HAL Id: tel-02592804
https://hal.inrae.fr/tel-02592804
Submitted on 15 May 2020HAL is a multi-disciplinary open access archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
multi-échelle par la relaxation et la diffusion par RMN
M. Adam Berret
To cite this version:
M. Adam Berret. Étude de la cristallisation des lipides : Approche multi-échelle par la relaxation et la diffusion par RMN. Sciences de l’environnement. Doctorat de l’Université de Rennes 1, Mention Physique, 2009. Français. �tel-02592804�
THÈSE
présentée
DEVANT L’UNIVERSITÉ DE RENNES 1
pour obtenir
le grade de :
DOCTEUR DE L’UNIVERSITÉ DE RENNES 1
Mention Physique
PARADAM-BERRET Matthieu
Équipe d’accueil : IRM-Food, UR TERE, CEMAGREF de Rennes
École Doctorale : Vie-Agro-Santé
Composante universitaire : UFR S.V.E.
Étude de la cristallisation des lipides :
Approche multi-
échelle par la relaxation et
la diffusion par RMN
SOUTENUE le 2 avril 2009 devant la commission d’ExamenCOMPOSITION DU JURY :
Mme Sylviane LESIEUR Rapporteur
M. Jean-Pierre KORB Rapporteur
M. Bertrand TOUDIC Examinateur
M. Wouter MEEUSSEN Examinateur
M. François MARIETTE Directeur de thèse
CemOA
: archive
ouverte
d'Irstea
Ce projet a été réalisé dans les équipes IRM-Food du Cemagref de Rennes et BIA-ISD de l’INRA de Nantes entre Février 2006 et Mars 2009. Il a fait l’objet d’une collaboration scientifique et financière entre le Cemagref et l’Inra, fait rare, mais comme l’a démontré cette thèse, peut apporter beaucoup à ces deux organismes de recherche. Je tiens donc à remercier les deux parties pour leur soutien financier qui m’ont permis de travailler dans de bonnes conditions.
Il commence à être de notoriété publique que travailler au sein du Cemagref de Rennes est un réel plaisir, et je ne peux pas déroger à la règle : travailler pour l’équipe IRM-Food fut un pur bonheur, et l’ambiance chaleureuse et la bonne humeur de tous y sont certainement pour beaucoup. Il ne faut pas non plus oublier les compétences de recherche de chacun, tant sur le point technique que scientifique. S’il est parfois difficile de se confronter à la démarche qualité entreprise au sein de l’équipe, elle m’a permis d’avoir une rigueur dans mon travail de recherche qui me servira inévitablement par la suite. Je tiens donc à remercier Mr Nicolas Petit, directeur du groupement de Rennes, et Mr Armel Davenel, responsable de l’unité TERE, de m’avoir accueilli dans leurs locaux et permis de réaliser cette thèse.
Je voudrais également remercier tous les membres du jury, qui ont accepté de lire et de juger ce travail. Tout d’abord je remercie Mme Sylviane Lesieur qui en tant que rapporteur s’est beaucoup investie alors qu’elle n’était pas dans son domaine. Egalement comme rapporteur, je voudrais remercier Mr Jean-Pierre Korb pour son haut niveau de compétence scientifique et ses remarques très intéressantes. Je remercie également Mr Wouter Meeussen pour s’être déplacé de Belgique et qui a dû beaucoup apprécier de voir le travail scientifique qui peut être effectué dans l’équipe IRM-Food. Enfin, je remercie Mr Bertrand Toudic, président du jury et examinateur pertinent.
Je tiens à remercier tout particulièrement deux personnes qui ont beaucoup contribué au projet. Tout d’abord François Mariette qui a dirigé ces travaux. Il a su me conseiller, m’apprendre beaucoup de choses, faire preuve de patience dans les moments plus durs. J’ai apprécié l’évolution de nos conversations au fur et à mesure de la thèse pendant lesquelles mon avis est devenu de plus en plus important, et la confiance que tu avais en moi pour
CemOA
: archive
ouverte
d'Irstea
triglycérides de la meilleure des manières. Je le remercie également de m’avoir laissé tranquillement travailler au Cemagref à Rennes, et de ne pas avoir eu à faire les trajets jusqu’à Nantes trop souvent. Travailler avec vous a été une expérience enrichissante qui m’a permis d’apprendre beaucoup sur la recherche.
J’adresse également mes remerciements à Corinne Rondeau-Mouro pour son aide précieuse dans le domaine de la RMN haut champ, et pour les nombreuses discussions que nous avons pu avoir sur le projet. Sa relecture poussée sur les aspects de la RMN permet à ce manuscrit de n’en être que meilleur.
Je voudrais remercier Christelle Lopez qui pu apporter un regard critique sur le travail en cours. J’ai également apprécié sa présence pour le congrès à Athènes qui m’a permis de ne pas être tout seul perdu au milieu de cette grande foule de scientifiques.
Merci à Bruno Pontoire pour son aide lors des expériences de diffraction des rayons X.
Je remercie chaleureusement Marine Boulard pour son très bon travail en tant que stagiaire. Sa rigueur et ses compétences scientifiques lui ont permis d’apporter sa pierre à l’édifice de cette thèse et m’ont permis à moi de rendre le travail d’encadrement très facile.
Je voudrais remercier tous mes collègues du Cemagref de Rennes pour l’environnement privilégié qu’ils m’ont offert lors de ce projet. Que ce soit les membres de l’équipe ou les autres, j’y ai rencontré de nombreuses personnes qui m’auront permis de m’épanouir. Je préfère ne pas faire de listes tellement il y aurait de gens à citer, mais je pense bien évidemment à tous les membres actuels et passés de l’équipe, à mes partenaires de foot, squash, tandem, tennis, karting, aux joueurs du midi jeux, aux personnes rencontrées lors des activités du groupe… Si je dois n’en citer qu’un, ce sera évidemment François avec qui nous avons traversé ensemble ces 3 années de thèse. Il fut mon collègue de bureau, mon partenaire multi-sport et un ami. Je vais également remercier Franck, qui par ses connaissances nous a permis d’avoir des discussions très intéressantes, que ce soit sur le travail scientifique ou sur la vie de chercheur en général.
CemOA
: archive
ouverte
d'Irstea
alimentaire, cela permet de grands moments de dégustation privilégiée.
Je tiens également à remercier mes collègues de l’Inra. Passant l’essentiel de mon temps à Rennes, je fus très rarement présent là-bas, mais j’y fus toujours bien accueilli.
Je voudrais également remercier ma famille pour m’avoir supporté pendant ces 3 années, tant d’un point de vue matériel que psychologique, et de m’avoir permis d’en arriver là aujourd’hui.
Merci à Guilaine, qui a toujours cru en moi et a su traverser ces épreuves avec moi. Son amour est une source de joie quotidienne qui m’a permis d’avancer tout au long de ce projet. CemOA : archive ouverte d'Irstea / Cemagref
A la mémoire de mon grand-père,
CemOA : archive ouverte d'Irstea / CemagrefSOMMAIRE
Introduction... 1
Chapitre I. Etude Bibliographique et Objectifs
1. Généralités sur les lipides...4
1.1
Définition ...4
1.2
Propriétés physiques des lipides ...5
2. La cristallisation des lipides...9
2.1
Généralités sur la cristallisation...9
2.1.1 La surfusion ...10
2.1.2 La nucléation ...11
2.1.3 La croissance des cristaux ...12
2.1.4 Le murissement d’Ostwald...13
2.2
Le polymorphisme des triacylglycérols...14
2.2.1 Organisations transversales des chaînes hydrocarbonées ...16
2.2.2 Organisation longitudinale des chaînes aliphatiques...18
2.2.3 Propriétés microscopiques des triacylglycérols ...19
2.2.4 Organisation des triacylglycérols à l’état liquide ...22
2.3
Les triacylglycérols hétérogènes et les mélanges de triacylglycérols ...24
2.3.1 Triacylglycérols saturés mixtes...24
2.3.2 Triacylglycérols insaturés...25
2.3.3 Les mélanges de triacylglycérols à l’état solide ...27
2.3.4 Les diagrammes ternaires...28
2.4
Les émulsions...29
3. Caractérisation des lipides ...29
3.1
La calorimétrie ou Analyse Enthalpique Différentielle...30
3.2
La diffraction des rayons X ...32
3.2.1 Principe...32
3.2.2 Description des sous-cellules par diffraction des rayons X...34
CemOA
: archive
ouverte
d'Irstea
3.2.3 Etude des systèmes en dynamique...34
3.2.4 Mesure de la taille des cristaux ...35
3.3
La Résonance Magnétique Nucléaire (RMN) ...36
3.4
Les autres techniques ...37
3.4.1 La spectroscopie infrarouge ...37
3.4.2 Les ultrasons ...37
3.4.3 La microscopie à lumière polarisée...38
3.4.4 Autres techniques ...39
4. La Résonance magnétique nucléaire (RMN) ...40
4.1
Principe Général...40
4.2
Le déplacement chimique ...41
4.3
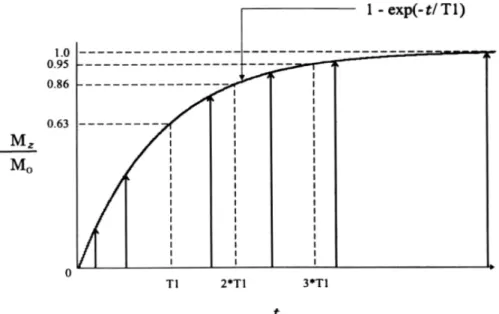
La relaxation ...42
4.3.1 La relaxation longitudinale...43
4.3.2 La relaxation transversale ...45
4.3.3 Le second moment dipolaire M2...46
4.3.4 Le temps de relaxation T1ρ...47
4.4
Interprétations phénoménologiques des paramètres de relaxation ...48
4.4.1 Les mécanismes de relaxation ...49
4.4.2 Interprétations dynamiques et moléculaires ...49
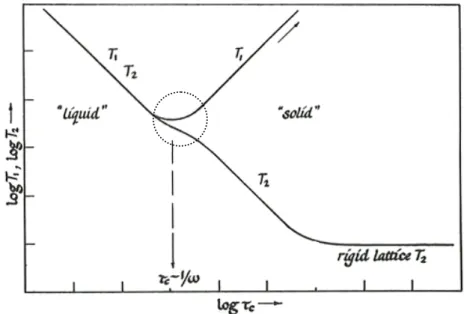
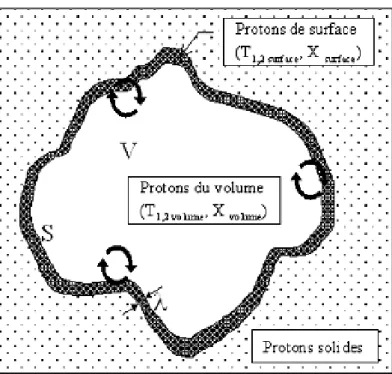
4.4.3 L’effet du confinement sur les temps de relaxation ...53
4.5
Mesure du coefficient de diffusion par RMN...56
4.5.1 Principe...56
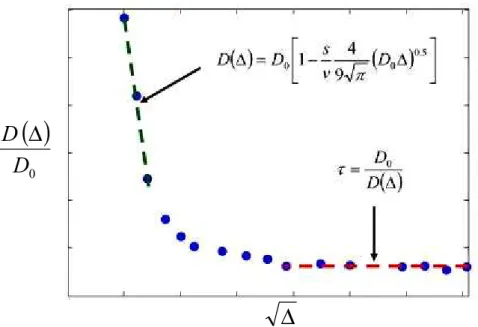
4.5.2 Diffusion libre et diffusion restreinte ...59
4.6
Applications de la RMN aux triacylglycérols ...62
4.6.1 La détermination du taux de solide par RMN...62
4.6.2 Caractérisation du polymorphisme...66
4.6.3 Mesure de la taille des gouttelettes dans les émulsions...73
5. Description du projet...74
6. Références...76
CemOA : archive ouverte d'Irstea / CemagrefChapitre II. Matériels et Méthodes
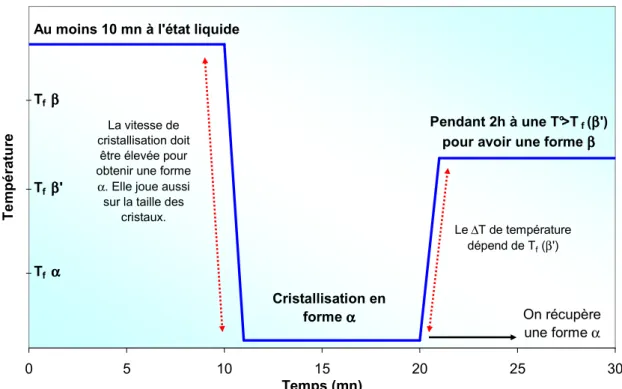
1. Préparation des échantillons...88
1.1
La cristallisation des triacylglycérols ...88
1.2
Stabilité des formes polymorphiques...90
1.3
La préparation des mélanges binaires de triacylglycérol...91
2. Mesures de spectres de poudre par diffraction des rayons X ...92
2.1
Conditions de mesures ...92
2.2
Mesure de l’épaisseur des cristaux par DRX...93
3. La microscopie à lumière polarisée...95
4. L’analyse enthalpique différentielle...95
5. Les mesures par RMN...95
5.1
Les mesures à bas champ...95
5.1.1 La mesure du FID ...96
5.1.2 La mesure du T1...99
5.1.3 La mesure du T2 liquide ...100
5.1.4 Mesure du coefficient de diffusion ...101
5.2
Les mesures à haut champ ...103
5.2.1 Spectre de la tricaprine liquide...104
5.2.2 Spectres CP/MAS des triacylglycérols à l’état solide...104
5.2.3 Mesures du T1...107
5.2.4 Mesure du T2...108
6. Références...110
Chapitre III. Etude du polymorphisme des triacylglycérols par RMN
Publication n°1:
Study of triacylglycerol polymorphs by Nuclear Magnetic Resonance:
effects of temperature and chain length on relaxation parameters...111
CemOA
: archive
ouverte
d'Irstea
Chapitre IV. Relation entre la taille des cristaux et le T
1Publication n°2:
Effects of crystal growth and polymorphism of triacylglycerols on NMR
relaxation parameters. Part 1: Evidence of a relationship between crystal
size and spin-lattice relaxation time...137
Chapitre V. Une étude solide – solide
Publication n°3:
Effects of crystal growth and polymorphism of triacylglycerols on NMR
relaxation parameters. Part 2: A solid-solid study...165
Chapitre VI. Evolution de la microstructure
Publication n°4:
Evolution of fat crystal network microstructure followed by NMR ...192
Chapitre VII. Discussion générale et perspectives
1. Discussion générale ...224
1.1
Le M
2et le T
2de la fraction cristallisée et le polymorphisme...226
1.1.1 M2 et polymorphisme : influence de la phase liquide ...229
1.1.2 M2 et polymorphisme : influence des mélanges solide/solide...230
1.2
Le T
1de la fraction cristallisée ...233
1.2.1 Le T1 et la taille des cristaux...234
1.2.2 Influence des mélanges solide/solide ...237
1.3
Evolution de la cristallisation du mélange binaire...238
1.4
Temps de relaxation T
2et T
1de la phase liquide ...240
CemOA
: archive
ouverte
d'Irstea
1.4.2 Influence du taux de solide sur le T2 et le T1 liquide ...242
1.4.3 Influence du stockage sur le T1 et le T2 liquide ...243
1.5
Diffusion de la phase liquide ...245
1.5.1 Influence du temps de diffusion sur le coefficient de diffusion ...245
1.5.2 Influence du taux de solide et du stockage sur le coefficient de diffusion ...246
1.6
Formation et évolution du réseau de cristaux de triacylglycérols ...248
2. Perspectives ...250
2.1
Perspectives de recherche plus fondamentales ...250
2.1.1 Mobilité locale et RMN à haut champ. ...250
2.1.2 Comportement de la forme
β
' ...2512.1.3 Quantification du polymorphisme...252
2.1.4 Estimation de l’épaisseur des cristaux ...252
2.1.5 Caractérisation de la topologie du réseau de cristaux...253
2.2
Extrapolation à des systèmes complexes ...254
2.3
La RMN un outil pour expliquer les propriétés mécaniques ? ...256
3. Conclusion générale...257
4. Références...260
CemOA : archive ouverte d'Irstea / CemagrefCCC = TG10 : Tricaprine LLL = TG12 : Trilaurine MMM = TG14 : Trimyristine PPP = TG16 : Tripalimitine SSS = TG18 : Tristéarine
TG = TA = TAG : Triacylglycérol = Triglycéride
RMN = NMR : Résonance Magnétique Nucléaire DRX = XRD : Diffraction des Rayons X
WAXS : Wide Angle X-Rays SAXS : Small Angle X-Rays
AED = DSC : Analyse Enthalpique Différentielle
FID : Free Induction Decay
C.P.M.G. : Carr-Purcell-Meiboom-Gill FSR : Fast Saturation Recovery
T1 : Temps de relaxation longitudinal = Temps de relaxation spin-réseau
T2 : Temps de relaxation transversal = Temps de relaxation spin-spin
M2 : Second Moment Dipolaire
T1,2vol = T1,2b : Temps de relaxation T1 ou T2 de la phase liquide pure
ρρρρ1,2 : Relaxivité de surface
D0 : Coefficient de diffusion du triacylglycérol pur
∆∆∆∆ : Délai entre les deux impulsions de gradients (ms) δδδδ: Durée de l’impulsion de gradient (ms)
g : Intensité du gradient appliqué (T/m)
SFC = Solid Fat Content : Taux de solide Tf = Tm : Température de fusion ηηηη : Viscosité cinématique CemOA : archive ouverte d'Irstea / Cemagref
Introduction
CemOA
: archive
ouverte
Introduction
Les lipides sont des constituants essentiels de nombreux produits alimentaires. Les triacylglycérols sont les éléments présents majoritairement dans des produits tels que le beurre, le chocolat, les crèmes ou les viennoiseries. Dans tous ces produits, la fraction solide de ces lipides, liée à la présence d’une forte fraction d’acides gras saturés, joue un rôle important sur leur texture. Sachant qu’actuellement les recommandations actuelles vont dans le sens de la réduction des acides gras saturés au profit des acides gras insaturés, il est important de pouvoir maîtriser la structure physique du réseau de cristaux de la matière grasse. En effet, la structure du réseau joue un rôle capital sur les propriétés physiques et rhéologiques du produit fini. Ainsi, la connaissance du réseau de cristaux permettrait de procéder au remplacement sans altérer les caractéristiques mécaniques et sensorielles des produits alimentaires. Le réseau de cristaux couvre plusieurs niveaux d’échelle et chacun d’entre eux doit être caractérisés. Il est donc important de connaître la quantité de cristaux, ainsi que leur organisation à l’échelle moléculaire (polymorphisme), microscopique (taille des cristaux) et macroscopique (taille des assemblages dans le réseau).
Les différentes techniques utilisées actuellement pour caractériser le réseau sont la diffraction des rayons X (DRX), pour déterminer le polymorphisme, la calorimétrie, pour connaître les propriétés de fusion et de cristallisation de la matière grasse, la Résonance Magnétique Nucléaire (RMN) bas champ pour déterminer le taux de solide, et la rhéologie et la microscopie pour déterminer la dimension fractale du réseau, seul paramètre utilisé pour le moment pour caractériser la microstructure. Toutes ces techniques n’apportent qu’une information parcellaire de la complexité du réseau cristallin et donc leur combinaison est nécessaire pour décrire les mécanismes. Des démarches ont été développées telles que le couplage de la DRX et de la DSC. Pour les autres techniques le recoupement est souvent délicat car il est nécessaire de valider que les cycles thermiques appliqués sont les mêmes pour toutes les techniques, ce qui dans de nombreux cas est difficile du fait des contraintes expérimentales imposées par les techniques elles-mêmes. Dans ce contexte, la RMN apparaît comme une technique prometteuse pour apporter des informations à différentes échelles. En effet, la RMN bas champ a déjà montré de nombreuses aptitudes quant à la caractérisation de la structure de produits agro-alimentaires tels que les produits laitiers. Cette caractérisation peut se faire par la mesure des temps de relaxation et des coefficients de diffusion. Les temps
CemOA
: archive
ouverte
d'Irstea
de relaxation sont sensibles à la mobilité interne des molécules, tandis que la diffusion est sensible aux déplacements des molécules. La mesure du coefficient de diffusion est actuellement la méthode utilisée pour déterminer la taille des gouttelettes dans une émulsion.
Quelques études ont montré qu’il était possible de déterminer le polymorphisme à partir des spectres 13C, des signaux de décroissance du signal, de la mesure du second moment dipolaire M2 ou du temps de relaxation spin-réseau T1. Ces mesures se basent sur les
différentes mobilités existant dans les formes polymorphiques. Le T1 s’est d’ailleurs révélé
être le paramètre le plus sensible au polymorphisme. Cependant, malgré ces études, la RMN n’est actuellement pas utilisée pour obtenir des informations sur la structure du réseau de cristaux de la matière grasse. De plus, les mécanismes qui participent à la relaxation sont encore méconnus. Le T1 et le M2 sont-ils réellement liés au polymorphisme ou bien à un autre
paramètre du réseau ? L’écart important entre les paramètres de relaxation des différentes formes polymorphiques existe t’il pour tous les triacylglycérols et toutes les températures ? N’est-il pas possible d’obtenir d’autres informations sur le réseau de cristaux de la matière grasse par des mesures de relaxation et de diffusion ?
Ce travail vise donc à essayer de répondre à l’ensemble de ces questions. Cette thèse a tout d’abord pour objectif de comprendre à quelles caractéristiques du réseau sont réellement sensibles les paramètres de relaxation. Dans un premier temps, le travail s’est focalisé sur des systèmes purs dont les mécanismes de cristallisation étaient décrits dans la littérature. Dans un deuxième temps, les résultats obtenus pour les systèmes purs ont été extrapolés à un mélange binaire de triacylglycérols sous forme cristallisé puis dans le cas de mélanges partiellement liquides. Enfin, les mesures des temps de relaxation et du coefficient de diffusion de la phase liquide ont été utilisées pour déterminer les paramètres dimensionnels du réseau de cristaux de la matière grasse.
Ce projet de thèse a été réalisé au sein du Cemagref de Rennes et de l’Inra de Nantes. Le manuscrit de la thèse est organisé entre différents chapitres :
Le chapitre I est une revue bibliographique présentant les triacylglycérols avec leurs propriétés physiques, leur cristallisation et leurs méthodes de caractérisation actuelles. La Résonance Magnétique Nucléaire est présentée dans la deuxième partie de ce chapitre, ainsi
CemOA
: archive
ouverte
d'Irstea
Le chapitre II est consacré à la description des méthodes et des protocoles utilisés.
Puis les résultats de ce travail qui ont fait l’objet de quatre publications sont présentés :
Le chapitre III est constitué de la publication n°1. Il présente l’étude du polymorphisme des triacylglycérols saturés purs et des effets de la température et de la longueur de chaine sur les paramètres de relaxation RMN.
Le chapitre IV traite lui des effets de la croissance des cristaux et du polymorphisme sur les paramètres de relaxation dans un mélange solide-liquide de triacylglycérols. Cela permit de mettre en évidence l’existence d’un lien entre la taille des cristaux et le temps de relaxation T1. Ce chapitre est constitué de la publication n°2.
Le chapitre V est lui la suite logique du chapitre précédent puisqu’il utilise cette relation entre le T1 et la taille des cristaux pour obtenir des informations sur la cristallisation d’un
mélange solide de triacylglycérols (publication n°3).
Le chapitre VI est lui composé de la publication n°4 et se concentre sur l’étude de la partie liquide du réseau de cristaux de triacylglycérols avec des mesures de temps de relaxation et de coefficient de diffusion.
Enfin, le chapitre VII présente une discussion générale reprenant l’ensemble des résultats obtenus au cours de cette étude et les différentes perspectives qui s’en sont dégagées.
CemOA : archive ouverte d'Irstea / Cemagref
Chapitre I
Etude bibliographique et
objectifs
CemOA : archive ouverte d'Irstea / CemagrefLes lipides sont une des trois classes de molécules organiques biologiquement essentielles avec les glucides et les protéines. Ils se trouvent dans tous les organismes vivants et jouent un rôle capital pour le métabolisme. Ce sont également les constituants majeurs des matières grasses naturelles et donc souvent des produits alimentaires. Il existe de nombreux livres et revues traitant des propriétés physiques des lipides ainsi que de leur cristallisation. Toutes les informations suivantes sur les lipides sont extraites de ces publications. Pour les propriétés physiques, il est possible de citer Small (Small, 1986), Larsson (Larsson, 1994), Ghotra (Ghotra et al., 2002), Marangoni et pour la cristallisation Sato et Garti (Garti & Sato, 2001; Sato, 2001) et Himawan (Himawan et al., 2006).
1 Généralités sur les lipides
1.1 Définition
Il n’existe pas de définition précise pour les lipides. Ce sont des molécules de poids moléculaire intermédiaire (entre 100 et 5000 g.mol-1) qui contiennent des acides gras à chaînes aliphatiques pouvant posséder une ou plusieurs insaturations (Small, 1986). Il existe deux grandes classes de lipides :
- Les phospholipides. Ce sont des lipides de structures qui constituent les membranes cellulaires.
- Les triacylglycérols (ou triglycérides). Ce sont des lipides de réserve de nombreuses plantes et animaux. Ceux-ci sont constitués de trois chaînes linéaires d’acides gras estérifiées sur les différentes fonctions alcools du glycérol. Ils possèdent différentes organisations à l'état solide.
Les stéroïdes, les savons, ou encore les détergents sont également des lipides. Ils sont aussi très présents dans le domaine de la pharmaceutique ou encore de la cosmétique. Ces molécules sont donc très présentes dans notre environnement.
Les triacylglycérols sont les constituants majeurs des huiles et des matières grasses naturelles animales et végétales et de ce fait ils sont à la base de nombreux produits alimentaires. C’est le cas du chocolat, du beurre, de la margarine, des viennoiseries ou encore
CemOA
: archive
ouverte
d'Irstea
Figure 1. Formule générale des triacylglycérols.
La fabrication de ces produits fait intervenir des matières grasses animales ou végétales majoritairement saturées comme la matière grasse du lait, le beurre de cacao ou l’huile de palme. Les caractéristiques structurales des réseaux formés par les cristaux de triacylglycérols dans la matière grasse sont importantes car elles confèrent au produit fini ses propriétés d’usages comme la texture, le goût ou la tartinabilité.
1.2 Propriétés physiques des lipides
Une des plus intéressantes qualités des lipides est leurs comportements physiques très variés. Ces comportements dépendent de la taille et de la structure des parties polaires et apolaires (acides gras) des molécules. Les lipides présentant une partie polaire importante, vont interagir avec l’eau pour former de nombreuses structures (lyotropisme) qui peuvent être trouvées dans le Tableau 1. La nature et la structure de la partie apolaire va influencer la structure des cristaux en phase solide (polymorphisme).
Acide gras CemOA : archive ouverte d'Irstea / Cemagref
Classe de lipide Solubilité dans l’eau Propriétés de surface Exemple
Non Polaire Insolubles Ne s’étalent pas sur l’eau
Alcanes, carotène, ester de cholestérol,
cires
Polaire
Classe I Insolubles
S’étalent pour former des mono couches stables
Triacylglycérols, diacylglycérols,
alcools gras
Polaire Classe II
Insolubles mais gonflent en présence d’eau
S’étalent pour former des mono couches stables
Phospholipides, monoacylglycérols,
glycolipides
Polaire Classe IIIa
Solubles, forment des micelles dans l’eau et des
cristaux liquides pour de faible concentration en eau
S’étalent mais forment des mono couches
instables
Savons, détergents, lysophospholipides
Polaire Classe IIIb
Soluble, forment des micelles mais pas de cristaux
liquides
S’étalent mais forment des mono couches
instables
Sels biliaires, pénicilline
Tableau 1. Comportement des lipides suivant leur polarité.
Les triacylglycérols sont peu polaires et insolubles dans l’eau, ils forment des émulsions en présence de celle-ci. Pour ces molécules, le polymorphisme cristallin est plus important que les interactions avec l’eau. Du fait de la grande diversité d’acides gras, les triacylglycérols ont des propriétés physiques très différentes comme leur température de fusion ou leur mécanisme de cristallisation. Ce dernier point est très important car ce sont souvent les caractéristiques du réseau cristallin qui influent le plus sur les propriétés physiques des triacylglycérols et qui confèrent aux aliments leurs propriétés d’usages.
CemOA
: archive
ouverte
d'Irstea
Ces dernières peuvent être :
- la tendreté et la texture ; - le goût en bouche ;
- les propriétés mécaniques ; - l’incorporation de l’air ; - le transfert de chaleur ; - la durée de conservation.
Toutes ces caractéristiques sont très importantes pour le domaine agroalimentaire. Deux facteurs jouent sur les capacités d’une matière grasse à présenter ces propriétés : ce sont d’une part la quantité de cristaux, appelé communément « taux de solide », et la structure de ces cristaux. La structure des cristaux est en fait un assemblage qui dépend :
- de l’organisation des triacylglycérols les uns par rapport aux autres à l’échelle moléculaire (≈ Angstrom) qui suivant la composition de ceux-ci et des conditions thermo-mécaniques peut présenter des structures différentes qui à leur tour ont des points de fusion différents ;
- de la forme et de la taille des cristaux formés (≈ µm) ;
- et de la forme et de la taille des agrégats cristallins formant un réseau tridimensionnel (≈ 100 µm).
La taille moyenne des cristaux a des conséquences importantes sur la rhéologie et les propriétés organoleptiques des produits alimentaires (Narine & Marangoni, 1999b). Il apparaît donc essentiel de pouvoir connaître la structure physique des triacylglycérols, afin de pouvoir donner au produit final les caractéristiques désirées. Le réseau de cristaux présente une large gamme d’échelles qui peut se décomposer de la façon suivante (Tang & Marangoni, 2007) :
CemOA
: archive
ouverte
d'Irstea
Figure 2. Représentation du réseau de cristaux aux différentes échelles.
Dans une lamelle, dont l’épaisseur d varie en fonction de la longueur de la chaine latérale, il y a un assemblage de triacylglycérols. A cette échelle, c’est le polymorphisme qui permet de caractériser la matière grasse. Un domaine est composé d’un ensemble de lamelles et un cristal est formé par un ensemble de domaines. Le nombre de domaines composant le cristal permet de déterminer l’épaisseur des cristaux. Les cristaux ont tendance à s’assembler pour former des clusters. A ce niveau, c’est l’échelle micrométrique qui est pertinente. Ces assemblages de cristaux forment des flocs puis un réseau. La structure de ces ensembles est maintenue par différentes forces liées aux interactions entre les cristaux. La taille de ces ensembles est d’environ 100µm.
Sachant que l’organisation du réseau joue un rôle prépondérant sur les propriétés physiques des produits finis, de nombreuses tentatives ont été réalisées pour modéliser le réseau. Un premier modèle mécanique fut développé par Van Den Tempel dans lequel des attractions de Van der Walls permettait de maintenir agréger les particules de la matière grasse (Van Den Tempel, 1979). Cependant, les propriétés du réseau ne purent être décrites quantitativement par ce modèle. L’introduction de la dimension fractale pour caractériser les gels colloïdaux dans les années 90 a permis d’ouvrir de nouvelles perspectives pour la
CemOA
: archive
ouverte
d'Irstea
modélisation du réseau de cristaux des matières grasses. Vreeker fut le premier à suggérer que le réseau de cristaux pouvait avoir une nature fractale (Vreeker et al., 1992). Il utilisa la dimension fractale pour quantifier la relation entre la masse d’un agrégat et sa taille. Quelques années plus tard, Marangoni développa les applications du modèle fractal pour le réseau de cristaux en deux puis en trois dimensions (Narine & Marangoni, 1999a; Tang & Marangoni, 2006; Tang & Marangoni, 2008). Il réussit à relier le module élastique G’ des réseaux de cristaux de matière grasse à la fraction volumique de la phase cristallisée grâce à la dimension fractale du réseau. La dimension fractale du réseau de cristaux peut donc être déterminée par des mesures de G’ en fonction du taux de solide, qui peut être rapproché à la fraction volumique de la phase solide, ou par des observations microscopiques en lumière polarisée. Cependant, ce paramètre n’est pas suffisant pour caractériser complètement la microstructure des réseaux de cristaux de la matière grasse. En effet, ce réseau s’est révélé fractal uniquement pour une échelle supérieure au cluster. Il est donc nécessaire d’étudier le réseau à chaque échelle inférieure pour essayer d’obtenir une caractérisation plus complète. La première étape pour y parvenir consiste à étudier la cristallisation qui est relativement complexe pour les triacylglycérols.
2 La cristallisation des lipides
2.1 Généralités sur la cristallisation
La cristallisation est une transition de premier ordre d’un ensemble de molécules de l’état liquide à l’état solide de telle sorte que les molécules à l’état solide s’assemblent de manière régulière pour former une structure solide.
Lorsqu’un liquide est refroidi à une température inférieure à son point de fusion, le processus de cristallisation devient thermodynamiquement favorable puisque l’énergie libre de l’état solide devient moins importante que celle de l’état liquide. Pour un processus isobare, la variation de l’énergie libre de Gibbs entre les états solide et liquide est donnée par la relation :
S
T
H
G
=
∆
−
∆
∆
où ∆H et ∆S sont les modifications d’enthalpie et d’entropie associées à la transition de phase
CemOA
: archive
ouverte
d'Irstea
∆G<0, l’état solide est favorisé. La différence d’enthalpie entre les états solide et liquide est majoritairement due aux différences d’organisations moléculaires et à l’importance des interactions intermoléculaires. La cristallisation est un processus exothermique qui s’accompagne d’un dégagement de chaleur (∆H<0).
Plusieurs paramètres ont une influence sur la cristallisation comme :
- le transfert de chaleur ; - l’agitation mécanique ; - les additifs et impuretés ; - la viscosité de l’ensemble ;
- la composition chimique des triacylglycérols.
Ces paramètres jouent sur la thermodynamique et la cinétique du processus de cristallisation. Ce processus peut-être divisé en trois étapes que sont la surfusion, la nucléation et la croissance des cristaux (Boistelle, 1988).
2.1.1 La surfusion
La cristallisation ne peut se produire que si le liquide est refroidi en dessous de son point de fusion. Néanmoins, même si la cristallisation est thermodynamiquement favorable, l’état liquide peut persister pendant une période de temps considérable, car une importante barrière d’énergie libre doit être dépassée avant que la transition de phase liquide-solide se produise. La différence d’énergie libre ∆G négative entre les états liquide et solide favorise la cristallisation, mais il existe une barrière d’énergie ∆G* qui doit être dépassée avant que la cristallisation ait réellement lieu. L’importance de l’énergie d’activation de cristallisation, qui est fortement liée à la température, dépend de la capacité d’un nucléon stable à se former dans le liquide.
Le degré de surfusion est défini de la manière suivante : ∆T = T – Tf, où T est la
température du matériel et Tf son point de fusion. La valeur de ∆T à laquelle la cristallisation
est observée dépend des paramètres énoncés auparavant. Cette valeur permet également de déterminer la vitesse de refroidissement ou de cristallisation définie comme :
CemOA
: archive
ouverte
d'Irstea
c c
t
T
V
=
∆
Avec tc le temps nécessaire pour atteindre T.2.1.2 La nucléation
Les molécules à l’état liquide suivent un mouvement chaotique, qui est déterminé par un compromis entre l’entropie et l’adoption de paquets arrangés. Au dessus du point de fusion, des ensembles de petites molécules se regroupent pour former un cristal et se dissocient de manière dynamique, il y a un équilibre entre la forme cristalline et le liquide, les cristaux ne se forment pas. Lorsque la température diminue, les agrégats de petites molécules sont de plus en plus gros jusqu’à former un nucléus stable. La formation de nucléi dans le liquide conduit à une diminution de l’énergie libre du système, mais leurs structures exactes dans le liquide ne sont pas connues. La taille minimale d’un nucléus pour qu’il soit stable dépend de l’énergie libre du système. L’équation de Gibbs-Thomson permet de caractériser ce phénomène dans le cas de la cristallisation de la matière grasse :
( )
( )
2 2 2 2 33
16
T
H
T
V
G
f f s m r∆
∆
=
∆
πδ
Avec : ∆Gr l’énergie libre nécessaire pour former un nucléus stable de rayon r (J), δ l’énergie libre de surface solide-liquide par unité de surface (J.m-2), Vms volume molaire du solide
(m3.mol-1), ∆Hf l’enthalpie de fusion (J.mol-1), Tf et ∆T étant décrits dans la partie précédente.
Ainsi, plus le ∆T sera important, plus il y aura de nucléi formés.
Il existe différents types de processus de nucléation qui sont la nucléation homogène et la nucléation hétérogène. Lorsque la nucléation n’est pas catalysée par la présence d’interfaces ou de particules extérieures, il s’agit d’une nucléation homogène primaire. Cependant, ce cas est rare car il y a toujours la présence de surfaces qui vont induire une orientation et un ordre. Deux types de mécanismes de nucléation hétérogène existent. La nucléation
hétérogène primaire se produit lorsque la nucléation est catalysée par du matériel ayant une
structure chimique différente de celle du liquide qui cristallise. La nucléation hétérogène
secondaire se produit lorsque des cristaux du même matériel sont présents dans le liquide. La
CemOA
: archive
ouverte
d'Irstea
présence d’impuretés peut induire la nucléation à des degrés de surfusion plus faibles que ceux requis pour la nucléation homogène.
Des impuretés typiques susceptibles de catalyser la nucléation des matières grasses sont les particules de poussière, la surface interne du récipient contenant la matière grasse, l’interface huile-eau d’une gouttelette d'émulsion et différents constituants chimiques présents dans la matière grasse. Il a été montré que des monoacylglycérols, des diacylglycérols ou même des triacylglycérols pouvaient agir comme des impuretés catalytiques pour la cristallisation des triacylglycérols (Skoda & van den Tempel, 1963).
2.1.3 La croissance des cristaux
La croissance des cristaux se produit une fois que des nucléi stables se sont formés dans le liquide en surfusion. Les cristaux croissent par incorporation de molécules du liquide à l’interface solide-liquide. La vitesse de croissance des cristaux dépend de la diffusion d’une molécule du liquide vers l’interface solide-liquide puis de son incorporation dans le cristal. Ces facteurs sont dépendants de la température. En effet, la viscosité du liquide augmente lorsque la température diminue, ce qui retarde la diffusion des molécules à l’interface liquide-cristal. La vitesse de croissance des cristaux d’un triacylglycérol augmente initialement avec l’augmentation du degré de surfusion, atteint un maximum pour certaines températures puis décroît pour des degrés de surfusion plus importants (Dickinson & McClements, 1996), tout ceci étant relié à l’équation de Gibbs-Thomson.
La taille et la morphologie des cristaux formés dépendent d’un certain nombre de facteurs à la fois interne (comme l’organisation moléculaire et les forces intermoléculaires) et externes (la température, le refroidissement, le cisaillement et les impuretés). La vitesse de refroidissement d’une matière grasse à une température inférieure à son point de fusion a une influence sur le nombre et la taille des cristaux formés.
Le degré d’organisation des molécules dans les cristaux dépend également de la vitesse de refroidissement. Lorsqu’une matière grasse est refroidie lentement, ou lorsque le degré de surfusion est faible, les molécules ont suffisamment le temps d’être incorporées dans le cristal, conduisant à la formation de gros cristaux. Avec des vitesses de refroidissement
CemOA
: archive
ouverte
d'Irstea
suffisamment de temps pour pouvoir s’organiser de manière la plus compacte possible avant qu’une autre molécule soit incorporée à la surface. En conséquence un refroidissement rapide tendra à produire des cristaux moins bien organisés, plus petits et dans lesquels les molécules sont arrangées d’une manière moins compacte (Lopez, 2001).
Il existe donc une compétition entre la nucléation et la croissance des cristaux selon le mode de refroidissement puisqu’à vitesse de cristallisation élevée, la nucléation est favorisée. Cela conduit à différentes structures cristallines qui auront des propriétés différentes comme le polymorphisme ou la taille des cristaux.
Il a été montré que la vitesse de croissance des cristaux de triacylglycérols augmentait lorsque la longueur de chaine diminuait. Ainsi, pour un même degré de surfusion, les cristaux de triacylglycérols à petite longueur de chaine sont plus gros que les cristaux de triacylglycérols à grande longueur de chaine (Himawan et al., 2006).
2.1.4 Le murissement d’Ostwald
Après la nucléation, de nombreux cristaux de tailles et de formes différentes se développent dans la phase liquide. Cependant, tant que l’équilibre thermodynamique n’a pas été atteint, le système reste instable. Vu que les cristaux ont des tailles différentes, la condition d’énergie libre minimale ne peut pas être atteinte pour tous les cristaux. C’est ainsi que les cristaux de plus petites tailles fondent en solution pour recristalliser sur de plus gros cristaux. De cette manière, le système finira par se rapprocher de l’équilibre. Ce phénomène de dissolution et de recristallisation s’appelle le murissement d’Ostwald. Il est également possible d’observer ce phénomène dans les mousses et certaines émulsions où la taille des bulles et gouttelettes tend à augmenter et à s’homogénéiser au cours du temps. La cinétique de croissance de cristaux dû au murissement d’Ostwald a déjà été étudiée et il a été démontré que la taille des cristaux obéissait à une loi de puissance au cours du temps de la forme (Lifshitz & Slyozov, 1961; Binder, 1977; Donhowe & Hartel, 1996) :
n
t
t
L
(
)
∝
Avec L (t) la taille moyenne des cristaux et n l’exposant qui dépend de la géométrie du système. CemOA : archive ouverte d'Irstea / Cemagref
Cette relation montre que le réseau de cristaux de matière grasse continue à évoluer au cours du stockage conduisant à l’augmentation de la taille des cristaux.
2.2 Le polymorphisme des triacylglycérols
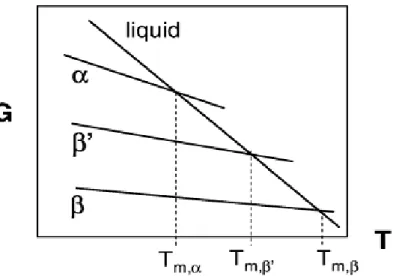
Le polymorphisme d’une molécule se définit par son aptitude à pouvoir cristalliser sous plusieurs formes cristallines correspondant à des arrangements moléculaires différents. Le polymorphisme est associé aux différents assemblages des chaînes hydrocarbonées. Le comportement à la cristallisation et à la fusion diffère entre les différentes formes ou structures cristallines car elles correspondent à des énergies libres différentes. En effet, la cristallisation et les transformations polymorphiques des triacylglycérols sont majoritairement déterminées par une stabilité thermodynamique, exprimée par l’évolution de l’énergie libre en fonction de la température (Figure 3). Les formes polymorphiques possédant une température de fusion faible révèlent des énergies libres plus importantes et une plus faible stabilité.
Figure 3. Relation entre l’énergie libre et la température pour les trois principales formes
polymorphiques des triacylglycérols (Himawan et al., 2006).
Le polymorphisme des triacylglycérols est de type monotropique, c’est-à-dire que les transitions cristallines sont irréversibles et s’effectuent toujours des formes cristallines les moins stables vers celle thermodynamiquement la plus stable.
Les triacylglycérols saturés purs cristallisent selon trois variétés polymorphiques principales notées α, β’ et β, correspondant à des arrangements de chaînes de plus en plus
CemOA
: archive
ouverte
d'Irstea
trans. Les deux premières variétés sont généralement instables et se transforment sous certaines conditions en forme stable β.
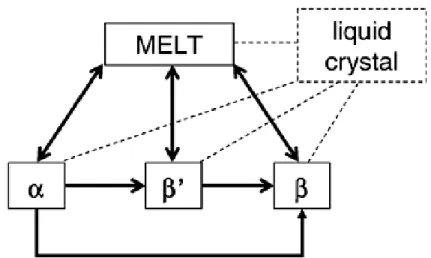
Pour les triacylglycérols, les transformations possibles pour obtenir une forme polymorphique obéissent au schéma suivant (Himawan et al., 2006) :
Figure 4. Transformations polymorphiques possibles pour les triacylglycérols en prenant en
compte l’état cristal liquide.
La forme β est la plus stable et est celle qui a la température de fusion la plus élevée. Il est nécessaire de distinguer l’état fondu et l’état cristal liquide. L’état fondu correspond aux triacylglycérols liquides dont la température de fusion de la forme la plus stable a été dépassée. L’état cristal liquide est un état fondu transitoire observé lors de transition solide-solide entre les formes polymorphiques. Dans ce cas, la transition est dite « melt-mediated ».
Les interactions entre les chaînes hydrocarbonées composées de groupe CH2 se font par
les forces de dispersion de Van Der Walls – London, s’exerçant seulement aux petites distances (Lopez, 2001). L’assemblage des chaînes dans un cristal est avant tout conditionné par ce type d’interaction, conduisant en premier lieu à des assemblages de chaînes parallèles entre elles, et à la formation de strates dans un deuxième temps.
Les chaînes hydrocarbonées aliphatiques ont la possibilité de s’empiler de différentes manières avec une organisation transversale dans un plan perpendiculaire aux chaînes, et une organisation longitudinale parallèle aux chaînes. Cette propriété est à l’origine du polymorphisme des triacylglycérols.
CemOA
: archive
ouverte
d'Irstea
2.2.1 Organisations transversales des chaînes hydrocarbonées
La sous-cellule cristalline est définie comme l’unité de base de l’organisation des lipides. Les différents types de sous-cellules rencontrés dans les différentes variétés cristallines des lipides peuvent se diviser en deux classes principales (Ollivon & perron, 1992).
La première classe est constituée de chaînes aliphatiques en arrangement compact dans lequel s’exercent des interactions spécifiques entre les chaînes. Ces sous-cellules présentent des réseaux bidimensionnels, normaux aux axes de chaînes, de type rectangulaire droit, orthorhombiques parallèles et perpendiculaires, ou obliques, triclinique parallèle et monoclinique parallèle. Les chaînes aliphatiques peuvent être soit parallèles entre elles soit perpendiculaires les unes aux autres.
B
Figure 5. Différentes configurations des sous-cellules cristallines. (A) Hexagonale. (B)
Orthorhombique perpendiculaire O ┴. (C) Triclinique parallèle O //.
A B C CemOA : archive ouverte d'Irstea / Cemagref
La seconde classe de sous-cellules comprend des assemblages de chaînes moins compactes. Les interactions entre les chaînes sont très atténuées en raison de leur inclinaison partielle autour de leur axe longitudinal ou celle de groupes CH2, qui conduit à un désordre
statistique autour des axes des chaînes. Il en résulte un réseau bidimensionnel hexagonal centré, dénommé généralement forme α. Les sous-cellules cristallines montrent des différences dans leur compacité, ce qui induit des différences de stabilité.
Dans le cas des triacylglycérols, trois principales formes de sous-cellule sont adoptées (Figure 5):
La forme α
Cette forme correspond à une organisation hexagonale. C’est la forme la moins stable et la moins dense car ses chaînes aliphatiques sont perpendiculaires au plan basal formé par les groupes méthyles terminaux (CH3) et sont parallèles entre elles. Elle s’obtient
uniquement par refroidissement rapide du liquide et il n’y a pas d’orientation privilégiée des zigzags plans.
La forme β’
La forme β’ est plus stable que la forme α et est réalisée en procédant d’abord au refroidissement du liquide jusqu’à la température de fusion de la forme α, puis en maintenant l’échantillon à cette température jusqu’à cristallisation complète. Elle peut aussi être obtenue dans certains cas par fusion d’α, suivie de recristallisation. La maille est orthorhombique, à sous-cellule O┴. Les chaînes sont inclinées sur leur plan de base.
La forme β
C’est généralement la forme la plus stable. Elle s’obtient soit par fusion de α ou de β’, soit par cristallisation du liquide aux environs de la température de fusion de β’, soit encore par cristallisation dans un solvant. La maille est triclinique à sous-cellule T //. Les chaînes sont encore plus inclinées sur leur plan de base que les chaînes de la forme β’. Les températures de fusion des différentes variétés polymorphiques des triacylglycérols sont croissantes dans l’ordre α, β’, β.
CemOA
: archive
ouverte
d'Irstea
Pour les triacylglycérols purs, la variété polymorphique la plus stable est la variété β. Cependant, pour des triacylglycérols contenant des acides gras de différentes longueurs ou des insaturations, la variété β’ peut être la forme la plus stable.
L’identification des différentes formes cristallines est obtenue par mesure des dimensions des mailles cristallines à partir des diagrammes de diffraction des rayons X aux grands angles (WAXS).
2.2.2 Organisation longitudinale des chaînes aliphatiques
L’organisation longitudinale des chaînes hydrocarbonées aliphatiques résulte de l’arrangement des molécules sous forme de strates. L’épaisseur de ces strates est proportionnelle à la longueur des chaînes (L) qui elle même est approximativement proportionnelle à leur nombre d’atomes de carbone (n) : L= 271. ×n+b (b est fonction de la nature des groupements polaires et terminaux de leurs empilements). L’épaisseur des strates reflète le nombre et l’inclinaison des chaînes aliphatiques qui sont superposées dans la strate.
Il existe trois principaux types d’empilement de chaînes : 2L (deux longueurs de chaînes), 3L (trois longueurs de chaînes) ou plus rarement 6L (six longueurs de chaînes). Les chaînes d’acide gras dans la molécule de triacylglycérols seraient arrangées en configuration chaise, c’est-à-dire qu’il y a deux chaînes adjacentes et la troisième chaîne d’acide gras est orientée dans la direction opposée.
CemOA
: archive
ouverte
d'Irstea
Figure 6. Exemple de strates à 2 ou 3 longueurs de chaîne adoptées par les triacylglycérols selon la nature des acides gras qui les constituent (Small, 1986).
Les organisations longitudinales des chaînes d’acides gras, principalement de types 2L et 3L, dépendent essentiellement de la composition en acides gras du triacylglycérol considéré. Les triacylglycérols possédant des acides gras similaires en longueur de chaînes et en insaturations, c’est-à-dire homogènes, donnent des strates de type 2L (Larsson, 1994). En revanche, s’il y a une ou plusieurs insaturations ou des acides gras de longueurs de chaînes différentes, les triacylglycérols ont tendance à former des strates 3L, avec ségrégation de la chaîne insaturée ou courte au sein de la même strate. Cette organisation longitudinale est susceptible d’être convertie de 2L en 3L ou de 3L en 2L au cours de transformations polymorphiques pour conduire à des formes plus compactes (Sato, 1996). Ces variations dans l'organisation longitudinale sont identifiées par des diagrammes de diffraction des rayons X aux petits angles (SAXS) et des températures de fusion différentes. Dans ce cas, la notation des différentes variétés se fera par ordre croissant de la plus stable à la moins stable, c’est-à-dire dans l’ordre décroissant de leurs points de fusion.
2.2.3 Propriétés microscopiques des triacylglycérols
Il existe donc une organisation des chaînes à l'échelle moléculaire (Å), qui conduit à certaines propriétés du réseau de cristaux. Cependant, il y a également une organisation de ces
CemOA
: archive
ouverte
d'Irstea
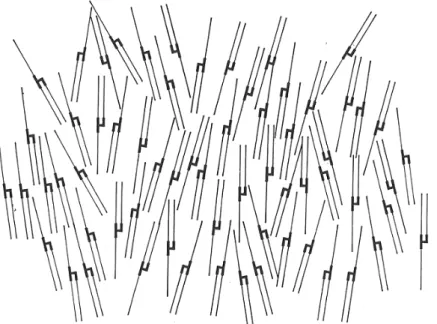
cristaux à l'échelle microscopique (≈ µm). Chaque type de polymorphes mène à une forme microscopique différente :
- La forme α conduit à un arrangement de cristaux de type informes et petit, - La forme β' conduit à un arrangement en forme de sphérulites,
- La forme β conduit à un arrangement en forme de lamelles.
Informe (αααα) Sphérulite (aiguille) (ββββ’)
Lamelles (ββββ)
Figure 7. Les trois différents types d'arrangement de cristaux de triacylglycérols en fonction de la forme polymorphique.
Du fait de cette diversité, les propriétés physiques des formes polymorphiques sont différentes et il est essentiel de pouvoir connaître les formes des cristaux formés. En analysant la cinétique de formation des cristaux, il est possible de mettre en évidence des mécanismes de cristallisation différents. Ces cinétiques sont souvent décrites par le modèle d’Avrami qui relie le taux de solide et le temps. (Avrami, 1941; Campos, 2005). Ce modèle pour la matière grasse prend la forme suivante :
CemOA
: archive
ouverte
d'Irstea
( )
n kte
SFC
t
SFC
− ∞−
= 1
Avec SFC (t) : SFC (%) en fonction du temps SFC∞ : SFC limite à l’infini
k : constante d’Avrami qui représente la constante du taux de cristallisation
n : exposant d’Avrami ou indicateur de cristallisation qui indique le mécanisme de croissance des cristaux.
L’indicateur d’Avrami est une fonction combinée de la dépendance au temps de la nucléation et du nombre de dimensions dans lesquelles la croissance a lieu. Différentes valeurs de n sont obtenues en fonction des conditions d’opération, celles-ci variant de 1 pour une croissance en forme de cylindre à 4 pour une croissance en forme de sphérulites. Cependant, suivant le type de systèmes étudiés, il peut être nécessaire de modifier le modèle d’Avrami (Narine et al., 2006).
Dans le même temps, il a été montré que la croissance des cristaux de triacylglycérols se faisait selon une direction privilégiée (Skoda & Van Den Tempel, 1967; Boistelle, 1988; Hollander et al., 1999; Hollander et al., 2002; Hollander et al., 2003). Ainsi, les aiguilles des sphérulites ont tendance à augmenter en longueur mais pas en épaisseur. Il en est de même pour les lamelles. Celles-ci grossissent dans un plan uniquement, ce qui donne des cristaux très fin et très long. La vitesse de croissance des cristaux de triacylglycérols saturés purs peut être trouvée dans la revue d’Himawan (Himawan et al., 2006).
En général, dans le domaine de l’agroalimentaire, la forme engendrée par les cristaux de β’ est la forme la plus recherchée car c’est celle qui a le plus de qualités gustatives. De plus, plus la structure est stable, plus la température de fusion est élevée et moins il y a de propriétés de mélange. C’est pourquoi souvent la transformation β’→β est considérée comme une dégradation du produit qu’il faut essayer d’éviter (Yano & Sato, 1999). L’exemple le plus connu pour ce phénomène est le blanchiment du chocolat qui est lié à une transformation polymorphique et se passe à taux de solide constant. Ainsi, au cours du temps il peut se passer deux différents phénomènes que sont les transformations polymorphiques, lorsque le système est instable, et une augmentation de la taille des cristaux due au murissement d’Ostwald. Ce
CemOA
: archive
ouverte
d'Irstea
dernier évènement s’accompagne d’une diminution de la surface spécifique comme il a pu être observé pour les triacylglycérols (Knoester et al., 1968).
Les différents facteurs qui influencent la stabilité du réseau de cristaux de la matière grasse sont donc:
- La longueur de la chaîne d’acide gras et sa diversité (insaturation, ramification) ; - L'organisation longitudinale du triacylglycérol ;
- La quantité de liquide présent dans le système gras ; - Les variations de la température pendant le stockage ; - La présence de triacylglycérols en phase liquide.
Tous ces facteurs, en conjonction avec l’histoire du procédé, touchent le comportement macroscopique des produits gras, qui est essentiel dans le domaine de l'agroalimentaire (Narine & Marangoni, 1999b).
2.2.4 Organisation des triacylglycérols à l’état liquide
L’organisation des triacylglycérols à l’état liquide fait l’objet d’un débat depuis de nombreuses années. A ce jour, il est possible de dénombrer trois modèles différents pour l’organisation de la phase liquide. Le premier modèle, le modèle smectique (Figure 8), a été proposé par Larsson suite à des études de diffraction des rayons X (DRX). Il observait une raie large pour les triacylglycérols à l’état liquide. Il en a donc conclu qu’en phase liquide, les triacylglycérols conservaient la même structure longitudinale que dans les cristaux, mais que les chaînes se trouvaient à l’état fondu (Larsson, 1992; Larsson, 1994). Des études de DRX avec une source synchrotron menées par Sato arrivèrent à la même conclusion (Sato et al., 1999). CemOA : archive ouverte d'Irstea / Cemagref
Figure 8. Représentation du modèle smectique de l’organisation des triacylglycérols dans l’état liquide selon Larsson et Sato.
En 1992, Cebula mena des études de diffraction de neutrons sur de la trilaurine deutérée, et arriva à la conclusion, très controversée par Larsson, que n’observant pas de signal sur le diagramme de diffraction, il ne pouvait exister d’organisation structurée pour les triacylglycérols liquides (Cebula et al., 1992). Il proposa alors le modèle nématique qui existe déjà pour certains cristaux liquides :
Figure 9. Représentation du modèle nématique proposé par Cebula pour la trilaurine liquide.
Plus récemment, un nouveau modèle discotique a été proposé par Corkery sur la base d’observations générales dans le domaine des cristaux liquides et par des simulations informatiques (Corkery et al., 2007). Ces résultats ont été appuyés par une étude par spectroscopie Raman qui, sans confirmer que le modèle était exact, démontra qu’il était au
CemOA
: archive
ouverte
d'Irstea
moins plausible (Da Silva & Rousseau, 2008). Ce dernier modèle permettrait d’expliquer les différents résultats observés par Larsson et Cebula.
Figure 10. Représentation du modèle discotique pour les triacylglycérols à l’état liquide. L’organisation des triacylglycérols à l’état liquide reste donc incertaine. Cependant, la conclusion de Corkery, et de Da Silva et Rousseau est de dire que Larsson et Cebula auraient tous les deux raison quant à leurs modèles. L’organisation smectique serait présente juste après la fusion de la phase cristalline, et cette organisation serait perdue à une température plus élevée pour privilégier une organisation nématique.
2.3 Les triacylglycérols hétérogènes et les mélanges de triacylglycérols
Les triacylglycérols sont rarement homogènes et coexistent en mélange dans la nature. Leur comportement est sensiblement différent lorsqu’il y a plusieurs triacylglycérols en présence, surtout en ce qui concerne la cristallisation. De nombreuses études ont été menées afin de pouvoir déterminer empiriquement le comportement d’un mélange de triacylglycérols, car actuellement, il est toujours impossible de modéliser ce comportement.
2.3.1 Triacylglycérols saturés mixtes
Les triacylglycérols saturés constitués par des acides gras de longueurs de chaînes différentes ont un polymorphisme plus complexe que celui des triacylglycérols homogènes. La compensation des excès de matière ou des vides créés par les variations relatives des longueurs de chaînes se fait soit grâce à un changement des sous-cellules cristallines (β ou β’) soit par des empilements des chaînes différents correspondant à des arrangements 3L, 4L ou 6L (Small, 1986). Il est nécessaire de distinguer plusieurs cas selon les différences de
CemOA
: archive
ouverte
d'Irstea
Si les triacylglycérols ont des acides gras de longueurs de chaînes peu différentes, il y a soit un arrangement 2L, soit un arrangement 3L. Si les triacylglycérols possèdent un acide gras très différent des deux autres, c’est-à-dire avec un écart entre les longueurs de chaîne supérieur ou égal à 4 carbones, l’observation par rayons X aux grandes distances montrent que l’empilement choisi est systématiquement 3L avec ségrégation de la chaîne possédant un défaut. Les vides ou les contraintes générés dans un empilement 2L seraient énergétiquement défavorables par rapport à un empilement 3L. La structure longitudinale dépend alors de la position de l'acide gras différent.
Figure 11. Possibles variations dans l’organisation des cristaux de la trilaurine causées par la variation de la longueur de la chaîne différente dans la position 1 ou 2.
2.3.2 Triacylglycérols insaturés
La présence d’une double liaison dans une chaîne hydrocarbonée provoque la rigidification locale de cette dernière en imposant, dans le cas des insaturations cis, un changement de direction aux zigzags plans de CH2. L’insaturation crée une perturbation dans
l’organisation des chaînes qui s’accompagne généralement d’une séparation de phase à l’échelle moléculaire (Ollivon & perron, 1992). Les chaînes insaturées cis se rassemblent en strates différentes de celles des chaînes saturées.
CemOA
: archive
ouverte
d'Irstea
Figure 12. Organisation longitudinale des chaînes possédant une insaturation.
Pour les chaînes insaturées trans, l'organisation longitudinale est peu différente du cas où les chaînes d'acides gras sont saturées.
Les triacylglycérols insaturés symétriques comme SOS (1,3-distéaroyl-2-oléoyl-sn-glycérol) ou POP (1,3 dipalmitoyl-2-oléoyl-sn-(1,3-distéaroyl-2-oléoyl-sn-glycérol) peuvent adopter un empilement de type 3L dans leur état thermodynamique le plus stable. Cependant il existe de nombreuses formes polymorphiques pour cette classe de triacylglycérols que Sato a résumé dans un article (Sato, 1996). Du fait de l’asymétrie des empilements formés, les mailles élémentaires peuvent contenir 6 chaînes inclinées selon l’axe des chaînes CH2 (6L) (Larsson, 1994).
Pour les triacylglycérols insaturés non symétriques, la conformation en forme de « chaise » n’est plus possible lorsque l’acide gras différent des autres est placé en position externe, 1 ou 3, sur le glycérol. Les diagrammes de diffraction des rayons X aux grandes distances mesurés par exemple pour les formes β’ de OPP et OSS (Sato et al., 1989) suggèrent un empilement de type 3L avec ségrégation de l’acide oléique en position 1 dans une monocouche et les chaînes en position 2 et 3 dans la bicouche. Dans ce cas, la conformation du glycérol doit être altérée (Small, 1986).
CemOA
: archive
ouverte
d'Irstea
2
1
2.3.3 Les mélanges de triacylglycérols à l’état solide
La Figure 13 présente un diagramme de phase de systèmes binaires formés par deux triacylglycérols homogènes de longueurs de chaînes différentes (SSS-PPP) (Small, 1986). Ce type de diagramme est obtenu en combinant des analyses thermiques comme la calorimétrie et des analyses structurales de diffraction des rayons X. Le diagramme ci-dessous montre la présence d’un eutectique à environ 80% de PPP et à 64°C (1). Cette température de fusion est inférieure à la température de fusion du triacylglycérol le plus fusible, ce qui traduit bien la présence d’un eutectique. Les extrémités droites de ce diagramme binaire montre que la forme β de la tristéarine est capable d’incorporer une quantité non négligeable de triacylglycérols monoacides à chaîne plus courte, puisqu’il y a environ 20 % de PPP (2). L’autre extrémité de ce diagramme montre que la solubilité de SSS dans des chaînes plus courtes est beaucoup plus faible (5%) (3). A la vue de ces résultats, il apparaît probable que la résorption de vides dans les structures de SSS est énergétiquement moins coûteuse que la perturbation causée par les excès de matière dans les gradins des méthyles terminaux. Notons également que les triacylglycérols sont miscibles dans les formes α et β' (4).
Figure 13. Diagramme binaire de SSS dans PPP.