BDNF and gp145trkB in multiple sclerosis brain lesions: neuroprotective interactions between immune and neuronal cells?
11
0
0
Texte intégral
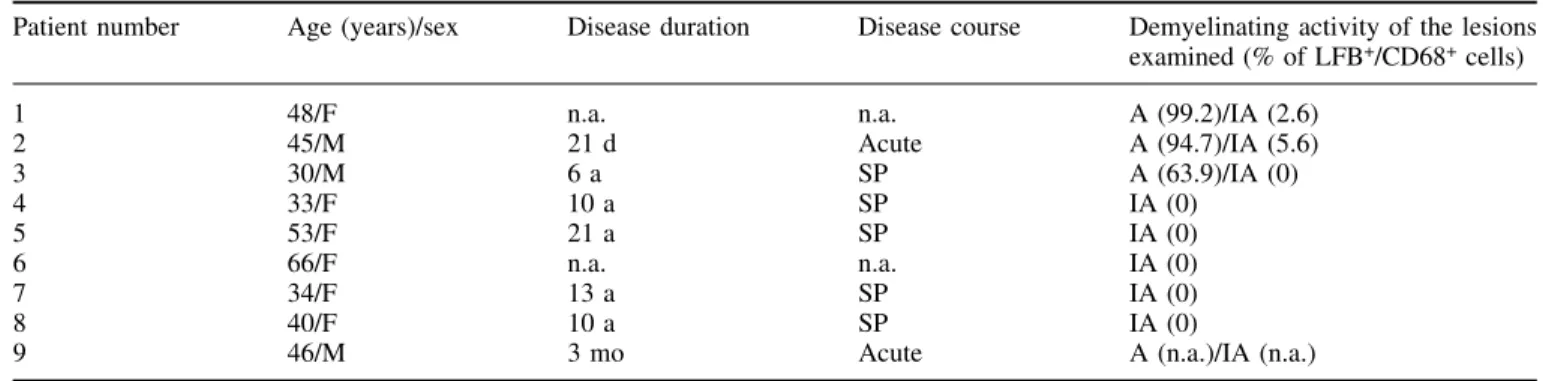
(2) 76. C. Stadelmann et al.. damage to neurones in the course of multiple sclerosis (Davie et al., 1995; Jeffery et al., 2000; Rovaris et al., 2000). In line with these ®ndings, a recent study demonstrates a pronounced loss of retinal ganglion cells in a rat model of optic neuritis (Meyer et al., 2001). However, the majority of neurones and a signi®cant number of axons seem to be preserved. It is unclear so far which factors govern the demise and preservation of neurones and their axons in in¯ammatory CNS diseases. In¯ammation is considered to be the key feature in multiple sclerosis pathogenesis (Lassmann et al., 1998). The neurotoxic properties of in¯ammation are well established and thought to be at least partially responsible for the observed axonal damage (Piani et al., 1992; Brosnan and Raine, 1996; Bitsch et al., 2000; Gimsa et al., 2000). However, a number of recent studies have proposed that autoimmune in¯ammation can have a neuroprotective role in the CNS (Moalem et al., 1999; Hammarberg et al., 2000; Hauben et al., 2000). The factors involved in immune-mediated neuroprotection remain to be clari®ed. Members of the nerve growth factor (NGF) neurotrophin family such as NGF, brain-derived neurotrophic factor (BDNF), neurotrophin (NT)-3 and NT-4/5 are important regulators of neuronal death and survival (Lewin and Barde, 1996). Besides their well established role in neuronal development, they have the capability to protect neurones against various pathological insults (Cheng and Mattson, 1991; Frim et al., 1994; Lindvall et al., 1994; Holtzman et al., 1996). Notably, BDNF can rescue CNS neurones, e.g. spinal and bulbar motor neurones, after axonal transection, and can promote axonal preservation and regeneration (Weibel et al., 1995; Gravel et al., 1997; Kobayashi et al., 1997). Traditionally, neurones have been considered the major cellular source of BDNF in the CNS (Hofer et al., 1990; Lewin and Barde, 1996). Recently, we and others have been able to demonstrate the production of substantial amounts of BDNF in immune cells (Besser and Wank, 1999; Braun et al., 1999; Kerschensteiner et al., 1999). Preliminary observations have shown that immune cell-derived BDNF can be detected in human immune-mediated CNS in¯ammation.. BDNF binds two different types of receptors (Bothwell, 1995): the tyrosine kinase receptor B (trkB), which exists in two isoformsÐthe full-length receptor (gp145trkB) and the truncated receptor (gp95trkB) lacking the tyrosine kinase domain (Klein et al., 1990, 1991)Ðand the p75 neurotrophin receptor. Most, if not all, known BDNF functions are transmitted via the full-length gp145trkB receptor (Bothwell, 1995). Furthermore, it is known that gp145trkB is upregulated after brain insults (Frisen et al., 1992; Merlio et al., 1993; Kobayashi et al., 1997). BDNF and gp145trkB, therefore, are attractive candidates for mediating neuroprotective effects in multiple sclerosis. The focus of the present study was to further characterize the cellular sources of BDNF in multiple sclerosis lesions and to study the expression pattern of its receptor gp145trkB. Our present data show that immune cells are a major source of BDNF in multiple sclerosis lesions. Furthermore, in actively demyelinating lesions, a higher percentage of BDNFimmunoreactive cells is detected compared with inactive lesions. Additionally, neurones in the direct vicinity of active multiple sclerosis lesions, as well as reactive astrocytes in the lesions, express high levels of gp145trkB. These observations underscore the potential importance of BDNF-mediated neuroimmune interactions in multiple sclerosis.. Material and methods Tissue. Formalin-®xed and paraf®n-embedded autopsy brain tissue from nine multiple sclerosis patients [age 43.9 6 11.3 years (mean 6 standard deviation)] (Table1) was examined. Five control brains were included in this series from patients without evidence of neurological disease or neuropathological alterations (age 56 6 13.9 years). Clinical and pathological data of the multiple sclerosis cases are summarized in Table 1. Routine neuropathology (haematoxylin and eosin, luxol fast blue, periodic acid±Schiff-reaction and Bielschowsky silver impregnation) and immunohistochemistry for myelin proteins (myelin basic protein, proteolipid. Table 1 Clinical and pathological features of patients with multiple sclerosis included in the study Patient number. Age (years)/sex. Disease duration. Disease course. Demyelinating activity of the lesions examined (% of LFB+/CD68+ cells). 1 2 3 4 5 6 7 8 9. 48/F 45/M 30/M 33/F 53/F 66/F 34/F 40/F 46/M. n.a. 21 d 6a 10 a 21 a n.a. 13 a 10 a 3 mo. n.a. Acute SP SP SP n.a. SP SP Acute. A (99.2)/IA (2.6) A (94.7)/IA (5.6) A (63.9)/IA (0) IA (0) IA (0) IA (0) IA (0) IA (0) A (n.a.)/IA (n.a.). SP = secondary progressive multiple sclerosis; A = active lesion; IA = inactive lesion; n.a. = not available; in parentheses: percentage of LFB+ cells per CD68+ macrophages/microglia..
(3) BDNF and trkB in multiple sclerosis. 77. Fig. 1 BDNF-immunoreactivity present in neurones, in¯ammatory cells and astrocytes in multiple sclerosis brains. (A, B and C) Serial sections demonstrate a perivascular in¯ammatory in®ltrate immunoreactive for BDNF (A), CD68 (B) and CD3 (C) (Case 2). (D and E) Hippocampal neurones contain BDNF in the subiculum (D) and the dentate gyrus (E). (F) Reactive astrocytes within multiple sclerosis lesions display weak to moderate BDNF-immunoreactivity. D±F Case 6. Magni®cation: all parts 3400. protein, myelin oligodendrocyte glycoprotein, cyclic nucleotide phosphodiesterase and myelin-associated glycoprotein) and immune cells [common leucocyte antigen (CLA): leucocytes; CD68: macrophages/microglia; CD3: T cells; Ig (immunoglobulin): plasma cells] were performed for diagnostic purposes. Lesional demyelinating activity was classi®ed according to the presence of luxol fast blue-positive myelin degradation products in macrophages/microglia indicating ongoing demyelination. The percentage of luxol fast blue-positive macrophages/microglia (CD68+ cells) was calculated for each lesional area (Table 1).. Immunohistochemistry. Immunohistochemistry was performed applying antibodies against BDNF (4.FL.1A1; c = 10 mg/ml, IgG1; kindly provided by R. Kolbeck and Y.-A. Barde, Martinsried, and I.. Bartke, Penzberg, Germany) and trkB [TrkB (794), cat sc-12, rabbit polyclonal, c = 0.2 mg/ml; Santa Cruz Biotechnology, Santa Cruz, Calif., USA]. The TrkB (794) antibody exclusively recognizes full-length trkB. Brie¯y, after deparaf®nization and blocking with 10% FCS/PBS (foetal calf serum/ phosphate-buffered saline), primary antibodies were applied at the concentrations indicated and permitted to bind overnight at 4°C. Control sections were incubated with isotype control antibodies, rabbit serum or without primary antibody. A standard avidin-biotin-peroxidase technique with 3,3¢-diaminobenzidine hydrochloride as the chromogenic substrate was used to visualize bound antibody. Alternatively, an alkaline-phosphatase/anti-alkaline-phosphatase-based technique (APAAP; Dako, Glostrup, Denmark) with Fast Red (Sigma, St Louis, Mo., USA) was used. All antibodies were applied after pre-treatment of the sections three times for 5 min in the microwave in citric acid.
(4) 78. C. Stadelmann et al.. Table 2 Expression of BDNF in active and inactive multiple sclerosis lesions Case number. Active area BDNF+ cells. CLA+ cells. % BDNF+/CLA+. Inactive area BDNF+ cells. CLA+ cells. % BDNF+/CLA+. 1 2 3 4 5 6 7 8. 290 80 190 ± ± ± ± ±. 1823 856 1118 ± ± ± ± ±. 15.9 9.3 17.0 ± ± ± ± ±. 30 17 10 7 20 60 9 0. 1170 405 425 1353 373 477 340 935. 2.6 4.2 2.4 0.5 5.4 12.6 2.6 0. BDNF+ cells = BDNF-immunoreactive cells/mm2; CLA+ cells = CLA-immunoreactive cells/mm2; % BDNF+/CLA+ = percentage of BDNF-immunoreactive cells/CLA-immunoreactive cells. Cases 4±8 do not contain active areas; from multiple sclerosis Case 9 only trkB immunostaining was available.. buffer, 10 mmol/l, pH 6.0. BDNF-, CD68-, CLA- and CD3immunoreactive cells were counted in corresponding areas on serial sections. Quanti®cation and photodocumentation were performed on sections stained with the above-mentioned antibodies against BDNF and trkB. The patterns of immunostaining were con®rmed applying the anti-BDNF antibody N20 (cat sc-546, 2 mg/ml; Santa Cruz Biotechnology) and the polyclonal anti-trkB T16030-050 (6 mg/ml; Transduction Laboratories, San Diego, Calif., USA), which recognizes full-length and truncated trkB. For multiple sclerosis Cases 1±8, BDNF and trkB immunohistochemistry were performed, whereas from multiple sclerosis Case 9, only sections for trkB immunostaining were available.. Morphometry. The number of BDNF-, CLA-, CD68-, CD3- and LFBpositive cells was determined in at least 10 standardized microscopic ®elds of 10 000 mm2 each, de®ned by an ocular morphometric grid at a total magni®cation of 31000. Cells from perivascular in¯ammatory in®ltrates were not included in these counts. Corresponding lesional areas were counted in serial sections. In sections stained for BDNF, cells with astrocytic morphology and neurones were not counted. Cell numbers are given as cells/mm2. BDNF-immunoreactive in¯ammatory cells in perivascular in®ltrates were determined separately counting at least 150 perivascular in¯ammatory cells/case except for Case 6, where only 66 cells were available. Statistical analysis was performed applying the non-parametric Mann±Whitney U-test; a P value of P < 0.05 was considered signi®cant.. Results BDNF is detected in in¯ammatory cells in multiple sclerosis lesions. In the multiple sclerosis patients and control cases examined, BDNF immunoreactivity was detected in various neuronal. populations including cortical, hippocampal and brainstem neurones (Fig. 1D and E). No other BDNF-expressing cells were found in control cases. In contrast, in multiple sclerosis plaques a proportion of immune cells was found to contain BDNF immunoreactivity. This was especially pronounced perivascularly, but also found in the lesion parenchyma (Fig. 1A). In Cases 1±3 with actively demyelinating lesions, 48.8 6 6.6% (mean 6 standard deviation) of perivascular in¯ammatory cells were BDNF+, and in Cases 4±8 with inactive lesions 30.6 6 10.5% were BDNF+ (P = 0.07). The immunostaining of serial sections indicated that CD68+ macrophages/microglia and CD3+ T cells are BDNFimmunoreactive (Fig. 1A±C). Occasional BDNF-immunoreactive plasma cells were seen in the in®ltrate. BDNF+ in¯ammatory cells were also detected in other conditions, such as acute disseminated encephalomyelitis and ischaemic brain lesions (C. Stadelmann, M. Kerschensteiner, unpublished results). Reactive astrocytes showed immunoreactivity for BDNF, which was more pronounced in active multiple sclerosis lesions (Fig. 1F). In contrast, no BDNF-immunoreactivity was detected in oligodendrocytes in tissue from multiple sclerosis patients or controls.. BDNF expression in in¯ammatory cells correlates with lesional demyelinating activity. BDNF-immunoreactive immune cells were detected in lesions with ongoing demyelination as well as in inactive lesions (Table 2). However, comparisons between the numbers of BDNF+ cells in actively demyelinating areas and inactive areas in the same lesion (Cases 1±3) reveal that 3.1 6 1.0% (mean 6 standard deviation) of the total number of CLA+ immune cells are BDNF+, whereas this rises to 14.1 6 4.2% in actively demyelinating areas (Table 2 and Fig. 2). Analysing the total number of inactive multiple sclerosis lesions (Cases 1±8), 3.8 6 4.0% of CLA-expressing cells show BDNF-immunoreactivity (two-tailed P = 0.02, Mann±Whitney U-test). However, a single multiple sclerosis.
(5) BDNF and trkB in multiple sclerosis. 79. Fig. 2 BDNF is present in a higher number of cells in active compared with inactive lesional areas in multiple sclerosis plaques (Case 1). (A, C and E) Actively demyelinating zone. (A) Macrophages contain LFB+ myelin degradation products indicating ongoing myelin degradation (LFB-PAS staining). (C) CLA+ in¯ammatory cells in a corresponding area on a serial section. (E) A large number of in®ltrating in¯ammatory cells display BDNF-immunoreactivity. (B, D and F) inactive area. (B) No myelin degradation products are seen in phagocytes. (D) CLA+ in¯ammatory cells in a serial section. (F) Rare BDNF+ cells are detected. A, C, E, B, D and F are serial sections. A±F 3400.. case with an inactive, chronic lesion containing relatively high numbers of BDNF+ cells was observed (Case 6). In line with previous observations, the composition and cell numbers of in®ltrating immune cells were comparable. between active and inactive multiple sclerosis lesions. However, actively demyelinating lesions were characterized by a very high percentage of CD68+ macrophages/microglia containing LFB+ myelin degradation products (Table 1). In.
(6) 80. C. Stadelmann et al.. Fig. 3 Immunostaining for full-length trkB in in¯ammatory demyelinating multiple sclerosis lesions. (A) trkB-immunopositive neurones in a pontine in¯ammatory demyelinating lesion. (B) Single neurones within or close to lesions show enhanced trkB immunostaining. (C) In¯ammatory cells do not display immunoreactivity for full-length trkB. (A, B and C) Case 1. (D) Reactive astrocytes within multiple sclerosis lesions show prominent immunostaining for full-length trkB (Case 9). Magni®cation: all parts 3400.. all cases, CD68+ macrophages/microglia were the predominant in¯ammatory cell population in the lesions. In active areas, 1160 6 594 (mean 6 standard deviation) CD68+ cells/ mm2 were observed compared with 758.4 6 359 CD68+ cells/ mm2 in inactive lesions (P = 0.38). In contrast, numbers of CD3+ cells were low in the lesional areas examined, ranging from 31.6 6 19.5 T-lymphocytes/mm2 in inactive areas to 39.2 6 30 T-lymphocytes/mm2 in actively demyelinating areas (P = 0.8). Thus, macrophages/microglia are most likely. to constitute the predominant BDNF-containing in¯ammatory cells in the lesions.. gp145trkB is detected in neurones in the immediate vicinity of multiple sclerosis plaques. Immunohistochemistry for full-length trkB showed abundant immunoreactivity in various neuronal populations including cortical, hippocampal and brainstem neurones in multiple.
(7) BDNF and trkB in multiple sclerosis sclerosis patients as well as in controls (Fig. 3A and B). In multiple sclerosis lesions, reactive astrocytes showed strong, distinct staining for full-length trkB (Fig. 3D). Only rare astrocytic staining was observed in control brains. However, immune cells in the lesions, as well as perivascularly, were consistently negative independent of lesional activity (Fig. 3C). In addition, no immunoreactivity for full-length and truncated trkB was observed in oligodendrocytes in multiple sclerosis cases and controls. In some multiple sclerosis cases, staining for trkB was more abundant in neurones in the immediate vicinity than far away from the lesion, although the irregular distribution of neurones in and close to multiple sclerosis lesions precluded a quantitative evaluation. Immunohistochemistry applying both antibodies against both full-length and truncated trkB revealed a similar staining pattern. Again, immune cells were negative. In some multiple sclerosis lesions, immunoreactivity for trkB was found in neurones immediately adjacent to BDNF+ in¯ammatory cells.. Discussion. In the present study, we extend our previous observation that immune cells can be a potent source of the neuroprotective factor BDNF in neuroin¯ammatory disease (Kerschensteiner et al., 1999). We ®nd BDNF-immunoreactivity in T cells and macrophages/microglia in active and inactive multiple sclerosis lesions. Lesions with ongoing demyelination show higher ratios of BDNF+ immune cells. Furthermore, BDNF is found in neurones and astrocytes. The BDNF receptor trkB is present equally in neurones and astrocytes, but not in in¯ammatory cells or oligodendroglial cells. Our results demonstrate the presence of BDNF and trkB in multiple sclerosis lesions, and suggest possible neurotrophic interactions between in®ltrating immune cells and resident cells of the CNS. The notion that in®ltrating immune cells could provide BDNF to support neuronal survival has only emerged recently (Kerschensteiner et al., 1999), and since then has been con®rmed by a number of studies (Batchelor et al., 1999; Besser and Wank, 1999; Braun et al., 1999; Hammarberg et al., 2000; Kipnis et al., 2000; Barouch et al., 2001). In this study, we detect a signi®cant number of immune cells containing BDNF, mainly macrophages/ microglia and T-lymphocytes in active and inactive multiple sclerosis lesions. We have shown previously that these cells are capable of releasing bioactive BDNF in vitro (Kerschensteiner et al., 1999). A relevant question is whether BDNF is induced by immune cell activation in vivo. In vitro data show an increase of BDNF mRNA and protein after activation of immune cells (Braun et al., 1999; Kerschensteiner et al., 1999; Barouch et al., 2001). In vivo, an upregulation of BDNF mRNA expression is demonstrated in mouse alveolar macrophages after repeated allergenic challenge (Braun et al., 1999). In the present study, we show that a higher percentage of in¯ammatory cells in actively demyelinating areas of multiple sclerosis lesions contains. 81. BDNF compared with areas without ongoing myelin breakdown. Thus, the observations presented here con®rm that in vivo activation also upregulates BDNF in immune cells. In addition, a high percentage of BDNF+ macrophages and lymphocytes is found in perivascular in®ltrates. Taken together, these observations indicate that relatively high numbers of BDNF-containing immune cells are present at early stages of lesion development. In general, in chronic inactive lesions, only very few BDNF+ cells are encountered. However, exceptional multiple sclerosis cases contain relatively high numbers of BDNF+ in¯ammatory cells in inactive lesions. Astrocytes, which have been shown to store and release exogenous BDNF (Rubio, 1997), also show increased BDNF-immunoreactivity in actively demyelinating areas. All these factors result in the highest number of BDNF+ cells being found in the actively demyelinating edge early in the development of a multiple sclerosis lesion, just in the vicinity of axons that are not directly subject to the in¯ammatory insult, but still at high risk of in¯ammation-induced bystander damage. Outside the lesion area neurones are the major sources of BDNF in multiple sclerosis brains as well as in controls. It is well established that BDNF can be transported anterogradely and released by neurones (von Bartheld et al., 1996; Zhou and Rush, 1996; Canals et al., 2001). Notably this anterograde transport of BDNF is upregulated after axotomy (Tonra et al., 1998). Autocrine and paracrine interactions between neurones releasing BDNF and bearing gp145trkB receptors have been reported (Kokaia et al., 1993; Acheson et al., 1995; Marini et al., 1998) suggesting that neuronal BDNF may further enhance the endogenous neurotrophic support in multiple sclerosis lesions. However, we did not ®nd BDNF in dystrophic axonal spheroids of multiple sclerosis lesions. Thus, expression levels in dystrophic axons are possibly below the detection limit of our immunohistochemical methods. This observation further supports the view that in¯ammatory cells are the major source of BDNF in multiple sclerosis lesions. In addition to BDNF itself, we characterized the expression pattern of the major BDNF receptor, trkB. Two major species of trkB protein have been described: the full-length gp145trkB, capable of transducing BDNF signalling, and a truncated variant, pg95trkB, which is devoid of an active tyrosine kinase domain (Klein et al., 1990, 1991; Bothwell, 1995). In addition, BDNF, like the other neurotrophins of the NGF family, binds to the p75 neurotrophin receptor. Recent studies have demonstrated that the p75 neurotrophin receptor is primarily expressed in oligodendroctes and oligodendrocyte precursors, as well as macrophages/microglia in multiple sclerosis lesions (Dowling et al., 1999; Chang et al., 2000). As most, if not all, known BDNF functions are transmitted via the the full-length gp145trkB receptor, we have focused on the expression of gp145trkB. We ®nd a robust expression of gp145trkB in neurones in the immediate vicinity of multiple sclerosis plaques. Single neurones with clearly pronounced trkB-immunoreactivity.
(8) 82. C. Stadelmann et al.. close to multiple sclerosis lesions are observed, suggesting an upregulation of trkB in a proportion of damaged neurones. Additionally, we ®nd full-length trkB immunoreactivity in reactive astrocytes within the lesions, as has previously been described for reactive astrocytes in a rodent chronic brain injury model (McKeon et al., 1997). Thus far, no relevant function of BDNF signalling in astrocytes has been identi®ed, and so the signi®cance of our observation of full-length trkB expression in astrocytes in multiple sclerosis lesions remains elusive. In contrast to the expression of full-length trkB in neural cells, we were unable to detect full-length trkB in immune cells within multiple sclerosis lesions. This contrasts with in vitro observations that demonstrate full-length trkB mRNA expression in antigen-speci®c T-cell lines from human donors (Besser and Wank, 1999), an observation that we were able to con®rm in individual human T-cell lines. However, until now, we have been unable to ®nd full-length trkB protein in isolated human peripheral immune cells (M. Kerschensteiner, unpublished data). Whether immune cells can or cannot express full-length trkB currently remains an open question, nevertheless, our data clearly show that in neuroin¯ammatory lesions, autocrine or paracrine BDNF signalling to immune cells via trkB is unlikely to occur. The restriction of trkB expression to neural cell types in multiple sclerosis underscores the possibility that there may be BDNF signalling from in®ltrating cells to neurones in neuroin¯ammatory lesions, as would be necessary for immune cells to support neuronal survival or to provide axonal protection. The appeal of neurotrophin-mediated neuroprotection in multiple sclerosis lies in the pleiotropy inherent to neurotrophin actions (Levi-Montalcini et al., 1996; Lewin and Barde, 1996). It is well established that BDNF can prevent neuronal cell death after various pathological insults including experimental transection of axons in the spinal cord (Gravel et al., 1997; Kobayashi et al., 1997; Liu et al., 1999). Moreover, BDNF can also protect axons against elimination during development, as well as against degeneration after axotomy or in experimental neurodegenerative diseases (Mitsumoto et al., 1994; Ikeda et al., 1995; Weibel et al., 1995; Sagot et al., 1998). Furthermore, the preservation of axons provides the basis for neuroregenerative attempts including axonal regeneration and sprouting, which are directly supported by BDNF (Kobayashi et al., 1997; Gallo and Letourneau, 1998; Mamounas et al., 2000). Apart from in¯uencing survival and regeneration of neuronal elements, BDNF supports remyelination after both peripheral and CNS injury (McTigue et al., 1998). Finally, BDNF has been shown to downregulate the expression of MHC (major histocompatibility complex) molecules in hippocampal slices, and may thus also act as an immunomodulator (Torcia et al., 1996; Neumann et al., 1998). The neuroprotective capacity of immune cells is not restricted to BDNF: a panel of other potentially neuroprotective and neurotrophic mediators is released by immune cells, e.g. NGF, NT-3, transforming growth factor b and platelet-derived growth factor (Ehrhard et al., 1993; Blotnick. et al., 1994; Diemel et al., 1998; Letterio and Roberts, 1998; Hammarberg et al., 2000; Kipnis et al., 2000). Moreover, immune-derived cytokines have been demonstrated to exert neuroprotective effects (Cheng et al., 1994; Bruce et al., 1996). This is relevant in the context of observations showing that neuroin¯ammation directed to sites of traumatic injuries to the optic nerve (Moalem et al., 1999) or the spinal cord (Hammarberg et al., 2000; Hauben et al., 2000) can protect neurones from secondary degeneration. Moreover, immune cells in®ltrating spontaneously into sites of neural injury seem to protect brainstem neurones after experimental trauma (Serpe et al., 1999) and could potentially do so in neurodegenerative diseases (Batchelor et al., 1999), which show substantial immune activation in foci of pathological change (McGeer and McGeer, 1995). In summary, we demonstrate here that immune cells, reactive astrocytes and neurones contain BDNF, a potent neuroprotective agent, in multiple sclerosis. Within the multiple sclerosis lesion immune cells seem to be the major source of BDNF. They are likely to release this substance in the immediate vicinity of nerve cell processes, whichÐ according to the trkB expression that we observedÐare likely to be responsive to the neuroprotective effects of BDNF. This neurotrophin-mediated neuroimmune signalling network could be a major factor that helps to preserve axons in a microenvironment that is clearly capable of exerting signi®cant neurotoxicity. Thus, it should be considered as a bene®cial aspect of neuroin¯ammation that could be worth preserving therapeutically, or even reinforcing using tailored immunomodulatory treatment strategies (Hohlfeld, 1997; Hohlfeld et al., 2000).. Acknowledgements. We wish to thank Marianne Leisser, Petra Tassotti, Andrea Keller and Diana WoÈllner for expert technical assistance, and R. Kolbeck, Y.-A. Barde and I. Bartke for providing the antibody against BDNF (4.FL.1A1). This work is part of the MD thesis of C.S. and the study was funded by BM:BWK (Austria), Project GZ 70.056/2-Pr/4/99. The Institute for Clinical Neuroimmunology is supported by the Hermann and Lilly Schilling Foundation. References Acheson A, Conover JC, Fandl JP, DeChiara TM, Russell M, Thadani A, et al. A BDNF autocrine loop in adult sensory neurons prevents cell death. Nature 1995; 374: 450±3. Barouch R, Appel E, Kazimirsky G, Brodie C. Macrophages express neurotrophins and neurotrophin receptors. Regulation of nitric oxide production by NT-3. J Neuroimmunol 2001; 112: 72±7. Batchelor PE, Liberatore GT, Wong JY, Porritt MJ, Frerichs F, Donnan GA, et al. Activated macrophages and microglia induce dopaminergic sprouting in the injured striatum and express brainderived neurotrophic factor and glial cell line-derived neurotrophic factor. J Neurosci 1999; 19: 1708±16..
(9) BDNF and trkB in multiple sclerosis Besser M, Wank R. Cutting edge: clonally restricted production of the neurotrophins brain-derived neurotrophic factor and neurotrophin-3 mRNA by human immune cells and Th1/Th2polarized expression of their receptors. J Immunol 1999; 162: 6303±6. Bitsch A, Schuchardt J, Bunkowski S, Kuhlmann T, BruÈck W. Acute axonal injury in multiple sclerosis. Correlation with demyelination and in¯ammation. Brain 2000; 123: 1174±83. Blotnick S, Peoples GE, Freeman MR, Eberlein TJ, Klagsbrun M. T lymphocytes synthesize and export heparin-binding epidermal growth factor-like growth factor and basic ®broblast growth factor, mitogens for vascular cells and ®broblasts: differential production and release by CD4+ and CD8+ T cells. Proc Natl Acad Sci USA 1994; 91: 2890±4. Bothwell M. Functional interactions of neurotrophins and neurotrophin receptors. [Review]. Annu Rev Neurosci 1995; 18: 223±53. Braun A, Lommatzsch M, Mannsfeldt A, Neuhaus-Steinmetz U, Fischer A, Schnoy N, et al. Cellular sources of enhanced brainderived neurotrophic factor production in a mouse model of allergic in¯ammation. Am J Respir Cell Mol Biol 1999; 21: 537±46. Brosnan CF, Raine CS. Mechanisms of immune injury in multiple sclerosis. [Review]. Brain Pathol 1996; 6: 243±57. Bruce AJ, Boling W, Kindy MS, Peschon J, Kraemer PJ, Carpenter MK, et al. Altered neuronal and microglial responses to excitotoxic and ischemic brain injury in mice lacking TNF receptors. Nat Med 1996; 2: 788±94. BruÈck W, Schmied M, Suchanek G, Bruck Y, Breitschopf H, Poser S, et al. Oligodendrocytes in the early course of multiple sclerosis. Ann Neurol 1994; 35: 65±73. BruÈck W, Porada P, Poser S, Rieckmann P, Hanefeld F, Kretzschmar HA, et al. Monocyte/macrophage differentiation in early multiple sclerosis lesions. Ann Neurol 1995; 38: 788±96. Canals JM, Checa N, Marco S, Akerud P, Michels A, Perez-Navarro E, et al. Expression of brain-derived neurotrophic factor in cortical neurons is regulated by striatal target area. J Neurosci 2001; 21: 117±24. Chang A, Nishiyama A, Peterson J, Prineas J, Trapp BD. NG2positive oligodendrocyte progenitor cells in adult human brain and multiple sclerosis lesions. J Neurosci 2000; 20: 6404±12. Cheng B, Mattson MP. NGF and bFGF protect rat hippocampal and human cortical neurons against hypoglycemic damage by stabilizing calcium homeostasis. Neuron 1991; 7: 1031±41. Cheng B, Christakos S, Mattson MP. Tumor necrosis factors protect neurons against metabolic-excitotoxic insults and promote maintenance of calcium homeostasis. Neuron 1994; 12: 139±53. Davie CA, Barker GJ, Webb S, Tofts PS, Thompson AJ, Harding AE, et al. Persistent functional de®cit in multiple sclerosis and autosomal dominant cerebellar ataxia is associated with axon loss. Brain 1995; 118: 1583±92. DeStefano N, Matthews PM, Fu L, Narayanan S, Stanley J, Francis GS, et al. Axonal damage correlates with disability in patients with relapsing-remitting multiple sclerosis. Results of a longitudinal magnetic resonance spectroscopy study. Brain 1998; 121: 1469±77.. 83. Diemel LT, Copelman CA, Cuzner ML. Macrophages in CNS remyelination: friend or foe? [Review]. Neurochem Res 1998; 23: 341±7. Dowling P, Ming X, Raval S, Husar W, Casaccia-Bonne®l P, Chao M, et al. Up-regulated p75NTR neurotrophin receptor on glial cells in MS plaques. Neurology 1999; 53: 1676±82. Ehrhard PB, Erb P, Graumann U, Otten U. Expression of nerve growth factor and nerve growth factor receptor tyrosine kinase Trk in activated CD4-positive T-cell clones. Proc Natl Acad Sci USA 1993; 90: 10984±8. Ferguson B, Matyszak MK, Esiri MM, Perry VH. Axonal damage in acute multiple sclerosis lesions. Brain 1997; 120: 393±9. Frim DM, Uhler TA, Galpern WR, Beal MF, Breake®eld XO, Isacson O. Implanted ®broblasts genetically engineered to produce brain-derived neurotrophic factor prevent 1-methyl-4phenylpyridinium toxicity to dopaminergic neurons in the rat. Proc Natl Acad Sci USA 1994; 91: 5104±8. Frisen J, Verge VM, Cullheim S, Persson H, Fried K, Middlemas DS, et al. Increased levels of trkB mRNA and trkB protein-like immunoreactivity in the injured rat and cat spinal cord. Proc Natl Acad Sci USA 1992; 89: 11282±6. Gallo G, Letourneau PC. Localized sources of neurotrophins initiate axon collateral sprouting. J Neurosci 1998; 18: 5403±14. Gimsa U, Peter SV, Lehmann K, Bechmann I, Nitsch R. Axonal damage induced by invading T cells in organotypic central nervous system tissue in vitro: involvement of microglial cells. Brain Pathol 2000; 10: 365±77. Gravel C, Gotz R, Lorrain A, Sendtner M. Adenoviral gene transfer of ciliary neurotrophic factor and brain-derived neurotrophic factor leads to long-term survival of axotomized motor neurons. Nat Med 1997; 3: 765±70. Hammarberg H, Lidman O, Lundberg C, Eltayeb SY, Gielen AW, Muhallab S, et al. Neuroprotection by encephalomyelitis: rescue of mechanically injured neurons and neurotrophin production by CNS-in®ltrating T and natural killer cells. J Neurosci 2000; 20: 5283±91. Hauben E, Nevo U, Yoles E, Moalem G, Agranov E, Mor F, et al. Autoimmune T cells as potential neuroprotective therapy for spinal cord injury. Lancet 2000; 355: 286±7. Hofer M, Pagliusi SR, Hohn A, Leibrock J, Barde YA. Regional distribution of brain-derived neurotrophic factor mRNA in the adult mouse brain. EMBO J 1990; 9: 2459±64. Hohlfeld R. Biotechnological agents for the immunotherapy of multiple sclerosis. [Review]. Brain 1997; 120: 865±916. Hohlfeld R, Kerschensteiner M, Stadelmann C, Lassmann H, Wekerle H. The neuroprotective effect of in¯ammation: implications for the therapy of multiple sclerosis. [Review]. J Neuroimmunol 2000; 107: 161±6. Holtzman DM, Sheldon RA, Jaffe W, Cheng Y, Ferriero DM. Nerve growth factor protects the neonatal brain against hypoxic-ischemic injury. Ann Neurol 1996; 39: 114±22. Ikeda K, Klinkosz B, Greene T, Cedarbaum JM, Wong V, Lindsay RM, et al. Effects of brain-derived neurotrophic factor on motor.
(10) 84. C. Stadelmann et al.. dysfunction in wobbler mouse motor neuron disease. Ann Neurol 1995; 37: 505±11.. neuroprotective effect of N-methyl-D-aspartate. J Biol Chem 1998; 273: 29394±9.. Jeffery DR, Absher J, Pfeiffer FE, Jackson H. Cortical de®cits in multiple sclerosis on the basis of subcortical lesions. Mult Scler 2000; 6: 50±5.. McGeer PL, McGeer EG. The in¯ammatory response system of brain: implications for therapy of Alzheimer and other neurodegenerative diseases. [Review]. Brain Res Brain Res Rev 1995; 21: 195±218.. Kerschensteiner M, Gallmeier E, Behrens L, Leal VV, Misgeld T, Klinkert WE, et al. Activated human T cells, B cells, and monocytes produce brain-derived neurotrophic factor in vitro and in in¯ammatory brain lesions: a neuroprotective role of in¯ammation? J Exp Med 1999; 189: 865±70. Kipnis J, Yoles E, Porat Z, Cohen A, Mor F, Sela M, et al. T cell immunity to copolymer 1 confers neuroprotection on the damaged optic nerve: possible therapy for optic neuropathies. Proc Natl Acad Sci USA 2000; 97: 7446±51. Klein R, Conway D, Parada LF, Barbacid M. The trkB tyrosine protein kinase gene codes for a second neurogenic receptor that lacks the catalytic kinase domain. Cell 1990; 61: 647±56. Klein R, Nanduri V, Jing SA, Lamballe F, Tapley P, Bryant S, et al. The trkB tyrosine protein kinase is a receptor for brain-derived neurotrophic factor and neurotrophin-3. Cell 1991; 66: 395±403. Kobayashi NR, Fan DP, Giehl KM, Bedard AM, Wiegand SJ, Tetzlaff W. BDNF and NT-4/5 prevent atrophy of rat rubrospinal neurons after cervical axotomy, stimulate GAP-43 and Talpha1tubulin mRNA expression, and promote axonal regeneration. J Neurosci 1997; 17: 9583±95. Kokaia Z, Bengzon J, Metsis M, Kokaia M, Persson H, Lindvall O. Coexpression of neurotrophins and their receptors in neurons of the central nervous system. Proc Natl Acad Sci USA 1993; 90: 6711± 15. Lassmann H, Raine CS, Antel J, Prineas JW. Immunopathology of multiple sclerosis: report on an international meeting held at the Institute of Neurology of the University of Vienna. J Neuroimmunol 1998; 86: 213±17. Letterio JJ, Roberts AB. Regulation of immune responses by TGFbeta. [Review]. Annu Rev Immunol 1998; 16: 137±61. Levi-Montalcini R, Skaper SD, Dal Toso R, Petrelli L, Leon A. Nerve growth factor: from neurotrophin to neurokine. [Review]. Trends Neurosci 1996; 19: 514±20. Lewin GR, Barde YA. Physiology of the neurotrophins. [Review]. Annu Rev Neurosci 1996; 19: 289±317. Lindvall O, Kokaia Z, Bengzon J, Elmer E, Kokaia M. Neurotrophins and brain insults. [Review]. Trends Neurosci 1994; 17: 490±6. Liu Y, Kim D, Himes BT, Chow SY, Schallert T, Murray M, et al. Transplants of ®broblasts genetically modi®ed to express BDNF promote regeneration of adult rat rubrospinal axons and recovery of forelimb function. J Neurosci 1999; 19: 4370±87. Mamounas LA, Altar CA, Blue ME, Kaplan DR, Tessarollo L, Lyons WE. BDNF promotes the regenerative sprouting, but not survival, of injured serotonergic axons in the adult rat brain. J Neurosci 2000; 20: 771±82. Marini AM, Rabins SJ, Lipsky RH, Mocchetti I. Activity-dependent release of brain-derived neurotrophic factor underlies the. McKeon RJ, Silver J, Large TH. Expression of full-length trkB receptors by reactive astrocytes after chronic CNS injury. Exp Neurol 1997; 148: 558±67. McTigue DM, Horner PJ, Stokes BT, Gage FH. Neurotrophin-3 and brain-derived neurotrophic factor induce oligodendrocyte proliferation and myelination of regenerating axons in the contused adult rat spinal cord. J Neurosci 1998; 18: 5354±65. Merlio JP, Ernfors P, Kokaia Z, Middlemas DS, Bengzon J, Kokaia M, et al. Increased production of the TrkB protein tyrosine kinase receptor after brain insults. Neuron 1993; 10: 151±64. Meyer R, Weissert R, Diem R, et al. Acute neuronal apoptosis in a rat model of multiple sclerosis. J Neurosci 2001; 21: 6214±20 Mitsumoto H, Ikeda K, Klinkosz B, Cedarbaum JM, Wong V, Lindsay RM. Arrest of motor neuron disease in wobbler mice cotreated with CNTF and BDNF. Science 1994; 265: 1107±10. Moalem G, Leibowitz-Amit R, Yoles E, Mor F, Cohen IR, Schwartz M. Autoimmune T cells protect neurons from secondary degeneration after central nervous system axotomy. Nat Med 1999; 5: 49±55. Neumann H, Misgeld T, Matsumuro K, Wekerle H. Neurotrophins inhibit major histocompatibility class II inducibility of microglia: involvement of the p75 neurotrophin receptor. Proc Natl Acad Sci USA 1998; 95: 5779±84. Piani D, Spranger M, Frei K, Schaffner A, Fontana A. Macrophageinduced cytotoxicity of N-methyl-D-aspartate receptor positive neurons involves excitatory amino acids rather than reactive oxygen intermediates and cytokines. Eur J Immunol 1992; 22: 2429±36. Rovaris M, Filippi M, Minicucci L, Iannucci G, Santuccio G, Possa F, et al. Cortical/subcortical disease burden and cognitive impairment in patients with multiple sclerosis. AJNR Am J Neuroradiol 2000; 21: 402±8. Rubio N. Mouse astrocytes store and deliver brain-derived neurotrophic factor using the non-catalytic gp95trkB receptor. Eur J Neurosci 1997; 9: 1847±53. Sagot Y, Rosse T, Vejsada R, Perrelet D, Kato AC. Differential effects of neurotrophic factors on motoneuron retrograde labeling in a murine model of motoneuron disease. J Neurosci 1998; 18: 1132± 41. Serpe CJ, Kohm AP, Huppenbauer CB, Sanders VM, Jones KJ. Exacerbation of facial motoneuron loss after facial nerve transection in severe combined immunode®cient (scid) mice. J Neurosci (Online) 1999; 19: RC7 (1±5). Available from: http:// www.jneurosci.org Tonra JR, Curtis R, Wong V, Cliffer KD, Park JS, Timmes A, et al. Axotomy upregulates the anterograde transport and expression of brain-derived neurotrophic factor by sensory neurons. J Neurosci 1998; 18: 4374±83..
(11) BDNF and trkB in multiple sclerosis Torcia M, Bracci-Laudiero L, Lucibello M, Nencionci L, Labardi D, Rubartelli A, et al. Nerve growth factor is an autocrine survival factor for memory B lymphocytes. Cell 1996; 85: 345±56. Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mork S, Bo L. Axonal transection in the lesions of multiple sclerosis. New Engl J Med 1998; 338: 278±85. vanWaesberghe JH, Kamphorst W, De Groot CJ, van Walderveen MA, Castelijns JA, Ravid R, et al. Axonal loss in multiple sclerosis lesions: magnetic resonance imaging insights into substrates of disability. Ann Neurol 1999; 46: 747±54. vonBartheld CS, Byers MR, Williams R, Bothwell M. Anterograde. 85. transport of neurotrophins and axodendritic transfer in the developing visual system. Nature 1996; 379: 830±3. Weibel D, Kreutzberg GW, Schwab ME. Brain-derived neurotrophic factor (BDNF) prevents lesion-induced axonal dieback in young rat optic nerve. Brain Res 1995; 679: 249±54. Zhou XF, Rush RA. Endogenous brain-derived neurotrophic factor is anterogradely transported in primary sensory neurons. Neuroscience 1996; 74: 945±53. Received April 12, 2001. Revised June 30, 2001. Accepted August 30, 2001.
(12)
Figure
Documents relatifs
(A) Nerve Growth Factor (NGF), (B) Brain-Derived Neurotrophic Factor (BDNF) and (C) Neurotrophin-3 (NT-3) concentrations measured by ELISA in systemic sclerosis (SSc) patients
Our findings represent the first report concerning BDNF involvement in injured brain of non- mammalian vertebrates. Our results demonstrated that: a) the peak of BDNF
Artificial Neural Network Answer Set Programming Clinically Isolated Syndrome Convolutional Neural Network Central Nervous System Cerebrospinal Fluid Cortico-Spinal Tract
Suite aux expériences professionnelles et observations que nous avons toutes deux faites, nous sommes arrivées à la question suivante : « Comment soutenir les parents et instaurer
2 Frequencies (a) and relative fitness (b) of uninfected Daphnia magna immigrant genotypes during the 43 days after immigration plotted against the prevalence of Octosporea bayeri
- catégorie B : demandeurs d’emploi tenus de rechercher un emploi, ayant exercé une activité réduite courte (de 78 heures ou moins dans le mois) ; - catégorie C :
Major role in the acquisition, analysis, and interpretation of data Christophe Parizot, MS Sorbonne Universit´ e, Inserm, Centre d ’Immunologie et des Maladies
