N -Nitrosamine-{ cis -Re[CO] 2 } 2+ cobalamin
conjugates as mixed CO/NO-releasing molecules †
Giuseppe Santoro,
aRuben Beltrami,
bEmmanuel Kottelat,
bOlivier Blacque,
aAnna Yu. Bogdanova
cand Fabio Zobi*
bMixed CO/NO-releasing molecules were prepared by conjugation of the 17-electron rhenium dicarbonyl cis-[Re(CO)2Br4]2−complex to N-nitrosamine modified cyanocobalamin (B12) bio-vectors. The species were fully characterized by standard analytical techniques, including X-ray crystallography for cyanocobal- amin-5’-O-pyrazine and (1) and itsN-pyrazine nitrosylated derivative (1a). TheN-nitrosamine B12 deriva- tives are able to liberate low NO doses in buffer solutions and appear to be“activated” towards NO release if in contact with cultured cells. Coordination of the cis-[Re(CO)2Br4]2− complex on the axial cyanide of B12 allows for the combined loss of CO and NO from the conjugates. The mixed CO/NO- releasing molecules show cytoprotection in an ischemia–reperfusion model but no significant enhanced synergistic effects over the relative NORMs and CORMs building constituents.
Introduction
Nitric oxide (NO) and carbon monoxide (CO) are established gasotransmitters involved in multiple signalling pathways in cells and tissues. NO is formed enzymatically as a product of
L
-arginine cleavage by NO synthases (NOS) and upon non-enzy- matic reduction of NO
2−by deoxygenated haemoglobin and myoglobin. CO is produced during the degradation of heme by heme oxygenases (HOs). Crucial importance of NO and CO for living organisms was confirmed by the studies of HOs and NOS in knockout mice. Biochemical parallels between the two gases are particularly striking in the circulation and nervous system where both NO and CO may function as co-transmitters in the same neurons.
1,2Strong evidence supports the notion that interactions of CO and NO may influence the physiologi- cal responses to each of these gases at the enzyme biosynthetic level via a positive feedback loop mechanism whereby NO elevates the levels of heme oxygenase-1 mRNA and protein,
3–5and CO increases the steady state level of NO.
6Uncoupling or dysregulation of NO and CO production results in severe
pathologies such as oncotransformation, hypertension, chemo- reception, cardiovascular diseases etc.
7–9Restoration of NO/CO bioavailability upon exhaustion of their pools in diseased tissues may be achieved by adminis- tration of NO and CO releasing molecules.
10,11Nitric oxide releasing molecules (NORMs) have been used for more than 100 years for therapeutic use.
12–14More recently, CORMs (CO releasing molecules) have been the subject of intense scrutiny.
15–20Like their nitric oxide-releasing counterparts, CORMs are pro-drugs that upon activation release therapeutic doses of carbon monoxide and display beneficial e ff ects in e.g.
atherosclerosis, vascular constriction, inflammation, cellular protection, hyperoxia-induced injury, and other physiological situations.
21In light of the akin biological roles of NO and CO, synergistic cytoprotective e ff ects of two gases might be expected. Indeed these were demonstrated in an acute hepatic and vascular injury model.
22,23Intrigued by these reports, we hypothesized that mixed CO/NO-releasing molecules could provide an advantageous alternative to CORMs and NORMs, as such entities should be able to deliver both gasotransmitters and thus in principle promote, or exploit, the positive feedback loop mechanism now recognized for the two diatomic mole- cules in higher organisms. To the best of our knowledge, a chemical approach to these types of molecules has not yet been described.
To test our hypothesis we prepared mixed CO/NO-releasing molecules from the reaction of the 17-electron organometallic CORM cis-[Re(CO)
2Br
4]
2−complex with N-nitrosamine functio- nalized cyanocobalamin (B12) bio-vectors. The design of the molecules was based on previous findings published by our group. Indeed in the last few years we have shown that d
5†Electronic supplementary information (ESI) available: Crystal data and struc- ture refinement for1and1a;1H-NMR, UV-Vis, HPLC, IR and high-resolution MS spectra of molecules. CCDC 1054820 and 1054821. For ESI and crystallographic data in CIF or other electronic format
aDepartment of Chemistry, University of Zürich, Winterthurerstrasse 190, CH-8057 Zürich, Switzerland
bDepartment of Chemistry, University of Fribourg, Chemin du Musée 9, CH-1700 Fribourg, Switzerland. E-mail: [email protected]; Fax: (+41) 026 300 9738;
Tel: (+41) 026 300 8785
cInstitute of Veterinary Physiology, University of Zürich, Winterthurerstrasse 260, CH-8057 Zürich, Switzerland
Published in 'DOWRQ7UDQVDFWLRQV±
which should be cited to refer to this work.
http://doc.rero.ch
carbonyl Re complexes possess features which make them attractive as CORMs
24,25and if appended to B12 are able to aid the di ff erentiation of embryonic stem cells into energetically- e ffi cient cardiomyocytes which may contribute to functional repair in progressive degenerative cardiac diseases.
26,27In this contribution we report the synthesis and characterization of the above mentioned mixed CO/NO-releasing molecules and the cytoprotective e ff ects of the same tested on cultured 3T3 fibroblasts under conditions of ischemia – reperfusion mimick- ing settings. While all molecules show cytoprotection in our model, no significant enhanced synergistic e ff ects over the relative NORMs and CORMs building constituents were observed.
Results and discussion
Synthesis and characterization of compounds
With the long-term aim of applying the molecules in vivo, we selected as a biocompatible sca ff old for our chemistry, cyano- cobalamin. NO-releasing molecules (NORMs 1a, 2a and 3a, Scheme 1) were first obtained by N,N-carbonylditriazole (CDT) amide coupling of the desired macrocycle on the ribose 5 ′ -OH functionality, followed by NaNO
2nitrosation to the corres- ponding N-nitrosamines. Overall, this procedure a ff ords reasonable yields, but the e ffi ciency of the reaction is rather poor, particularly in the case of hexacyclen. Specifically, we found that the critical synthetic step is the first amide coupling with typical isolated yield of ca. 70, 40 and 15%
for pyrazine, tetraazacyclodecane and hexacyclen respectively.
Conversely, nitrosation a ff ords complete conversion of the pre- cursors to species 1a, 2a and 3a. Mixed CO/NO-releasing
molecules (1b, 2b and 3b in Scheme 1) were finally isolated fol- lowing conjugation of the 17-electron cis-[Re(CO)
2Br
4]
2−anion to the axial cyano group of derivatives 1a, 2a and 3a.
26Coordi- nation of the organometallic species was confirmed by the hypsochromic shift of the ν CN stretching frequency (typical of these metal conjugates)
28–31and the presence of characteristic ν CO bands in infrared spectra of 1b, 2b and 3b. Inductively coupled plasma/optical emission spectrometry further con- firmed the expected percentage concentration of rhenium in the species, calculated as the fraction of the weight of the samples.
All species were characterized by standard analytical tech- niques, including X-ray crystallography for 1 and 1a (see Fig. 1 and 2 respectively and ESI † for crystallographic details). Com- pound 1 crystallized in the P2
12
12
1space group (typical for B12 species) and showed no significant structural di ff erences when compared to B12. Due to low di ff raction of the crystals, and the presence of 11 disordered H
2O solvent molecules the R
1value for this species is relatively high when compared to other B12-based solid-state structures (10.4% vs. 7 – 9% on average, see the Experimental section for further details). Com- pound 1a crystallized as a dimer in the P2
1space group, a rare example of a vitamin B12 monomer whose solid state structure is not found in P2
12
12
1(Fig. 2).
32–35The pyrazine rings in both molecules of the unit cell were found to be disordered, prob- ably due to di ff erent conformations imposed by the high N – NO rotational barrier.
36,37Interestingly, the NO bond lengths are di ff erent in the two molecules. These are 1.096 (13) and 1.217 (14) for atoms labelled N17 – O16 and N34 – O32 respectively in Fig. 2. This dimer may persist in solution as revealed by the splitting of the pyrazine resonances evidenced by
1H-NMR spectroscopy (Fig. 2, ESI † ). In general, in comparison
Scheme 1 Synthetic procedure to mixed CO/NO-releasing molecules. Conditions are: (i) DMSO, CDT, 12 h, 60 °C followed by (ii) selected macro- cycle; (iii) AcOH, NaNO2, 2 h, RT; (iv) MeOH, [Et4N]2[Re(CO)2Br4], N2, 1 h, RT.
http://doc.rero.ch
with the free vitamin, the most significant feature observed in the
1H-NMR spectra of these species, is a progressive shift to lower magnetic field of the ribose R4, R5a and R5b protons as a consequence of amide coupling of the heterocycles and their subsequent nitrosation. Furthermore, resonances of the heterocycles appear as broad signals which can be attributed to their fluxionality and possibly to tautomerization and inter- conversion of the anchoring nitrogen between its amide and imidic acid forms. To illustrate these features Fig. 3 shows the comparison of the
1H-NMR spectra of B12, 2 and 3. The shifts of the ribose protons and the broad heterocycle resonances are clearly seen.
CO and NO releasing properties of 1a – 3a and 1b – 3b
The CO releasing profile of the cis-[Re(CO)
2Br
4]
2−species were previously reported and herein confirmed for 1b, 2b and 3b (see the ESI † for a typical spectrum).
26,38The nitric oxide releasing properties of molecules 1a, 2a and 3a were also evalu- ated via the well-known myoglobin assay.
39The assay consists of monitoring via UV-visible spectroscopy the conversion of deoxy-myoglobin (deoxy-Mb) with a maximum absorption peak of the Q band at 560 nm, to carboxy-myoglobin (Mb-CO) or nitrosyl-myoglobin (Mb-NO) which show two absorption maxima in the 540 – 548 and 575 – 580 nm for the same band.
40The reaction between deoxy-Mb and NO was initiated by adding a small aliquot of a freshly prepared concentrated aqueous solution of either 1a, 2a or 3a to a Mb solution in phosphate bu ff er (0.1 M, pH 7.4) previously treated with sodium dithionite
41(final concentrations: 12 μ M deoxy-Mb and 50 μM for NORM ’ s). Incubation of the deoxy-Mb solution with 1a caused little spectroscopic change over a 5 h monitor- ing period, indicating that the molecule is a poor NO releaser.
However, when 2a or 3a was used in the same assay, the deoxy- Mb spectrum evolved with the expected spectral changes associated with NO release (Fig. 4). Under the experimental conditions Mb-NO concentration reached a maximum within
Fig. 2 X-ray structure (top) of compound 1awith thermal ellipsoidsdrawn at 30% probability. Hydrogen atoms are omitted for clarity. Crys- tallographic details are summarized in the ESI.†1H-NMR (5.25–3.8 ppm, D2O, 298 K of (A) vitamin B12, (B) species1and (C) species1a. Peaks labeled Pz# refer to resonances of pyrazine, while R4, R5a and R5b are those of the ribose sugar (see the text further details).
Fig. 3 1H-NMR (5.25–3.8 ppm, D2O, 298 K) of (A) vitamin B12, (B) species2and (C) species 3. Signals enclosed in segmented boxes are those of 1,4,8,11-tetraazacyclotetradecane (tacd) and hexacyclen (hacd) respectively. Peaks labeled R4, R5a and R5b refer to resonances of the ribose sugar.
Fig. 1 X-ray structure of compound1with thermal ellipsoids drawn at 30% probability. Hydrogen atoms are omitted for clarity. Crystallo- graphic details are summarized in the ESI.†
http://doc.rero.ch
5 h of incubation. After this time, the two Mb-NO absorption maxima at 548 and 583 nm began to steadily decrease while peaks at 505 and 630 nm increased in intensity. The changes are associated with the conversion of Mb-NO to metmyoglobin (met-Mb), due to slow oxygen dissolution in the reaction medium.
In the presence of dioxygen, nitric oxide can catalyze the oxidation of Mb to met-Mb through the formation of a peroxy- nitrite derivative (referred to as met-Mb(OONO)).
42–44This species is also generally assumed to be the first intermediate of the reaction between oxy-myoglobin (oxy-Mb) and NO. In order to understand if 2a-, 3a-derived NO could also elicit the spectroscopic changes linked to oxy-Mb to met-Mb conversion, a fresh solution of oxy-Mb was prepared according to the method of Bauer and Pacyna and incubated with the mole- cules.
45As shown in Fig. 4 3a-derived NO provided a clean con- version between the two Mb species. In the presence of 3a, the
two absorption maxima of oxy-Mb at 542 and 579 nm steadily decreased over a 24 h period while the met-Mb peaks at 505 and 630 nm increased. A total of five isosbestic points at 350, 411, 478, 524 and 580 nm accompanied the conversion.
The total amount of NO released by 1a – 3a and 1b – 3b was determined by reductive gas-phase chemiluminescence detec- tion in PBS bu ff er and in cell culture medium overlaying the monolayer of 3T3 fibroblasts (data relative to 100 μ M solutions, see Table 1 and Fig. 5).
46,47The method entails the oxidation of all NO
xspecies in solution to NO
3−followed by its step-wise reduction to NO
2−and NO which is finally carried in a He flow and allowed to interact with O
3in a reaction chamber. The ensuing reaction is then followed by light emission detected by a photomultiplier (see Experimental for further details). For species 1a – 3a in PBS, 3a released the highest total detectable amount of NO, ca. 7 μ M, while 1a and 2a released about half the same amount. Similar results were found in cell culture. In this case, however, the total amount of NO released by 2a and 3a was twice that determined in PBS (ca. 7 and 15 μ M respecti- vely) while it was constant for 1a. Thus, it appears that 2a and 3a are activated by cultured 3T3 fibroblast towards NO liber-
Fig. 4 Spectrum changes of a deoxy-Mb (top) and an oxy-Mb (bottom) solution (both 12μM) incubated with a 50μM solution of3aover a 24 h period (1 h intervals). Arrows indicate the relative spectroscopic vari- ations. Insets detail changes of the Q band. In the topfigure insert, the initial growth of Mb-NO (overca.5 h) is followed by its disappearance and conversion to met-Mb (over remaining 19 h). See the text for further details.
Table 1 Amount of nitric oxide released by1a–3aand1b–3b(100μM solutions) in PBS buffer and 3T3 cell culture after 30 h of incubation
PBS total (μM)
Cell culture total (μM)
1a 3.7 ± 0.4 4.1 ± 0.9
2a 3.2 ± 0.6 6.9 ± 0.5
3a 7.1 ± 1.1 14.9 ± 2.0
1b 3.9 ± 0.3 4.4 ± 0.8
2b 3.6 ± 0.7 7.7 ± 0.4
3b 8.7 ± 0.9 19.0 ± 1.8
Fig. 5 Reductive gas-phase chemiluminescence detection of NO released by 100μM solutions: of1a(■),2a (●) and3a(▲) in (A) PBS buffer ( pH 7.4) and (B) 3T3 cell culture and of1b(■),2b(●) and3b(▲) in (C) PBS buffer ( pH 7.4) and (D) 3T3 cell culture.
http://doc.rero.ch
ation, with relative total concentrations of released NO com- parable to other N-nitrosamine derivatives.
36,37Mixed CO/NO molecules 1b – 3b released comparable amounts of NO, with 3b giving the highest total measured concentration of the gas (ca. 20 μ M, Table 1).
Cytoprotective properties of 1a – 3a and 1b – 3b
The cytoprotective properties of mixed CO/NO-releasing mole- cules 1b, 2b and 3b were tested in an ischemia – reperfusion model on the 3T3 fibroblast cell culture and compared to NORMs 1a, 2a and 3a and the CO-releasing molecule B12- ReCORM-2 (Fig. 6).
26The latter a ff orded the pure CORM-only model for direct comparison to the species herein described.
Compounds were administered at reperfusion at a 30 μ M con- centration and responses of cells to metabolic depletion were measured after 48 h as reflected in the percentage of dead cells in the monolayer. Identical sets of experiments were per- formed with two sets of samples, one of which was treated with freshly-dissolved compounds (blue bars in Fig. 6) and the other with compounds, which were in aqueous solution for more than 50 h to release all the gaseous molecules (orange bars in Fig. 6). B12-ReCORM-2 was used to assess the possible additivity of NO and CO for protection from ischemia – reperfu- sion injury. As followed from Fig. 6, the number of dead cells reduced when cells were treated with any of gas-emitting com- pounds compared to that in the non-treated control (grey bar).
The protective e ff ect was lost when compounds were adminis- tered after they released the gases (orange bars). The presence of NO did not result in any additional protection than that pro- vided by B12-ReCORM-2 alone. In agreement with our previous findings,
48protection accomplished by the combined CO/NO- releasing compounds was associated with prevention from de- attachment of 3T3 cells from the bottom of the petri-dish and
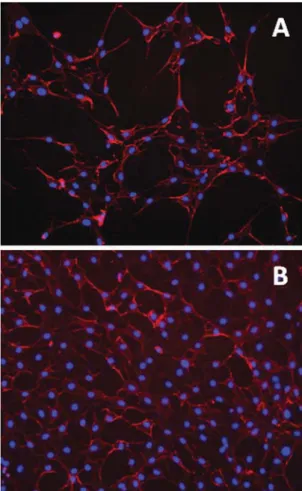
reduction of stress fiber formation. No other significant mor- phological changes could be observed in 3T3 cells treated with our compounds (Fig. 7). The lack of additional cytoprotective e ff ects of NO when applied along with CO suggests that both gases may share similar pathways of cytoprotection in our experimental settings.
49Conclusions
In summary, we have presented in this contribution the syn- thesis and characterization of the first examples of mixed CO/
NO-releasing molecules based on N-nitrosamine-{cis-Re- [CO]
2}
2+cobalamin conjugates. The derivatives were shown to be able to release both gases, albeit NO in relatively small doses. When tested in an ischemia – reperfusion model on 3T3 fibroblast cell cultures, all species a ff orded cytoprotection but we found no significant enhanced synergistic e ff ects over the relative NORMs and CORMs alone. Despite the lack of enhanced cytoprotection exhibited by the mixed CO/NO-releas- ing molecules in the specific model we selected, we believe this new class of compounds may be possibly considered as alternative therapeutic agents along classical NORMs and CORMs.
Fig. 7 (A) Representative histochemical staining of (A) untreated 3T3 fibroblasts (control) and (B) the cells treated with 30μM3b. Stained in blue (DAPI) are the nuclei, actinfilaments ( phalloidin) are shown in red.
Fig. 6 Effects of active (blue) and inactive (orange) B12 species1a,2a, 3aand1b,2b,3bon the viability offibroblasts in cell cultures under in an ischemia–reperfusion model. Non-treated control is shown in grey.
Data are means of 8 experiments ±SEM. B12 species concentration is 30μM; incubation time 48 h (see the ESI†for details). * and ** stand for p< 0.05 andp< 0.01 compared to the non-treated control.