Comparing quantum-chemical calculation methods for structural investigation of zeolite crystal structures by solid-state NMR spectroscopy
Texte intégral
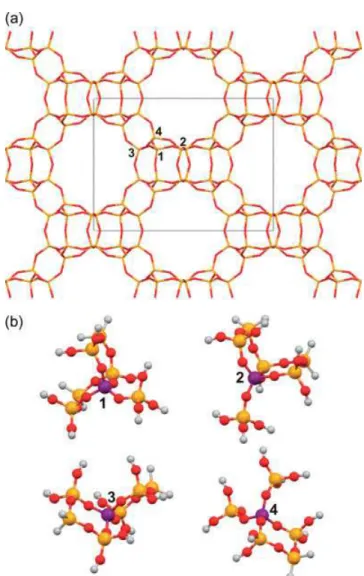
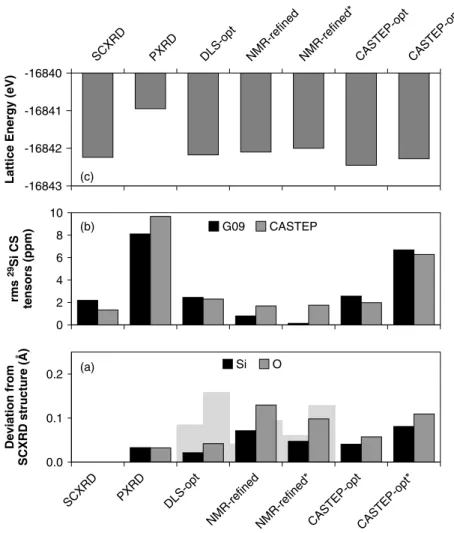
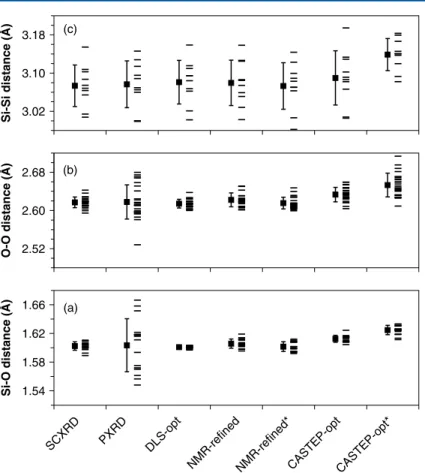
Figure


Documents relatifs
- Une lame cristalline uniaxe (( Z cut )) placee entre deux polariseurs circulaires identiques donne un reseau de franges qui est la transformke de Fourier du spectre
undoped spedmens) of constant oxygen stoichiometry (see TG results): large amounts of Cu on the Ga site (and the corresponding quantity of Ga on the Cu site) would probably lead to
describe the structure of KCdF3 by the parameter values determined from the intensity data excluding. those of R
The synthesis of a highly functionalized phenolic diaryl ether 5,5'-oxybis(4-hydroxy-3- methoxybenzaldehyde) (1) potentially interesting as new scaffold for drug design,
Inspired by Van Selm’s pioneering work, in the 1990s a number of Dutch scholars started to locate and photograph the extant Dutch library auction catalogues in
p-Type conducting transparent characteristics of delafossite Mg-doped CuCrO2thin films prepared by RF- sputtering.. Journal of Materials Chemistry C, Royal Society of Chemistry,
Comparison studies of ammonium sulfate particles showed that both instruments measured within 2 nm of classical K ¨ohler theory predictions as- suming complete solubility,
Prévoyez une démarche en étape (besoins individuels, analyse, présentation des résultats, mise en lien avec des besoins collectifs, concertation, priorisation, prise en compte
