Title
A C.
elegans
histone methyltransferase promotes
spermatocyte gene expression, spermatid production
and fertility
By
Christoph G. Engert
BSc. University of Heidelberg, 2007
Submitted to the Graduate Program in Computational and Systems Biology
in partial fulfillment of the requirements for the degree of
Doctor of Philosophy in Computational and Systems Biology
at the
MASSACHUSETTS INSTITUTE OF TECHNOLOGY SEvEMBER 2017
2017 Christoph Engert. All rights reserved.
The author hereby grants to MIT permission to reproduce and to distribute publicly
paper and electronic copies of this thesis document in whole or in part in any medium
now known or hereafter created.
Signature of Author:
...
Certified by:
...
Accepted by:
...
MASSACHUSETT"NSTITUTE OF TECHNOLOGYJUN 22 217
CO)Signature redacted
...
-
at~rogram
in Computational & Systems Biology
June 09, 2017
Signature redacted
.. .... . ... ...
... ...
H. Robert Horvitz
Professor of Biology
Thesis Advisor
Signature redacted
...
...
Christopher B. Burge
Computational and Systems Biology Ph.D. Program Director
A C. elegans histone methyltransferase promotes
spermatocyte gene expression, spermatid production
and fertility
By
Christoph G. Engert
Submitted to the Graduate Program in Computational and Systems Biology
on June
0 9th,2017, in partial fulfillment of the requirements for the degree of
Doctor of Philosophy in Computational and Systems Biology
Abstract
To better understand the tissue-specific regulation of chromatin state in cell-fate
determination and development, we defined the tissue-specific expression of all 36
lysine methyltransferase (KMT) genes by endogenous mRNA detection in C. elegans.
We found that most KMTs are expressed in only one or two tissues and that the
germline is the tissue with the most general KMT expression. We discovered that the
germline-expressed C. elegans ortholog of mammalian PRDM9, SET-1 7, promotes
fertility through gene regulation in primary spermatocytes. SET-17 drives transcription of
spermatocyte-specific genes from four genomic clusters to promote spermatid
production. SET-1 7 is concentrated in stable, chromatin-associated nuclear foci at
actively transcribed gene clusters, which we term spermatocyte transcription bodies.
Our results identify the spatially restricted function of a PRDM9 ortholog in
spermatocyte transcription and we propose that the spatial organization of chromatin
factors might be a conserved mechanism in tissue-specific control of transcription.
Thesis Supervisor: H. Robert Horvitz
Title: Professor
Biographical Note
I was born on March 16, 1985, in Aachen, Germany, where I grew up and went
to public school through graduation with the Abitur from Kaiser-Karls-Gymnasium.
I attended university in Heidelberg, Germany, and graduated with a Bachelor of
Science in Molecular Biotechnology. I conducted undergraduate thesis research in the
laboratory of Dr. Michael Boutros at the German Cancer Research Center on de novo
small RNA discovery from DNA sequence.
A Fulbright Scholarship allowed me to attend the University of California,
Berkeley. My research in the laboratory of Dr. Jennifer Doudna centered on repetitive
element RNA structure and function in eukaryotic genomes.
Following my time at Berkeley, I came to MIT for graduate school in
Computational and Systems Biology.
After graduate school, I will join the Boston Consulting Group to advance the
implementation of scientific knowledge.
Acknowledgments
"Dass ich erkenne, was die Welt [That / may know, what keeps the world Im Innersten zusammenha t." together at its innermost]
-- J.W. v. Goethe, Faust I
I have much to be grateful for over the time of this PhD that I was so fortunate to spend
at MIT. First, I must thank my advisor Bob Horvitz for inviting me into the lab, when it was clear that I knew nothing of experimental work. Above all, I have to thank Bob for his generous,
patient and forceful guidance throughout my time in the lab. The freedom to establish my own research program has been a tremendous privilege that has taken me deeper into science than
I ever imagined possible. The opportunity to go after my own mistakes has been humbling and
illuminating.
I would like to thank Alexander van Oudenaarden for letting me join his lab, which was
at the time one of the busiest places at MIT. I learned a lot about the simplicity of science that comes with relentless hard work, focus on the essentials and the irresistible creativity of many smart people working together.
I must also thank my committee members Rudolf Jaenisch and David Page. Their input
and perspective on this project have been a constant inspiration and instrumental to my
progress. It is worth noting that I became truly excited about developmental biology because of the seminar class Rudolf and David teach and which I attended in my second year at MIT.
My time in the Horvitz lab would not have been the same and this project would not
have made it without the fastidious support of Nikhil Bhatla. I have not shared the same intellectual connection with another person. Likewise, the friendship of Stefan Semrau has been an integral part of my PhD. Stefan deeply believes in science with the enthusiasm and intelligence that make any problem solvable.
I am grateful to each scientist in the Horvitz and van Oudenaarden labs that I
overlapped with in my time at MIT: Jeff Gore and Arjun Raj for making science fun and endlessly interesting; Kirk Burkhart and Akiko Doi for friendship and creating the most welcoming work environment in 68-411; Rita Droste for Germanship and her unwavering support; Tove Ljungars for showing me that belief can move mountains; Na An for moving mountains; and to Kaitlin
Driscoll, Cory Pender, Steve Sando, Holly Johnsen, Vivek Diwvedi and Eugene Lee for making the hardships of science brighter. Thank you also to my CSB class of 2008 for sticking it out through the first two years together, and the CSB community for shaping my understanding of what it
means to be a scientist. A special thank you to Yarden Katz for shining the light of intellectual creativity into many dark hours of this journey.
I would not have made it to MIT but for the support of my mentors at Heidelberg and
Berkeley: Michael Boutros was the first person to really take a chance on me and Jennifer Doudna taught me how to believe in and stick with scientific ideas.
Thank you to my parents for allowing me to disappear for long periods at a time and for always being supportive even though it is all happening really far away.
Thank you to Emily for her support throughout this writing process and for providing an outside perspective!
Table of contents
Title ...
...
1
Abstract
...
2
Biographical Note
...
3
Acknowledgments...4
Table of contents...6
Chapter 1- Histone methylation, chromatin organization and regulation of the
genome ...
12
Title ... 13
Sum m ary ... 14
M ain text...15
Chromatin regulation and the discovery of lysine methyltransferases... 15
Histone methylation patterns in development and disease ... 21
Histone methylation function in genome control ... 23
Table 1 - The most extensively studied methylation of lysine and arginine residues in histone proteins and their modifiers in human, D. melanogaster and C. elegans... 26
Chromatin function and nuclear organization ... 32
Spermatogenesis in C. elegans ... 38
Figure 1 - Spermatogenesis in C. elegans ... 40
C o n c lu sio n s...4 5 Acknowledgments ... 47
References ... 48
Chapter 2 - The C. elegans ortholog of PRDM9 promotes spermatocyte gene
expression, sperm production and fertility
...
72
Title ... 73
Sum m ary ... 74
Introduction ... 75
Results ... 78
Map of KMT endogenous mRNA expression reveals tissue-specificity and abundant KMT expression in the germ line ... 78
Figure 1 - Detection of endogenous expression of lysine methyltransferases by smFISH ... 81
Table 1 - Tissue-specific expression map of all C. elegans lysine methyltransferases...83
set-17 functions in the germline to promote sperm production and fertility...84
Figure 2 - set-17 functions in the germline to promote sperm-production and fertility...86
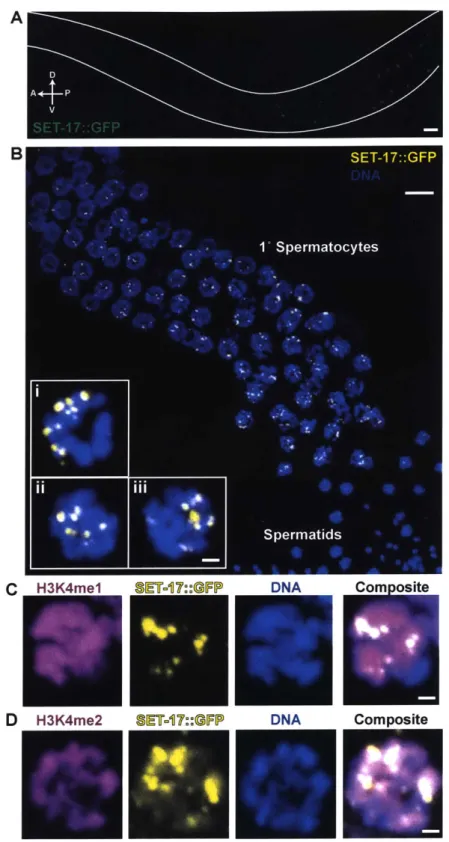
Figure 3 -set-17 promotes the production of major sperm protein vesicles in primary sp e rm ato cyte s ... 8 9 SET-17 promotes the production of fibrous-body membranous organelles in primary sp e rm ato cyte s ... 9 0 SET-17 is expressed in primary spermatocytes and localizes to nuclear, chromatin-asso ciate d fo ci...9 1 Figure 4 - SET-17 is expressed in the nuclei of primary spermatocytes and enriched in chrom atin-associated foci ... 93
Figure 5 -set-17 promotes expression of msp genes in spermatogenic gene clusters...97
SET-17 promotes the expression of all 28 msp genes ... 100
set-17 promotes expression of four clusters of spermatocyte-specific genes...101
Figure 6 - SET-17 promotes transcription at spermatogenic gene clusters...104
SET-17 promotes transcription of msp gene clusters in spermatocytes ... 106
m sp transcription sites localize to SET-17 foci ... 107
SET-17 foci are stable nuclear structures with features of liquid-like phase-separated d ro p le ts ... 10 8 ELT-1 promotes msp expression and fertility together with SET-17 ... 109
Figure 7 - ELT-1 promotes spermatogenic gene cluster expression and fertility downstream o f S ET -1 7 ... 1 1 3 D iscussion ... 114
Implications for the evolutionary origin and function of mammalian PRDM9 ... 114
Evidence for a SET-17-like transcriptional regulator in mammalian sperm production .... 115
Spermatocyte transcription bodies are analogous structures to histone locus bodies ... 116
Tissue-specific chromatin control and spatial restriction in transcriptional regulation...117
Experim ental Procedures...119
Single-molecule fluorescence in situ hybridization (smFISH)...119
Broodsize m easurem ents ... 119
RN A sequencing and analysis ... 119
Acknow ledgem ents ... 120
Supplem ental Figure Legends ... 128
Figure S1 ... 128
Supplem entary Table 1 ... 130
Figure S2 ... 132 Figure S3 ... 134 Figure S4 ... 136 Figure S5 ... 138 Figure S6 ... 141 Figure S7 ... 143
M aterials and M ethods ... 145
S tra in s ... 1 4 5 KM T identification ... 147
KM T probe sets for sm FISH ... 147
sm FISH on C elegans ... 148
sm FISH im age processing and tissue expression identification of KM Ts ... 149
Broodsize assays ... 149
Single-copy transgene strain construction ... 149
M ating assays ... 150
Sperm counts ... 150
Electron m icroscopy ... 151
Electron m icroscopy image analysis ... 151
Transcript analysis by RNA sequencing ... 153
Genome position analysis of spermatocyte-enriched genes ... 154
msp FISH imaging ... 154
msp FISH image processing ... 155
Transcription site analysis ... 155
Imaging of transcription sites and SET-17::GFP...156
Fluorescence recovery after photo-bleaching (FRAP)...156
Chapter 3 - Future directions for exploring SET-17 function, spermatocyte
chrom atin and tissue-specific chrom atin biology
...
158
Title ... 159
M ain Text ... 160
SET-17 function in spermatogenesis and chromatin structure ... 160
SET-17 function outside of spermatogenesis ... 163
SET-17 might regulate transgenerationally stable chromatin states ... 164
Regulation of msp gene expression and the function of clustering ... 165
Somatic KMT expression and function ... 166
C o n c lu sio n ... 1 6 7 References ... 168
Chapter 1- Histone methylation, chromatin organization and
regulation of the genome
Title
Histone methylation, chromatin organization and regulation of
the genome
Authors
Summary
The functional organization of the nucleus is an essential part of genome function. The nuclear genome contains the genetic information that instructs development of all tissues and cell-types. Every cell is defined by the particular genetic information it uses and must maintain the proper genomic context for its function. The term chromatin now encompasses all aspects of the physiological state of the genome. Access to genetic information for all aspects of biological function are enabled by chromatin regulatory mechanisms. Post-translational modification of histone proteins is an important component of chromatin regulation. Among many post-translational modifications, the methylation of lysine residues in the tails of histones has emerged as an important functional mediator of access to the genome. Lysine
methyltransferases deposit these modifications and most can be identified by their catalytic domain, termed SET-domain. This chapter introduces the emergence of lysine
methyltransferases and provides an overview of the current knowledge of histone methylation function in genome control in metazoans, with emphasis on the close connection of lysine methyltransferase functional discovery and oncology. We conclude with a brief overview of recent advances in tying the methylation state of histones at particular genes back into the spatial, physiological context of chromatin and the nucleus. New organizing principles, such as topologically associated domains and non-membranous nuclear organelles, have recently emerged to advance our understanding of the functional separation and hierarchical
organization of proper genomic context for cell-type specification and genome function. We propose that it will be important to understand the interplay between spatial organization and histone modification to be able to understand in detail what makes our genomes tick.
Main text
Chromatin regulation and the discovery of lysine methyltransferases The term "chromatin" has come to comprise all aspects of the physiological
manifestation of the genome. Its origin and functional analysis is tightly linked to the idea of "epigenetics" originally defined as the phenomenological separation of phenotype from genotype (Waddington, 1942). The early studies of chromatin and epigenetics were based on observation of nuclear structure and phenotypic variation. From the observation of
chromosomal banding patterns open euchromatin and closed heterochromatin were defined. Position-effect variegation and transposable element genetics by Muller and McClintock, respectively, provided the first suggestions of non-Mendelian genetics (McCLINTOCK, 1951; Muller and Altenburg, 1930). Important phenomenological observations on non-genetic modifications of phenotypic outcomes were made from studies of X-chromosome inactivation (LYON, 1961) and germline imprinting (McGrath and Solter, 1984; Surani et al., 1984). The first mechanistic understanding of the chemical modification of specific DNA sequences to alter their phenotypic impact came from analyses of methylation of cytosines in DNA and the
discovery of DNA methyltransferases (Gruenbaum et al., 1982; Li et al., 1993). DNA methylation and its importance in non-genetic inheritance and disease has been studied and reviewed extensively (Feinberg and Tycko, 2004; Jaenisch and Bird, 2003).
Studies of the regulation of hetero- and euchromatin then revealed the role of histone proteins in shaping genomic activity. The importance of histone proteins in constituting the nucleosome as the basic organizational unit of chromatin fibers, and by extension the genome, emerged from biochemical studies of nuclear proteins and gene activation (Kornberg, 1974).
Biochemical analysis of histone proteins also showed their modification by acetylation or methylation and hinted at the importance of these modifications for RNA polymerase activity (ALLFREY et al., 1964). Studies in yeast showed functional relevance of histone tail modification at specific residues in both gene activation and silencing. Genetic screens in Drosophila
identified several genes important for enhancing (E) or suppressing (Su) the epigenetic position effect variegation, such as Su(Var)3-9 and E(z). A third gene that shared a homologous domain, Trithorax, was identified in genetic studies of homeotic gene control (Elgin and Reuter, 2013). The shared -120 amino acid domain was termed SET-domain in their honor (Tschiersch et al., 1994).
The identification of enzymatic control of histone modification and its role in chromatin regulation came from studies in Tetrahymena thermophilia and utilized the separation of pro-and macronucleus - of euchromatic pro-and heterochromatic nature, respectively. The discovery of histone acetyl transferases followed and their conservation across eukaryotes was quickly confirmed. Histone acetylation had been identified as a hallmark of gene activity from studies in yeast and X-inactivation. The first histone acetyltransferase (HAT) was found in Tetrahymena.
p55 was cloned as the source of HAT activity and its homology to yeast Gcn5 discovered.
Combined with previous knowledge of Gcn5 function this work defined the gene activation function of nuclear HATs (Brownell et al., 1996). The first histone deacetylase was described shortly thereafter, using a small-molecule inhibitor of acetylation to purify the protein from mammalian cells (Taunton et al., 1996). The most important advance then followed when the first effector of histone acetylation was discovered, proving the signaling function of this modification, rather than it impacting nucleosome stability per se (Dhalluin et al., 1999).
Lysine methyltransferase activity was first shown in the human ortholog of Su(Var)3-9. The homology of the SET domain and its relationship to putative plant lysine
methyltransferases suggested the link between the SET domain, histone methylation and chromatin regulation. This motivated in vitro experiments to show that SUVAR39H1 specifically methylates lysine 9 on histone 3 into the di- and tri-methylated state (H3K9me2 and H3K9me3) (Rea et al., 2000). Heterochromatin protein 1 was a well-established factor in heterochromatin function. The discovery of its specific binding to SUVAR39H1 and, with its chromodomain, to H3K9me then established the function of histone lysine methylation in heterochromatin formation (Lachner et al., 2001). The histone code hypothesis was put forth to systematize emerging findings on the diverse roles post-translational modifications of histone tails could play (Jenuwein and Allis, 2001; Strahl and Allis, 2000). A further important step in establishing histone methylation as a dynamic contributor to chromatin regulation was the discovery of the first lysine demethylase from the family of deaminases (Shi et al., 2004).
The importance of histone methylation in oncology followed through the identification of the Polycomb complex as a marker for progressive prostate and breast cancers (Kleer et al., 2003; Varambally et al., 2002). The Polycomb complex of proteins consists of three core
subunits, Suz12, EED and EZH2, but myriad cofactors have been implicated in specific aspects of Polycomb function (Simon and Kingston, 2013). Polycomb function confers gene silencing through a combination of histone methylation, ubiquitination and nucleosome compaction. EZH2 is the catalytic core and deposits H3K27me3 in flies and humans (Cao et al., 2002; Czermin et al., 2002; Kuzmichev et al., 2002; Muller et al., 2002). From genetic studies in flies, Polycomb emerged as a powerful regulator of development of the fly body plan (Jurgens, 1985; Lewis,
1978; Struhl, 1981). By microscopy, Polycomb proteins were found to reside in heterochromatic
chromosome bands (Zink and Paro, 1989). These findings were confirmed genome-wide in Drosophila embryos and in mouse and human embryonic stem cells (Boyer et al., 2006; Lee et al., 2006; Schuettengruber et al., 2009). The importance of Polycomb function for
developmental plasticity is conserved in mammals and established the complex as a major arbiter of stem cell identity. The role of EZH2 as an oncogene and a tumor suppressor has been extensively researched (Comet et al., 2016). Importantly, somatic gain-of-function mutations in EZH2 have been identified as tumor growth drivers and clinical trials to study their therapeutic efficacy are under way (McCabe et al., 2012; Sneeringer et al., 2010). Loss of EZH2 function has also been described as a driver of oncogenesis in other malignancies (De Raedt et al., 2014; Kim and Roberts, 2016; Knutson et al., 2012). Recent discoveries of heterozygous somatic gain-of-function mutations in histone 3 lysine 27 in glioblastoma further support the decisive role of this residue and its targeting enzymes in oncology (Schwartzentruber et al., 2012). It is important to point out that the precise link between H3K27me3 and transcriptional control independently of Polycomb recruitment is not clearly established (Simon and Kingston, 2013).
The discovery of lysine methylation function in active transcription was directly motivated by studies of the genetic alterations that occur in hematological malignancies, specifically mixed lineage leukemia (Krivtsov and Armstrong, 2007; Rao and Dou, 2015). The founding member of the evolutionarily ancient KMT2 family was identified as a frequent
translocation partner in leukemias (Cimino et al., 1991; Djabali et al., 1992; McCabe et al., 1992; Tkachuk et al., 1992). The discovery of SET-domain catalytic activity of SUV39H1 motivated the identification of the H3K4-specific catalytic function of the trithorax-like SET-domain of KMT2
(Milne et al., 2002; Nakamura et al., 2002). These studies already noticed that MLL-fusion protein function in leukemia took place independently of H3K4me catalysis. MLL-fusion
proteins activate Hox gene expression inappropriately, conferring stem-cell-like properties onto cancer cells. MLL-fusion protein function in oncogenesis, however, depends on the recruitment of co-factors, such as Dot1L, another KMT, rather than the methyltransferase activity of MLL itself (Mishra et al., 2014; Okada et al., 2005). In this instance, small-molecule inhibition of Dot1L was found to be sufficient for the suppression of MLL-fusion protein oncogenesis (Daigle et al., 2011). This points to an important aspect of KMT function that has complicated
biochemical and genetic analysis of their in vivo function. Chromatin factors function in diverse and dynamic complexes with redundancies and extensive specialization. In the case of KMT2, additional co-factors are of paramount importance for its function. Some of these confounding dependencies have been studied in detail for the the KMT2 family. In vitro assays have been established for the KMT2 SET-domain and used to study its regulation by other factors (Dou et al., 2006). This work has been important in establishing a causal role of KMT2 function in transcription initiation. KMT2 complexes in yeast and mammals function synergistically with acetyltransferases in vitro (Tang et al., 2013). This couples H3K4me3 deposition and H3K9 acetylation at promoters and establishes a causal role for promoting transcription.
The SET domain of the KMT2 family targets H3K4 in all instances. However, the specificity of individual members has diversified, targeting H3K4mel, me2, me3 and their combinations. This differential biochemical activity translates into fundamentally different roles
differentiation (Lee et al., 2013). The detailed effects of individual KMTs on specific cellular processes remain poorly understood.
The discovery of Dot1L has been remarkable for its role in cell proliferation and oncogenesis, but also because it is the only known non-SET lysine methyltransferase in chromatin regulation (Min et al., 2003). Dot1p was discovered in telomere silencing in yeast (van Leeuwen et al., 2002) and subsequently shown to methylate the histone core (Ng et al., 2002). Its identification as a cofactor in MLL and further work have established its function in the maintenance of open chromatin and its role in cellular proliferation. Recently, it was discovered that a novel acetyl-lysine binding domain, termed YEATS, in the chromatin factor
AF9, recruits hDot1L to H3K9ac nucleosomes (Li et al., 2014). The deposition of H3K9ac is
mediated by cofactors of KMT2 at promoters. AF9 thus closes the loop on MLL-fusion protein recruitment of Dot1L through its cooperative function with acetyltransferases. This emerging understanding of the functional dependencies between various chromatin factors and their roles in development or oncogenesis are an important part on ongoing research.
The advent of genome sequencing has bolstered the importance of KMTs and other chromatin factors in oncogenesis in a variety of tumors and contexts (Cancer Genome Atlas Research Network et al., 2013; Fujimoto et al., 2012; Kandoth et al., 2013). Intriguingly, chromatin architecture can impact local mutation rates and thus also influence somatic mutation in specific tissues and cell-types (Schuster-Bockler and Ben Lehner, 2012).
Histone methylation patterns in development and disease
The biochemical and genetic analysis of chromatin regulation through post-translational histone tail modifications revealed the paramount importance of these complexes for most developmental processes and access to the genome overall. However, knowledge of the detailed effect of histone modifications on genome biology was limited to the analysis of individual instances of the interactions between chromatin factors and target genes, or the analysis of histone methylation state at specified loci (Litt et al., 2001; Noma K et al., 2001). The advent of genome-wide protein-DNA interaction analysis enabled the rapid expansion of knowledge of histone modification patterns (Bernstein et al., 2005; Johnson et al., 2007). This expansion revealed the extent of chromatin modification of the genome and its detailed correlation with cell-type-specific gene expression. The exact correspondence of transcription initiation and H3K4me3 was shown across all genes in human cells (Kim et al., 2005; Schmid and Bucher, 2007). The discovery of simultaneous labeling of the opposing H3K4me3 and
H3K27me3 at the promoters of developmental genes in embryonic stem cells, in which these genes are not expressed, suggested the prospective role of chromatin state for developmental decision making in differentiation (Bernstein et al., 2006; Boyer et al., 2006). Heterochromatic domains are denoted by H3K9me3 and were found to span several megabases. H3K9me3 covers gene-poor chromosomal regions, repetitive sequences and the ends of chromosomes proximal to telomeres, mimicking the distribution of DNA methylation in mammals (Du et al.,
2015). H3K9me3 is also associated with X-chromosome-inactivation in mammals and in the
The analysis of genome-wide histone modification patterns with greater tissue-specificity has revealed its adaptation to every cell-fate state and transition. Importantly, the histone methylation landscape of H3K4 and H3K27 at genes and in intergenic regions is adapted to cellular differentiation state and altered by reprogramming (Mikkelsen et al., 2008; 2007). Importantly, histone methylation was found to reliably identify cell-type specific enhancers. Enhancers in intergenic regions are marked by H3K4me1 and H3K27ac in mammalian genomes (Heintzman et al., 2007). Together, these results established histone modification signatures are associated with cell-type specific transcriptional regulation. These sites are tightly associated with transcription-factor binding motifs both at enhancers and promoters,
suggesting a close relationship between histone methylation and transcription factor activity. The effect on and consequence of histone methylation at transcription factor binding sites has been a subject of extensive analysis since (Long et al., 2016). Through the analysis of several specific functional contexts it was discovered that there is a class of transcription factors that can overcome a repressive chromatin environment to nucleate the establishment of active histone marks, facilitating the recruitment of additional factors in a chromatin- or DNA-sequence-dependent manner to establish the appropriate cell-type specific gene expression state. These factors have been termed pioneer factors. This model also highlights how the interplay between sequence specific factors and chromatin state can enable context-specific cell-fate specification.
Recent studies of enhancer malfunction in cancer have significantly enhanced our understanding of the importance of histone methylation clusters in driving gene expression and the oncogenic state (Bradner et al., 2017). The transcriptional drivers c-Myc and Brd4 can be
recruited to islands of histone methylation at oncogenes and hyper-activate their transcription in a variety of cancers (Chapuy et al., 2013; Hnisz et al., 2013; Tomazou et al., 2015). Curiously, the contributions of individual KMTs to the establishment and maintenance of these chromatin domains has not yet been defined.
Histone methylation function in genome control
Histone proteins can be post-translationally modified in a wide variety of ways. Among the modifications that have been detected and functionally analyzed are methylation,
acetylation, phosphorylation, ubiquitination, crotonylation and sumoylation (Kouzarides, 2007; Strahl and Allis, 2000; Tan et al., 2011). These post-translational modifications can take place at over 60 different amino acid residues, both in the core and in the tails of histone proteins. It has been thought that histone core modifications can regulate nucleosome stability through
modifying the affinity of the histone octamer to DNA (Tessarz and Kouzarides, 2014). Histone tail modification by contrast are thought to act as signaling modifications, involved in the recruitment of binding factors and additional complexes to effect chromatin state function (Taverna et al., 2007). Histone tails are predominantly modified at their basic residues by either methylation or acetylation, while phosphorylation of hydroxyl groups on tyrosines or serines also plays a role. As lysines can be both acetylated and methylated, it has emerged that
methylation or acetylation of a given lysine have opposite effects on access to the genome. For example, H3K9me is associated with silenced heterochromatin while H3K9ac is found at transcriptionally active loci. Similarly, H3K27me3 is associated with silenced developmental genes and H3K27ac is enriched at active enhancers. In general, surveys of histone tail
modifications have found groups of spatially correlated marks depending on the functional context of the cell (Wang et al., 2008). This suggests a high degree of redundant regulation by complexes of modifiers and effector proteins. The order of deposition of different marks in dynamic contexts is mostly unknown and the relationship between chromatin state and gene expression level (high, medium, low vs. on/off) is now beginning to be explored.
Methylation can occur on all basic residues in histone tails: arginines, lysines and histidines (Byvoet et al., 1972; Gershey et al., 1969; MURRAY, 1964). The terminal nitrogen of lysines can be mono-acetylated, altering its electrochemical potential from positive (at
physiological pH) to neutral. By contrast, methylation of lysines can define three distinct states of mono-, di- or tri-methylation of the epsilon nitrogen (Hempel et al., 1968; Paik and Kim,
1967). Methylation does not alter the electrochemical potential of the nitrogen but does affect
the hydrophobicity of the residue, creating a peculiar type of charged, hydrophobic residue. The stereochemistry of mono-, di- and tri-methyl is sufficiently distinct to enable their
differential detection. Arginine methylation can also define three states in mono-methylated, symmetrically dimethylated or asymmetrically dimethylated. It is less clear what the functional consequences of most arginine methylation events are. One well-established example of
Histone 3 Arginine (R) 2 methylation shows that H3R2me2a, mediated by PRMT6, can prevent methylation of the adjacent lysine 4 in human cells (Guccione et al., 2007). This type of
interference between the post-translational modification of adjacent histone tail residues has been observed before between DNA-replication associated H3S10 phosphorylation and H3K9 methylation by SUV39H1, as well as binding by HP1 (Lachner et al., 2001; Rea et al., 2000).
instance in the PhD finger of TRIM24 that mediates transcriptional regulation via the
simultaneous recognition of unmethylated H3K4 and non-canonically methylated H3K23 (Tsai et al., 2010).
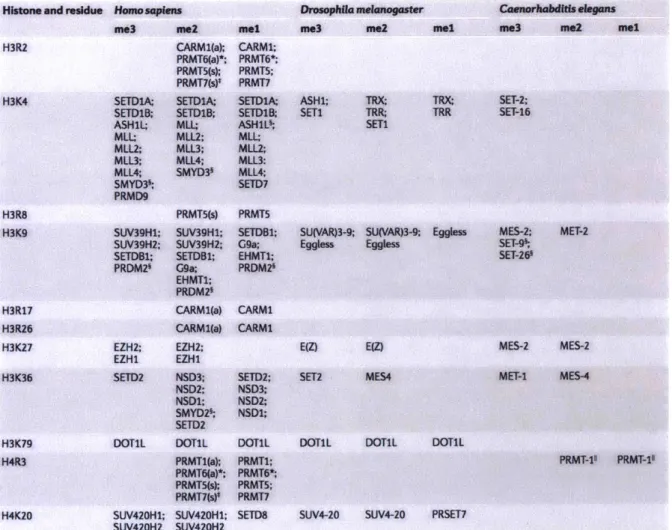
The best understood sites for histone lysine methylation are H3K4, H3K9, H3K27, H3K36, H3K79 and H4K20 (Table 1). Methylation of arginines has been observed on H3R2, H3R17,
H3R26 and H4R3 (Table 1). Mass spectrometric and other high-throughput chemical assays are rapidly identifying additional methylation events at basic amino acids, as well as additional types of modifications, as alluded to above (Tan et al., 2011). Their functional roles remain to be studied in vitro and in vivo. At least one enzyme catalyzing the methylation of the canonical residues have been identified in most organisms, though understanding of the biochemical processes is most thorough in mammalian systems due to the ready biochemical analysis cell culture systems enable (Table 1). The specificity of individual enzymes is difficult to define as subtle changes in conditions or cofactors can affect the target specificity of the SET domain. These factors have been studied most extensively for the KMT2 type family and for the EZH family (Kim and Roberts, 2016; Rao and Dou, 2015). Such complexities of allosteric regulation through complexes or other contexts are part of the reason for the difficulty in mapping biochemical properties to function in specific developmental processes, and vice versa, in vivo.
Table 1- The most extensively studied methylation of lysine and arginine residues in
histone proteins and their modifiers in human, D. melanogaster and C. elegans (adapted from
(Greer and Shi, 2012).
Histone and residue
H3R2 H3K4 H3R8 H3K9 H3R17 H3R26 H3K27 H3K36 H3K79 H4R3 H4K20 Homosapien me3 SETDlA; SETDIB. ASH1L; MLL: MLL2; MLL3; MLL4; SMYD36; PRMD9 ts mZ2 CARM1(a); PRMT6(a)*; PRMT5(s); PRMT7(s)* SETD1A; SETD1B; MLL; MLL2; MLL3; MLL4; SMYD3' PRMT5(s) SUV39H1; SUV39Hl; SUV39H2; SUV39HZ; SETDBl; SETDBl; PRDM2 G9a; EHMT1; PRDM2 CARM1(a) CARMl(a) EZH2; EZH2; EZHl EZH1 SETD2 NSD3; NSD2; NSDl; SMYD2' SETD2 DOTIL DOTIL PRMTl(a); PRMT6(a)*; PRMT5(s); PRMT7(s)* SUV420H1; SUV420HI; SUV420H2 SUV420H2 Drosophila melanogaster mel CARM1; PRMT6*; PRMT5; PRMT7 SETDlA; SETDIB ASHLS; MLL; MLL2; MLL3: MLL4; SETD7 PRMT5 SETDB1; G9a; EHMT1; PRDM2i CARMI CARMI SETD2; NSD3; NSD2; NSDl; DOTIL PRMT1; PRMT6*; PRMT5; PRMT7 SETD8 me3 ASHM; SETI
me2 mel me3 me2 mel
TRX TRR;
SETI
TRX; SET-2;
TRR SET-16
SU(VAR)3-9; SU(VAR)3-9; Eggless Eggless Eggless E(Z) EZ) SET2 MES4 MES-2; SET-9% SET-26 MET-2 MES-2 MES-2 MET-I MES-4
DOTIL DOTML DOTIL
PRMT-11 PRMT-1l
SUV4-20 SUV4-20 PRSET7
Histone methylation marks are associated with either euchromatin or heterochromatin. H3K4 methylation is generally associated with transcriptional activation as explored above. H3K4me3 is prevalent at all RNA Polymerase 11 promoters and in metazoans is required for transcriptional activation of gene expression. H3K4me2 is correlated with H3K4me3 in most instances, but can define broader domains of gene activation beyond the core promoter sequence upstream of the transcription start site. H3K4me is prevalent in non-coding elements that correlate with transcription-factor binding sites and transcriptional enhancer function that can take place over a variety of distances between the enhancer and the core promoter (Long et al., 2016). It is important to note that not only can the different valiances be deposited by different enzymes, they are also differentially affected by the removal enzymes (Greer and Shi, 2012). The exact relationship between H3K4me2 and H3K4me3 especially remains unclear and worthy of exploration.
H3K36 tri-methylation is associated with actively transcribed gene bodies. This pattern closely traces the DNA-association pattern of elongating, but not paused, RNA Polymerase II. Indeed, the methyltransferase that deposits H3K36 trimethyl in all eukaryotes, the SET2-type KMT, has been shown to be directly associated with the transcription elongation complex and to label histones co-transcriptionally with Pol 11 activity across eukaryotes (Li et al., 2003; 2002; Sun et al., 2005; Xiao et al., 2003). The core splicing machinery is also part of this
co-transcriptional complex and required for the deposition of H3K36me3 by SET2 (Yuan et al., 2009). Counter-intuitively, one of the most important functional consequences of H3K36 tri-methylation is the suppression of non-promoter transcriptional activation, or the activation of transcription along the opposite DNA strand, in addition to roles in RNA splicing (Carrozza et al.,
2005; Fang et al., 2010; Kaplan et al., 2003). This means that while H3K36 is associated with Pol
II elongation, it is specifically deposited to prevent spurious Pol II initiation in yeast and metazoans to prevent misguided gene expression. In specific tissue contexts, H3K36me3 has also been found at promoters of genes that will be activated in later developmental stages in zebrafish sperm production (Wu et al., 2011).
The relationship between H3K36 mono- and di-methyl states to the tri-methyl state remain complex and only specific cases of the interdependence are clearly understood. A class of methyltransferases that was first identified as associated with nuclear hormone receptors (nuclear hormone receptor associated SET Domain, NSD) has been found to deposit mono- and di-methylation of H3K36 in metazoans (Bell et al., 2007; Rechtsteiner et al., 2010). This
deposition of H3K36me2 affects gene expression and the later deposition of H3K36me3 by the SET2 complex. A general principle that can be deduced from these various observations is that
the dimethyl mark of both H3K4 and H3K36 presents an opportunity to prepare the chromatin state of particular genes for the rapid activation by additional down-stream signals by either recruiting supportive factors directly or by preventing the deposition of additional repressive signatures, such as H3K9 or H3K27 methylation. The detailed layers of this regulation remain to be explored.
The heterochromatic state then is defined by the methylation of H3K27, H3K9 and H4K20. The execution of heterochromatin at the level of histone methylation can be
deconstructed into several layers that are interlaced in different contexts. The first layer is the developmental regulation of specific genes in the tissues of their function. The second layer is the silencing of developmental genes in the tissues they are not expressed in. The third layer is
the broad silencing of chromosomal domains containing repetitive elements or proximal to telomeric sequences. The maintenance of silenced states through cell-division is also an area of active research with important implications for heterochromatin maintenance.
Methylation of H3K27 predominantly functions in the first layer of developmental gene expression regulation in the tissue context. The functional characterization of this histone mark is tightly linked with the only catalytically verified class of enzymes, the EZH-KMTs that are the catalytic core of the Polycomb group (PcG) complexes. As discussed above, H3K27me3 is important for developmental gene control and organismal tissue-specification. However, it is also been shown that its deposition and associated PcG complexes can trigger further and more permanent states of silencing (Simon and Kingston, 2013). This transition is hypothesized to occur when the composition of the Polycomb repressive complex (PRC) switches from predominantly H3K27me3 deposition and erasure of co-transcriptional marks such as H3K4me3, to H3K27me3 deposition and H2AK119 ubiquitination, triggering permanent
compaction of specific loci (Simon and Kingston, 2013). These mechanisms were first identified
in Drosophila but are conserved across Metazoans and their integrity harbors substantial
importance for human medicine.
The second layer of heterochromatin has been termed facultative heterochromatin and is identified by elevated H3K9 tri-methylation and in mammals DNA methylation (Fodor et al., 2010). There are many roles for H3K9 methylation in genome function and gene expression regulation. Many enzymes can deposit H3K9 methylation in a variety of contexts (Table 1). Facultative heterochromatin is found at repetitive sequences throughout the genome of most organisms. Heterochromatin plays an important role in defining centromeric regions in
organisms with pericentric chromosomal pairing. In mammalian genomes SUVAR39H1/2 is thought to deposit repeat-associated H3K9me3. These domains are maintained through cell-divisons and stable over long periods of time (Fodor et al., 2010). Developmental
heterochromatin is functionally important in differentiation processes. When embryonic stem cells differentiate, broad megabase domains of H3K9me2 are established over regions of silenced genes by G9a (Wen et al., 2009). H3K27me3 can be an entryway to this more
permanent layer of developmental heterochromatin via the recruitment of SUVAR39 and G9a
by either the PRC itself or by H3K27me3 deposition. Their recruitment then leads to the
deposition of H3K9me2 or H3K9me3 at the same loci and the recruitment of additional heterochromatin factors. The compensatory layers and redundant functions of several pathways have complicated the detailed elucidation of H3K9 methylation function in
organismal development and physiology. Recently, the PRDM-type KMTs PRDM3 and PRDM16 were identified as H3K9me1 depositors in adipocyte development (Pinheiro et al., 2012). The
broad defects in heterochromatin formation and maintenance in the absence of these genes suggest an important role for H3K9mel in recruiting various H3K9me2 and me3 enzymes as well as potentially the DNA methylation machinery.
The role of H4K20 methylation remains understood only in specific limited contexts. H4K20 is coregulated with H3K9 trimethylation and deposited by SUV420H1/2 in pericentric heterochromatin (Schotta et al., 2004). H4K20 methylation also functions in dosage
compensatipon mechanisms in several species, is involved in proper resolution of DNA damage and functions in proper cell-cycle progression. SUV420 appears to regulate broad aspects of gene expression in dosage compensation, telomeric stability and DNA damage (Benetti et al.,
2007; Schotta et al., 2004; Tuzon et al., 2014), while PR-SET7 (SETD8) is required for cell-cycle
progression, involved in replication-origin-initiation and for DNA repair pathway choice in mammals (Fang et al., 2002; Tardat et al., 2010; Tuzon et al., 2014). PR-SET7 also serves as an interesting example of the underexplored possibility of co-functional methylation of non-histone targets by non-histone KMTs. PR-SET7 function in DNA damage also depends on the methylation of P53BP1 to recruit it to sites of DNA damage (Shi et al., 2007). In the case of H4K20 it also appears that higher-order methylation states depend on the deposition of the mono-methylated form.
The functional association for the histone methylation marks themselves quickly becomes complicated beyond the broad strokes outlined above. There are several reasons for this complexity. Firstly, a lot of functional hypotheses are derived from correlational genome-wide analyses of samples of varying purity and without taking account of cell-to-cell variability
(Junker and van Oudenaarden, 2014; Schwartzman and Tanay, 2015). Secondly, independent measurements of histone methylation marks do not take into account specific biochemical context, such as multiple-labeling of individual tails or the combinatorial labeling between
histone proteins. Thirdly, we are only just beginning to understand the various effector proteins of specific marks to understand the changes they cause specifically (Taverna et al., 2007).
Fourth, most studies of histone methylation and their distribution are based on antibody
affinity purification. While antibodies are selected to be specific for the methylation state of the particular lysine, the interference from other post-translational modifications in the vicinity of these lysines or their occupation by effector proteins, remain structural issues in most instances of recent lysine methylation studies, with notable exceptions that employed
mass-spectrometry on in vivo histones (Towbin et al., 2012). Fifth, most studies of chromatin are focused on steady state outcomes from genetic manipulation and do not investigate functional roles of chromatin state regulation in dynamic developmental or genomic transitions. This surely masks many functional properties of individual layers important in the regulation of gene expression state or genome stability that might be relevant on very short (e.g. differentiation) or very long timescales (e.g. transgenerational inheritance).
Therefore, the most helpful basis of interpretation of particular histone methylation marks is to understand them as hallmarks of activity of a variety of different complexes and as a base-line reflection of genome structure and cell-type specification. The most important path forward in the systematic analysis of chromatin modifications is the continued analysis of the combinatorial effects of the many regulatory layers under specific circumstances of genome control, such as wide-ranging developmental remodeling or long-term genome stability. The elucidation of effector, adapter, writer and eraser proteins will shape our ability to pinpoint the specific signaling inputs and functional outputs of chromatin modification signatures and their relationship to cellular state.
Chromatin function and nuclear organization
Chromatin and the nuclear genome are organized in terms of their spatial arrangement to enable the physical association of specific genetic elements, the silencing of deleterious regions and to create specialized nucleic-acid processing "factories" (Bickmore and van
Steensel, 2013; Dekker and Mirny, 2016; Dekker et al., 2016). The important role of maintaining such spatial arrangements and the factors involved are only emerging. Several principles of the
important functional interplay between histone methylation of specific sequence elements and the spatially structured nuclear environment have been discovered and questions about their interaction are beginning to find answers.
The nuclear envelope is an important component of nuclear organization, as it is both the gate to the cytoplasm and the only definitive membranous structure. Heterochromatic structures are localized to the nuclear periphery, such as the ends of chromosomes and repetitive elements, via their interaction with nuclear lamin (Guelen et al., 2008). lamin is a structural component of the inner nuclear envelope that sequesters heterochromatic
chromatin at the nuclear periphery through the interaction with various proteins. The tethering of heterochromatin to the nuclear periphery is incompletely understood. Recent measurements of nuclear lamina interactions with DNA in single cells revealed wide-spread single-cell
heterogeneity in certain lamin-associated-domains, but also that gene-poor domains are constitutively found at the nuclear periphery (Kind et al., 2015). Lamin-associated domains are always low in transcription and depleted for chromatin marks that are associated with active transcription. However, the exact nature of the connection between histone methylation state and lamin association is unknown. The discovery of the wide-spread defects in heterochromatin and nuclear periphery organization in the absence of H3K9mel, suggests a role for this mark in organizing the localization of chromosomal domains in the nucleus (Pinheiro et al., 2012). Recent work in C. elegans identified a role for H3K9me3 in the anchoring of silenced repetitive regions to the nuclear periphery (Gonzalez-Sandoval et al., 2015). In C. elegans, a
chromodomain protein associates with the nuclear envelope in a lamin-independent manner to sequester H3K9me3 to the nuclear envelope. This mechanism appears to function in the
reinforcement of cellular differentiation in situations where cell-fate choice is disrupted, e.g. by the overexpression of the a trans-differentiating master transcription factor (Gonzalez-Sandoval et al., 2015). Interestingly, the tethering of H3K9me3 sequences does not affect gene
expression per se in C. elegans. The repression of transcription is mediated indpendently by the recruitment of H3K9me2 via the SETDB1 KMT met-2 (Towbin et al., 2012). The function of lamin and the nuclear periphery has important consequences in human health. Several devastating rare genetic diseases are caused by point mutations in different lamins, termed laminopathies, such as muscular dystrophies, cardiomyopathies and progeria (Butin-Israeli et al., 2012). The exact molecular mechanism of these lamin-associated diseases remains to be understood and the impact of heterochromatin misorganization may be limited as lamin proteins may function in many aspects of nuclear organization and mutations in known heterochromatic factors have not been found to phenocopy lamin mutations.
The nucleolus is a distinct compartment in the nucleus that contains the production of ribosomes from ribosomal RNA (rRNA), transcribed from rDNA clusters in the genome, and ribosomal proteins reimported from the cytoplasm (Boisvert et al., 2007). Transcription of rDNA is executed by RNA polymerase I and generally distinct from the chromatin environment of RNA polymerase 11 transcribed genes. The importance of ribosome biogenesis and the necessity to place rDNA loci specifically in the nucleolus gives the nucleolus an important role in organizing the nuclear genome. Indeed, it has been found that lamin associated domains can also be associated with the nucleolus (Nemeth et al., 2010; van Koningsbruggen et al., 2010). The nucleolus has also been found to be supported by an F-actin network in xenopus oocytes, suggesting that it has a nucleoskelletal surface for anchoring specific domains (Feric and
Brangwynne, 2013). The nucleolus stands out as a nuclear compartment because of its size and non-membranous structure. The absence of membranes to maintain its distinct composition and function highlights the power of phase-separation in creating spatially distinct structures (Banani et al., 2017; Courchaine et al., 2016). The relationship between such non-membraneous structures and the functional organization of the genome is an area of active research.
The genome is further organized in many thousands of smaller domains of various characteristics. The attempt to derive chromosomal domains from a linear analysis of many proteins and histone marks has been attempted in many different systems and anywhere from
6-51 chromatin domain types have been proposed (Bickmore and van Steensel, 2013; Filion et
al., 2010). It is therefore likely that much of the functional structure of chromatin remains to be explored. Known chromatin factors that establish functional domains across many genes famously include the Polycomb group complex, already discussed above. Polycomb deposits H3K27me3 to effect chromatin state and H3K27me3 can be distributed along domains of many genes. Polycomb group proteins remain associated with such domains and can further compact chromosomal regions (Francis et al., 2004), affecting their overall positioning in the nucleus (Eskeland et al., 2010). This compaction is mediated by CBX2 in mammals and functionally required for Polycomb-dependent gene silencing in mouse development (Lau et al., 2017). This Polycomb-dependent compaction mechanism is remarkable its physical properties. Volumetric imaging analysis of DNA labeling of Drosphila cells in situ revealed that Polycomb domains are the densest chromatin regions, stringently exclude active regions from nearby chromosomes and exhibit a remarkable increase in density with genomic size of the domain (Boettiger et al., 2016). Further imaging analysis of the distribution of Polycomb complexes in cells have
revealed that they cluster into many thousands of clusters in the genome to effect chromatin compaction and gene expression regulation (Wani et al., 2016). The establishment of Polycomb domains can be triggered by several distinct mechanisms, but importantly specialized sequence motifs have been found in Drosophila and in mammals that can nucleate the establishment of Polycomb domains (Negre et al., 2006; Woo et al., 2010). Non-coding RNAs have also been proposed to play a role in Polycomb recruitment, e.g. in the context of mammalian
X-chromosome inactivation, but also developmental Hox gene regulation (Simon and Kingston,
2013). The extensive analysis of Polycomb domains suggests that spatial organization is an
important component of chromatin function and extensive work on these questions is ongoing in many systems.
The tissue-specific expression of most developmental genes is governed by sequence elements in the vicinity of their transcription start site, either in the promoter or in enhancer elements (Long et al., 2016). Depending on the organism, enhancers can be separated from their target genes by many kilobases and even act across other genes (Hnisz et al., 2016). It has been hypothesized that enhancers interact physically with promoters of target genes through looping to drive expression (Schleif, 1992). The nature of this looping interaction is stochastic (Giorgetti et al., 2014), contributing to transcription frequency (Bartman et al., 2016).
Enhancers function additively to activate transcription, depending on their strength up to a maximum transcription rate (Bothma et al., 2015), consistent with a stochastic model of loop interaction. This model of spatial enhancer-gene interaction has raised many questions about the regulation of specificity and boundaries. Specificity has been evident from the beginning and boundaries have been known for an equally long time as well. Recent work on the spatial
interaction of the genome defined topologically associated domains of sequence stretches, that form areas of interaction. Boundaries of these topological domains correlate with many other features of the genome, such as heterochromatic or Polycomb domains, and frequently define boundaries of enhancer interaction (Dekker and Mirny, 2016; Hnisz et al., 2016; Lupi iez et al.,
2015; Rao et al., 2014). Intriguingly, a model emerges wherein all active genes occupy
nanoscopic domains of freely moving sequences, which are then modified and bound by protein complexes, consistent with the particular developmental state of the cell, leading to stochastic interactions that enable their transcriptional activation. Taking this spatial model for transcriptional control a step further, the spatial association of active genes has been observed in individual instances and speculated about for a long time. Recent work suggests, that
topological association is important, but that it is less significant which two loci are in proximity with one another. The ability to map the spatial association of enhancers and target genes in single cells has revealed that large tissue-specific enhancers are frequently clustered together spatially and that highly-expressed genes also frequently associate with other highly-expressed genes (Beagrie et al., 2017). This data and other observations have led to the hypothesis that tissue-specific transcriptional state depends on spatial association for fidelity and maintenance (Hnisz et al., 2017). Observations about the oncogenic consequences of ectopic formation or disruption of large enhancer domains support the idea that they are crucial in the maintenance of chromatin state and have led to the proposal that many cancers may be causally driven by the transcriptional consequences of such domains (Bradner et al., 2017; Chipumuro et al., 2014).
The exact nature of the spatial domains that are defined by this large enhancer groups and their associated target genes is at present unknown. Understanding the mechanisms of their establishment, maintenance and spatial distribution will be illuminating for the role domain association plays in development.
Spermatogenesis in C. elegans
The propagation of most metazoans depends on the production of sperm and oocytes for fertilization and the formation of progeny. The genome of any new organism is therefore founded by the oocyte and spermatid that fused at fertilization. Both sperm and oocytes are produced from diploid germ cells that differentiate and undergo reductive meiotic divisions to create haploid gametes (Ellis and Stanfield, 2014a).
In C. elegans, the hermaphrodite germline produces both sperm and oocytes, while the male germline only produces sperm. In the hermaphrodite, sperm production occurs before oocyte production during the fourth larval stage (L4). Sperm are then stored in the
spermatheca and the germline switches to oocyte production after the onset of adulthood. A population of mitotically dividing stem cells resides at the distal tip of the germline and cells differentiate as they migrate along the germline tubular structure. The cells remain connected via syncytium, whose central part is the rachis. As they migrate germ cells first exit from the mitotic cell cycle and then enter meiosis 1. At this stage of meiotic initiation differences between germ cells that will produce sperm or oocytes can already be found at the transcriptional level (Morgan et al., 2013).
A AToo&o Gern flinwamA nuclgus Spermatids B) most proximal germline 14 rachis progression to pachytene FB-MOs form Spermatocytes Spermi 2* wOr-buddi budding Residual body
meiosis asymmetric division,
completed unequal partitioning of organeiles and proteins SPERMATOGENESIS (seminal vesicle) @WormAtlas C) itid Spermatozoon pseudopod ng spermatids activated SPERMIOGENESIS (post-ejaculaion) FB-MO fheadliar MO co L or b ody @WrMMAtfas Evens Stage:
Figure 1 - Spermatogenesis in C. elegans A) A Nomarski image of the posterior part of a male C. elegans with the germline labeled, lateral view. Mitotic germ cells proliferate in the distal gonad near the DTCs and progress toward the proximal gonad. Meiosis is initiated as cell progress through the loop region. Spermatogenesis begins cells have migrated through the proximal arm and spermatids are stored in the seminal vesicle of the somatic gonad. DTCs: Distal tip cells; DG: Distal gonad ; PG: proximal gonad. Adapted from WormAtlas.
B) Schematic of the developmental stages of spermatogenesis and spermiogenesis. Primary spermatocytes mature and cellularize to bud off from the rachis. Rapid meiotic divisions then produce secondary spermatocytes and spermatids. Extraneous cellular material, such as ribosomes and most mRNAs are retained in the residual body. Spermatids are then stored until activation of spermiogenesis through external signals. Adapted from WormAtlas.
C) Progression of FB-MOs over spermatogenesis from right to left. MOs form in primary spermatocytes and then accumulate FBs over spermatocyte maturation. In spermatids FBs are resolubilized and only MOs remain. Upon spermiogenesis MOs fuse with the spermatid cell membrane and activate pseudopod formation. Adapted from WormAtlas.
In the male germline, these processes take place in an analogous manner, except that no switch occurs and sperm are produced throughout the reproductive life-span of the animal. Sex-specific features of spermatogenesis emerge in the activation process after ejaculation and at the morphological level in that male sperm are larger than hermaphrodite sperm and can outcompete them for fertilization of oocytes. There may also be transcriptional differences in spermatocytes of males versus hermaphrodites but this has not been conclusively explored.
Spermatocyte differentiation progresses from the initiation of meiosis and primary spermatocytes form when they cellularize and separate from the syncytial rachis of the distal germline (Fig 1C). A number of morphological transformations occur in spermatocytes, which
have been characterized extensively by electron microscopy. Mitochondria prepare of their important role in fertilization and change their structure in the cytoplasm. Spermatocytes also produce a prominent cytoplasmic vesicular structure the fibrous-body membranous organelle (FB-MO). FB-MOs consist of a membranous organelle with an octopus-like structure with a head and membranous extensions, which forms in spermatocytes. This MO is then filled with the FB that is made up of major sperm protein (MSP) paracrystals (Fig 1C). MSPs make up most of the spermatid by mass and are a multi-functional cytoskeletal-like protein that makes up the cytoskeleton of the pseudopod that drives spermatozoa crawling after activation. FB-MO formation is necessary for spermatid production as meiosis 11 cannot proceed in mutants that form MOs but do not accumulate FBs (L'Hernault, 2009).
A special feature of spermatogenesis is the structural transformation of the genome from a regular germ cell genome into the inert spermatid genome that can be protected from chemical or other mutagens that the genome might be exposed to between sperm production
and fertilization (Ellis and Stanfield, 2014b). In this process most nucleosomes are evicted and replaced with Sperm Nuclear Basic Proteins, the protamines in mammals and the SPCH proteins in C. elegans, which resemble protamines and also encode a histone 2A variant (Chu et al.,
2006). This exchange process begins with the diplotene stage of meiosis I in spermatocytes and
renders the spermatocyte genome refractory to transcriptional activity. This can be observed at the level of active RNA polymerase I staining in spermatocytes that reveals the global
down-regulation of transcriptionally active RNA Pol II (Shakes et al., 2009). This highlights the fact that transcriptional activity and regulation are only possible early on in spermatogenesis. While there are potentially many transcription factors involved in specifying spermatocyte cell fate,
one spermatocyte-specific transcription factor, SPE-44, has been found to be required for the spermatocyte cell-fate (Kulkarni et al., 2012). The ELT-1 GATA factor has also been implicated in the transcriptional regulation of spermatocyte gene expression (del Castillo-Olivares et al.,
2009).
The precise definition of genes that are transcribed in spermatocytes has been
complicated by a lack of meaningful techniques for their isolation. Over 2000 transcripts have been identified to be spermatocyte enriched from RNAseq studies for individual dissected germlines in the sperm- or oocyte-producing stage (Ortiz et al., 2014), consistent with previous analysis by microarray (Reinke et al., 2000). While many transcripts are enriched in sperm-producing germlines, post-transcriptional regulation and other confounding factors may also play an important role in this and it will be necessary to conduct detailed spatial analysis of transcriptional activity to identify which genes are specifically transcriptionally active in spermatocytes after the initiation of nucleosome replacement.