11 111 AComplex Subunits Protein Complex
B C ABC
A
W ChaperoneQC
Complex Subunits Protein Complex
B C ABC
V7~
Many protein complex subunits are unstable unless bound to their partners, and will often bind to cellular chaperones to remain soluble until they have formed the complex (Boulon et al. 2010). Several previous studies have indeed hinted to the fact that aneuploidy impacts protein quality-control systems. Budding yeast, mouse and human aneuploid cells exhibit a transcriptional signature that is reminiscent of a stress response and slow growth (Torres et al. 2007; Sheltzer et al. 2012; Stingele et al. 2012a). This transcriptional signature includes upregulation of protein chaperones (Sheltzer et al. 2012). Human aneuploidies generated by chromosome transfer were found to have a transcriptional stress signature that shows up-regulation of lysosome-mediated degradation and p62-dependent autophagy
(Stingele et al. 2012a; Stingele et al. 2013). Furthermore, many haploid S. cerevisiae strains harboring an additional chromosome (disomic yeast strains) were found to be sensitive to chemical compounds that impair protein quality control; many disomic yeast strains are sensitive to the proteasome inhibitor MG132, the ribosome poison cycloheximide and the Hsp90 inhibitors radicicol and geldanamycin (Torres et al. 2007). Mouse embryonic fibroblasts (MEFs) trisomic for any of chromosomes 1, 13, 16 or 19 are more sensitive to the Hsp90 inhibitor 17-AAG than are wild-type MEFs (Tang et al. 2011). These results can be interpreted in that that the aneuploid state causes proteotoxic stress leading aneuploid cells to rely more heavily on their protein quality control machinery. Thus, impairing chaperone function via use of chemical chaperone inhibitors is more detrimental to cells that are aneuploid than to cells that carry the appropriate number of chromosomes. This thesis directly tests this possibility.
PROTEIN QUALITY CONTROL MAINTAINS THE PROTEOME
At the core of cellular biology is the process of converting genetic information into proteins that both carry out the genetic program and provide structural integrity to the cell. The central dogma of molecular biology describes the lifecycle of each individual protein subunit. Protein coding genes are perpetuated in the genome as DNA. When necessary, the DNA is transcribed into mRNA molecules, which are then translated by the ribosome into polypeptides. In order to be functional the majority of polypeptides must acquire a well-defined three-dimensional structure (native structure) and, in many cases, bind to other protein subunits to form a functional protein complex. Once the protein is no longer necessary, it is degraded into individual amino acids that can then be recycled and used in the fabrication of new polypeptides. This process is highly dynamic and energetically costly and at the same time, is affected by almost all external cellular stressors; thus the process of maintaining protein homeostasis (or proteostasis) is one of extreme balance and precision. Protein synthesis is tightly controlled in cells, but in addition, protein folding and protein degradation play an important role in maintaining proteostasis.
Protein Folding
The information necessary to acquire the native structure is encoded in the primary amino acid sequence and thus, many proteins can fold unassisted in dilute solutions in
vitro.
In the cellular mileu where the total protein concentration can be as high as 300 mg per ml, acquiring native structure is much more challenging. Inter molecular interations are strongly favored in vivo and since folding intermediates often expose hydrophobic patches, the crowded cellular environment endangers newly synthesized proteins and unstable proteins with high propensity to misfold. Exposed hydrophobic regions constantly pose a threat and
non-productive interactions that can result in misfolding and/or aggregation compete with the formation of the native structure. Both protein misfolding and aggregation are detrimental and pose a significant burden to the cell and defects in these processes can result in human disease (Reviewed in (Young et al. 2004; Taipale et al. 2010; Tyedmers et al. 2010).
In order to maintain proteostasis and mitigate the effects of heat and other stresses on the proteome, cells have evolved a sophisticated network of protein chaperones. Protein chaperones are intricately involved in the folding and maturation of a protein - from a polypeptide exiting the ribosome acquiring the appropriate three-dimensional structure, to assembly into the appropriate complexes. Molecular chaperone proteins bind to folding intermediates, reducing the conformational space that can be explored and often times preventing aberrant interactions by sequestering hydrophobic patches. Chaperones exist in several structurally unrelated classes and have been classified into families according to their type of enzymatic activity, the co-chaperones they require and the clients that they aid in folding (Hard et al. 2011) and they are named according to their molecular size. Often times, a single polypeptide will interact with different chaperones sequentially, each aiding in a
specific aspect of protein folding or complex assembly. It is important to bear in mind that each chaperone family usually has multiple distinct members in each cellular compartment serving to both increase chaperone diversity and ensure redundancy. I will briefly discuss the specifics of the HSP90, HSP70, HSP60 (chaperonins) and small heat shock protein (sHSPs) families here (Figure 8).
Figure 8: Molecular Chaperone Mechanisms (adapted from (Richter et al. 2010)) Chaperone model: In general, proteins fold via increasingly structured intermediates (L, L) from the unfolded state (U) to the folded state (N). Protein chaperones bind proteins in nonnative conformations. The shift from the high-affinity binding state to the low-affinity release state is often triggered by ATP binding and hydrolysis. Hsp60/GroE: The GroE machinery consists of two identical rings that enclose a central cavity each. Nonnative protein is bound by the apical domains of the rings, and upon binding of ATP and the cochaperone GroES (caps), the protein is encapsulated and released into the cavity. ATP hydrolysis in one ring results in the release of GroES and substrate protein from the opposite ring. During encapsulation the protein may fold partially or completely. Hsp70: The Hsp70 system comprises two cochaperones, an activating protein (Hsp40/J-protein) and a nucleotide exchange factor (NEF). The activating protein can bind the nonnative protein and deliver it to Hsp70 forming a complex and stimulating its ATPase. The NEF will induce the exchange of nucleotide accelerating the ATPase cycle and the client protein is released Hsp90: In this chaperone system a large number of proteins work together. Often, Hsp70 delivers the substrates to Hsp90. Cochaperones (shown here in purple and yellow) modulate the system (shown here in purple and yellow). ClpB/Hsp104: This chaperone is able to dissolve aggregates by actively pulling proteins through a central channel of the hexameric structure. Refolding occurs upon release, and, to some extent, it can also occur in cooperation with other chaperones. sHsps: sHps are oligomeric complexes that are often activated, by heat or modifications. Many are believed to dissociate into smaller oligomers to become active. sHsps can bind many nonnative proteins per complex. Release requires cooperation with other ATP-dependent chaperones such as Hsp70.
Figure 8
chaperone model GroES/HspGQ Hsp7O
*rr
"Wegh% aLnt lowE affnit
Hsp9O ClpBIHsplO4 sHsp
AW~PI
Hsp9O is at the center of maintaining proteostasis, forming a hub that controls many important signaling pathways
(Figure
8, reviewed in (Tfaipale et al. 2010)). Hsp9O functions downstream of Hsp7O binding partially folded polypeptides and cooperating with many co-chaperones and regulatory subunits to ensure structural maturation. The activity of Hsp9O is ATP-dependent and closely coordinated with environmental perturbations. Hsp9O is poised to be a buffering force in protein quality control- under normal growth conditions, as it ispresent
in vast excess and can be reduced to 10% of its natural abundance without detrimental consequences to the cell (McClellan et al. 2007; Franzosa et al. 2011). Hsp9O client lists have been notoriously hard to define, perhaps because Hsp9O's essential folding roles seem to be in folding proteins that are central hubs of cellular processes such asregulatory subunits of signal transduction cascades or kinases. Recent genome wide studies have implicated Hsp90 in almost every cellular process from protein trafficking, secretion, RNA processing, signal transduction to telomere maintenance and immunity.
The constitutive and inducible forms of Hsp70 are core players in protein quality control (Figure 8, reviewed in (Richter et al. 2010)). Hsp70 (DnaK in E. coli) functions in concert with Hsp40 (DnaJ in E. coli) and nucleotide exchange factors to, in an ATP-dependent manner, aid in folding of nascent polypeptides and bind and release partially folded substrates. Hsp70 binds to substrates via small stretches of hydrophobic amino acids, exchanging rapidly in an ATP bound state and binding stably to substrates and Hsp40 after ATP hydrolysis. Rapid cycles of binding and release restrict the conformational folding space that the polypeptide is able to explore and allow for the rapid burial of hydrophobic patches that can partake in aberrant interactions and form aggregates. If the protein is not folded after interaction with the Hsp70/Hsp4O system it may be transferred to the specialized compartment of chaperonins to continue its folding trajectory.
Chaperonins are large cage-like ring complexes that function by enclosing the folding polypeptide (up to 60 kDa in size) and isolating it from all other proteins in the cell (reviewed in Hard and Hayer-Hartl 2011). Group I chaperonins, GroEL in bacteria, Hsp60 in eukaryotes are two component systems, with the barrel of the cage formed by the chaperonin and the lid being formed by GroES, in bacteria, or Hsp1O in the case of eukaryotes (Figure 8, reviewed in (Dunn et al. 2001)). Group II chaperonins, the TRiC/CCT system in eukaryotes function under the same premise, but instead of cooperating with another subunit to form a closed cage, they undergo conformational changes to enclose the structure. Chaperonins function in an ATP dependent manner, coordinating the encapsulation of the substrate with hydrolysis of the ATP molecule. The
encapsulated protein is free to fold in the chaperonin enclosure until it is released (10s in the GroEL/ES system, longer in the TrIC/CCT complex). Still unfolded substrates can re-bind and the process can be repeated until the protein has acquired its native fold or it is transferred to a different chaperone. Although chaperonins do not actively assist in folding, they have been shown to dramatically accelerate the speed of folding, probably by spatial confinement and the prevention of aberrant interactions and aggregation with other proteins. In S. cerevisiae the TrIC/CCT complex is essential for the folding of a small subset of proteins, but within these are proteins of high abundance and extreme importance in structural integrity of the cell such as actin and tubulin.
Small heat shock proteins (Figure 8, reviewed in (Richter et al. 2010)) are not as cohesive of a protein family as HSP90 or HSP70 are. sHSPs are usually monomeric proteins that bind to hydrophobic patches of amino acids. For the most part, their clients have not been well defined, but they are thought to be unstable folding intermediates and that the binding of sHSPs prevents aberrant interactions. sHSPs are thought to play a role in protein complex formation, binding to one protein subunit and occluding the binding interface (usually highly hydrophobic) until the binding partner is found and the complex is formed.
There is a vast network of proteins whose function is to ensure protein folding within the cell. Protein chaperones are both diverse and specialized, and while some assist in general folding of proteins, many have a defined subset of protein clients whose folding they aid. As protein homeostasis is a process of utmost importance, protein chaperones also maintain a large amount of redundancy, with many having obligate clients but being able to assist in folding of others if necessary. To cope with severe folding stress- many chaperones have two variants, one that is constitutively expressed at low levels and another whose expression is induced by proteotoxicity. In summary, the cell has developed a robust system
of protein folding factors to minimize aberrant interactions between proteins and ensure peptides acquire the appropriate 3-dimensional structure.
Controlling Protein Aggregation
When polypeptides cannot fold into their native structure and remain misfolded, if they partially unfold after being properly folded or if they are terminally damaged by oxidation or carbonylation, they become aggregate-prone. Assembly defects in protein complexes, as would happen when a required subunit is not expressed, can also lead to aggregation of the existing subunits as hydrophobic patches that would be buried within the complex remain exposed and form aberrant interactions. In addition to folding assistance provided by chaperones, the cell utilizes chaperones to solubilize protein aggregates and utilizes diverse mechanisms to prevent toxicity from aggregated proteins.
The Hsp10O chaperone family is comprised by members of the AAA ATPases, most notably ClpB in bacteria and Hsp104 in yeast. Both ClpB and Hsp104 have disaggregating capabilities, using ATP hydrolysis to break apart protein aggregates (Figure 8). The mechanism by which they do this is unclear, but they are thought to thread the misfolded proteins through a central pore of their hexameric ring, leaving the client protein in an unfolded state so that it can refold either on its own or assisted by the Hsp70/Hsp4O machinery (Richter et al. 2010, Mogk, 2004, Tyedmers, 2010). Although no homologues of Hsp104 have been found in higher eukaryotes, disaggregation activity has been attributed to the mammalian chaperone system comprised of Hsp110 and Hsp70/Hsp40 (Shorter 2011). Yeast cells also sequester certain types of protein aggregates, usually those that cannot be refolded, in special compartments The JUNQ (juxtanuclear quality control compartment) transiently accumulates aggregated proteins that are ubiquitinated and destined for
degradation whereas the IPOD (Insoluble protein deposit) houses insoluble terminatally aggregated proteins such as polyQ or carbonylated proteins(Kaganovich et al. 2008). When a cell is unable to disaggregate and refold aggregated proteins, degradation of the aggregated proteins is a viable alternative to alleviate toxicity.
Protein Degradation
In order to cope with alterations in protein homeostasis, cells degrade excess, misfolded and aggregated protein subunits and aberrant peptides by means of the Ubiquitin Proteasome System (UPS) or via autophagy.
The 26S proteasome is the central macromolecular machine responsible for the degradation of proteins and protein aggregates. Its functions are so essential to the cell that partially inhibiting its function can lead to neurodegeneration and complete inhibition is lethal (Bedford et al. 2008). The 26S proteasome is comprised of a core, barrel-like particle (20S subunit) and two regulatory complexes (19S) that function as lids. Proteins are recognized and targeted for degradation by E3 ubiquitin ligases that attach ubiquitin moieties. Specialized proteins that contain UBL (ubiquitin like) and UBA (ubiquitin associated) domains act as adaptors between the target protein (the ubiquitin moieties are bound by the UBA domain) and the 19S cap (binds the UBL domains). The proteins targeted for degradation are deubiquitinated, unfolded and threaded through the core particle. The recognition and binding of a substrate to the 19S cap is an ATP dependent process, and the ATP molecule is required for unfolding, but not translocation into the pore. Proteolysis occurs in the core particle through a threonine-dependent nucleophilic attack and results in short stretches of amino acids that can then be further processed by cytosolic proteases and recycled into new polypeptides.
Protein degradation is mediated not only by the proteasome but cells can additionally deploy autophagy as a means of protein quality control (Kubota 2009). Misfolded proteins are sequestered into aggregates and, in a p62-dependent manner, are targeted for autophagy. Autophagy utilizes double-membraned structures that engulph the cytosolic target proteins forming an autophagosome which then fuses with the lysosome for degradation of their content (Bukau et al 2010). A key player in autophagosome formation is the membrane
protein LC3/Atg8; upon autophagy induction, LC3 is conjugated to
phosphatidylethanolamine and recruited to the membranes of the nascent autophagosome. One of the many ways one can monitor autophagy is by assessing the number of LC3 foci, or by assaying the abundance of LC3-II, the autophagosome-specific, lipidated form of LC3.
Cellular responses to acute proteotoxic stressors
In order to maintain protein homeostasis under acute insults to proteostasis, there are transcriptional programs that cells implement when faced with abnormal quantities of misfolded proteins. These transcriptional programs are distinct according to which cellular compartment is being assaulted by protein misfolding but they are all transient, tailored to temporary stressors. The main goal of these programs is to reduce the folding burden
(by
reducing the number of polypeptides being produced) and to enhance the cell's folding capacity
(by
increasing the number of protein chaperones). The best studied is the program elicited by the general misfolding of cytosolic proteins elicited by exposure to high temperature and thus named the 'heat hock response' (HSR). High temperatures result in general protein misfolding which leads to the activation of the transcription factor HSF1 (heat shock factor 1) that then results in the up-regulation of a subset of genes enriched for protein chaperones and the down-regulation of genes involved in protein synthesis(reviewed in (Richter et al. 2010)). Hsfl is kept in an inactive complex together with components of the Hsp90 chaperone system. In a state of heat shock, the high abundance of misfolded proteins is thought to titrate away the chaperones bound to Hsfl. In complex with chaperone, Hsf1 is found as a monomer but its release leads to homotrimerization and transport into the nucleus. There, Hsf1 is hyperphosphorylated by several kinases (Holmberg et al. 2001). Further modification events, like sumoylation, regulate the activity of the final transcription factor complex (Hietakangas et al. 2003). Complex regulatory feedback ensures that the response is transient so as to return to normal levels of protein production and chaperone abundance once the proteotoxic stress has been relieved. Misfolded proteins in the endoplasmic reticulum (ER) result in a similar, but distinct response termed the Unfolded Protein Response (UPR). The UPR is also transient, and results in the up-regulation of ER specific chaperones and a general, temporary, reduction in protein synthesis (Walter and Ron 2011). Studies in mammalian cells have also recently described the mitoUPR (Mitochondria Unfolded Protein Response). Details are far less clear, but the essence of the response is the same: misfolded proteins in the mitochondria result in a signal that translates to a temporary decrease in protein production and an increase in protein quality control capacity (Haynes and Ron 2010). In summary, there are well-understood transcriptional programs that aid in coping with abrupt changes in misfolded proteins caused by disruptions of protein homeostasis.
ANEUPLOIDY, PROTEIN QUALITY CONTROL AND DISEASE
The connection between aneuploidy and disease has been at the forefront of the study of aneuploidy. David van Hansemann first described unbalanced mitoses in 1890. Theodor Boveri (1912) expanded upon his early description of aneuploid sea urchin
embryos to postulate that aneuploid cells could result in tumor formation. Aneuploidy of chromosome 21 was described as the cause of Down's syndrome by Lejeune in 1959
(Lejeune
et al. 1959). Recent studies have described associations between the aneuploid state and neurodegenerative diseases and aging. Here I expand upon the most common conditions associated with aneuploidy.Aneuploidy in Cancer
Aneuploidy is extremely prevalent in solid tumors, with 7
0-
90% estimated to havean unbalanced karyotype (Weaver and Cleveland 2006; Duijf and Benezra 2013). Cancer cells have also long been considered 'chaperone addicted' (Neckers 2002) and Hsp90 inhibitors are currently being developed as chemotherapeutics (Wagner et al. 2013). The dependency of tumors on chaperones has been attributed to the need to efficiently fold oncogene products, which are often kinases and thus Hsp90 clients. However, the high levels of aneuploidy in cancer cells, and the proteotoxic stress that stems from such aneuploidy, could provide an additional explanation for their chaperone addiction. Further investigation of compounds that increase chaperone burden or that inhibit the function of chaperones might lead to the discovery of new cancer therapeutics with efficacy in a broad spectrum of human tumors.
The high degree of aneuploidy observed in cancers also begs the question of whether cancer cells have evolved mechanisms that allow them to tolerate high levels of karyotypic imbalances. One aneuploidy-tolerating mutation appears to be loss of p53 function. In normal cells, chromosome mis-segregation leads to activation of the tumor suppressor p5 3;
the mechanisms whereby this occurs are still being elucidated and might be caused by multiple aspects of chromosome mis-segregation (Pavelka et al. 2010a; Thompson and Compton 2010; Janssen et al. 2011). Generating a comprehensive list of genetic alterations
that ameliorate the effects of aneuploidy and their characterization will shed light on tumor evolution. It will allow us to address important questions such as when such mutations arise with respect to aneuploidy and whether and how they contribute to tumorigenesis. Compounds that neutralize aneuploidy-tolerating mutations could also provide new avenues of cancer treatment.
Whole-organism aneuploidy
In addition to cancer, autosomal aneuploidy has been associated with numerous human conditions that result in impaired development. In humans, three viable trisomies have been described. An additional copy of chromosome 21 leads to Down syndrome, chromosome 18 to Edward's syndrome and a trisomy of chromosome 13 to Patau syndrome. Of these, only Down Syndrome individuals survive past childhood. It will be interesting to determine whether protein quality-control systems are affected in individuals with these constitutional aneuploidies. Chromosome 21 harbors the fewest genes of all human chromosomes and might thus not cause a significant burden on the cellular protein quality-control pathways. Determining the contribution of impaired protein homeostasis to the pleiotropic phenotypes of this syndrome could nevertheless be warranted because Down syndrome is strongly associated with a protein-folding disease. Individuals with Down syndrome are predisposed to early-onset Alzheimer's Disease (AD). Although the main cause of AD in Down syndrome individuals is likely to be the additional copy of the APP gene encoded by chromosome 21 (reviewed in (Kingsbury et al. 2006), mice overexpressing
APP
(which encodes amyloid beta A4 protein) do not fully recapitulate all the Alzheimer's-like phenotypes seen in Down syndrome mouse models (Cataldo et al. 2003). Conversely, mouse models of Down syndrome that lack theAPP
gene still exhibit some of theAlzheimer's-like pathologies (Table 1), suggesting that duplication of the A PP gene may not be the only cause of early-onset Alzheimer's disease in Down syndrome individuals. Thus, perhaps a reduced ability to maintain protein homeostasis contributes to the Alzheimer's disease pathology in individuals with Down syndrome.
Table 1. Comparison of the phenotypes associated with transgenic mouse models of
Down's syndrome or Alzheimer's disease
The two mouse models of Down syndrome are Ts65Dn and TslCje. Ts65Dn mice are trisomic for the distal region of chromosome 16 (92 genes homologous to human chromosome 21 from
APP
toMXJ);
this segment contains nearly two-thirds of the human chromosome 21 homologous genes, including the Down syndrome critical region (DSCR) and theAPP
gene. Ts65Dn mice are also trisomic for a segment of mouse chromosome 17 (60 genes) that is non-homologous to genes on human chromosome 21. TslCje mice are trisomic for a smaller region of chromosome 16 that includes the DSCR but notAPP
(67genes homologous to chromosome 21, from SODI to