Population genomics reveals additive and replacing horizontal gene transfers in the emerging pathogen Dickeya solani
Texte intégral
Figure

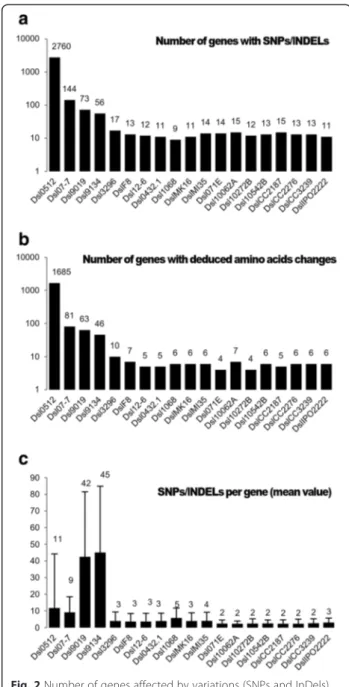
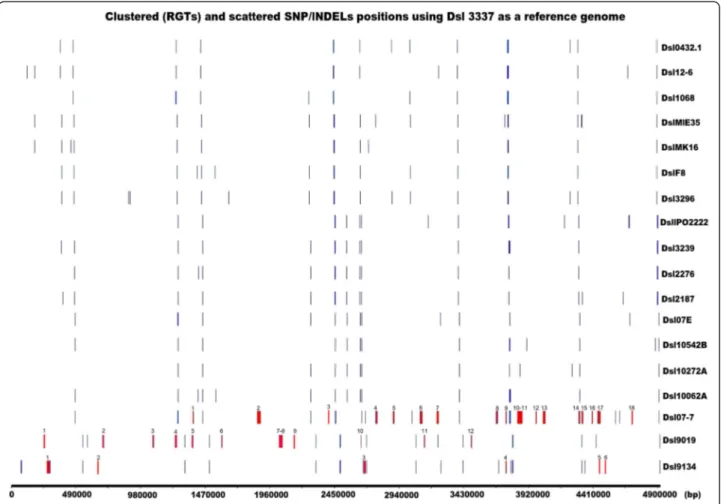


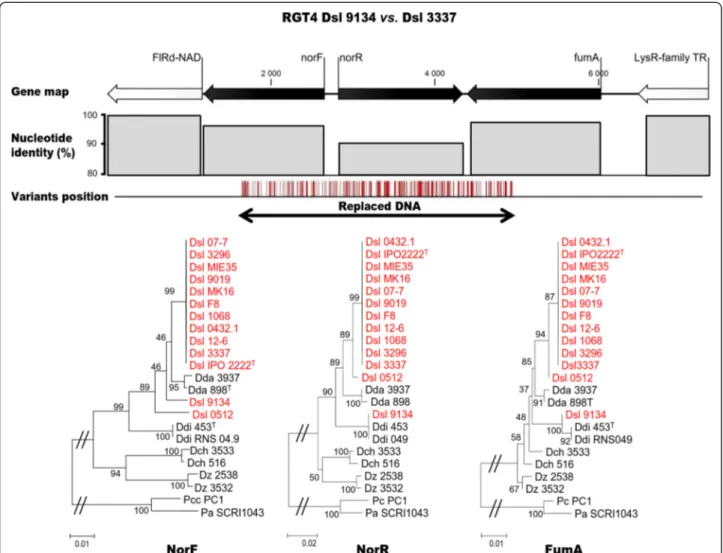

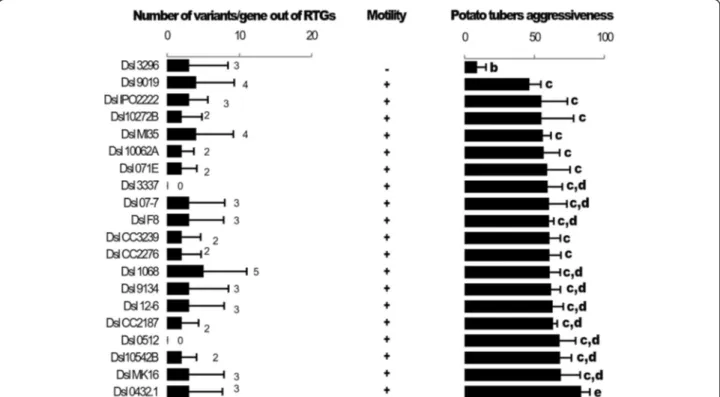
Documents relatifs
This paper fo- cuses on the simultaneity and succession relationships that organize actions on a timeline, the creation of multiple in- dependent timelines and on the notion of
Our solution D ISCUS is based on a distributed architecture using both physical and virtual probes, along with former security solutions (IDS and firewalls)..
Fig- ures 9 and 10 illustrate the performance of the RNLp model for different values of p on the two test images corrupted by impulse noise with parameter ρ = 0.3 and 0.5
However, for galaxies with very little or no bulge (specifically late types or irregular galaxies), the luminosity profiles were not decomposed, the disc component is then directly
In similar fashion to the UVW2 data, the UVW1 and UVM2 light curves are also observed to track the X-ray variations on long time-scales, displaying the characteristic drop in flux
Given the observation that some Xanthomonas strains across distinct taxa do not contain hrpG and hrpX, we speculate a stepwise evolution of pathogenicity (Figure 6), which involves
The collection involves 2.497 accessions maintained in an in vitro culture and divided into six sub-collections (Solanum tuberosum varieties, Solanum tuberosum
To find out what degree of synteny (conservation of gene order) exists between the Musa and rice genomes, 17 BACs representing 1.8 Mb of sequence have been selected using gene markers
