High-throughput detection of clinically targetable alterations using next-generation sequencing
Texte intégral
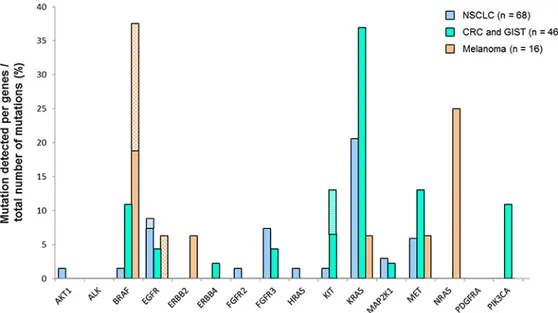
Figure

Documents relatifs
Funaki B, Rosenblum JD, Leef JA, Hackworth CA, Szymski GX, Alonso EM (1997) Angioplasty treatment of portal vein stenosis in children with segmental liver transplants: mid-term
The in vitro e ffects of 4 arylimidamides (DB811, DB786, DB750 and DB766) against the proliferative tachyzoite stage of the apicomplexan parasite Besnoitia besnoiti were
For each sample, ALK fusions were explored using IHC, a dual-color break-apart FISH, and NGS approaches using two different assays: Ion AmpliSeq RNA Lung Cancer Research Fusion
PCR amplification, library preparation and sequencing Each of our seven multiplexing experiment corresponds to an Illumina library composed of single-sequence or mock community
Many viruses infecting grapevine have been either confirmed or identified via HTS techniques, with at least five of which [grapevine asteroid mosaic-associated virus (GAMaV),
In this paper, we present an approach to the computa- tion of multirate symbolic models for incrementally sta- ble switched systems, where the period of symbolic tran- sitions is
This section includes complement-taking verbs expressing deontic and epis- temic modality, to the exclusion of verbs of volition (want, wish etc) and attitudinal verbs, which
Our aim is to control the transition graph of the PWA system to obtain an oscilla- tory behaviour, which is indeed of primary functional importance in numerous biological networks;
