Accelerating process development for biologics on an
automated, pharmacy-scale manufacturing system
by
Laura E. Crowell
M.S. Chemical Engineering Practice, Massachusetts Institute of Technology, 2015 B.S. Chemical and Biological Engineering, Tufts University, 2014
Submitted to the Department of Chemical Engineering in partial fulfillment of the requirements for the degree of
Doctor of Philosophy in Chemical Engineering
at the
Massachusetts Institute of Technology
May 2020
© Massachusetts Institute of Technology 2020. All rights reserved.
Author ... Laura E. Crowell Department of Chemical Engineering April 7th, 2020
Certified by ... J. Christopher Love Raymond A. and Helen E. St. Laurent Professor of Chemical Engineering Thesis Supervisor
Accepted by ... Patrick S. Doyle Robert T. Haslam Professor of Chemical Engineering Singapore Research Professor Chairman, Committee for Graduate Students
Accelerating process development for biologics on an automated,
pharmacy-scale manufacturing system
by
Laura E. Crowell
Submitted to the Department of Chemical Engineering in April 2020, in partial fulfillment of the requirements for the degree of
Doctor of Philosophy in Chemical Engineering
Abstract
The conventional large-scale, centralized, single-product manufacturing model for biologic drugs does not allow for the economical production of drugs for small patient populations or for the distribution of these drugs in developing countries. A decentralized model featuring small-scale, fully automated, multi-product manufacturing of biologics at the point-of-care could address some of these issues. To truly realize the benefits of such a manufacturing paradigm, it must also be paired with rapid process development methods for the production of new molecules. In this thesis, we describe the development of a bench-scale, automated, multi-product
manufacturing system for the end-to-end production of hundreds to thousands of doses of clinical quality protein medicines in about three days. We then demonstrate the application of this platform to the manufacture of a trivalent vaccine in a single campaign through
co-expression and co-purification. We further demonstrate new methodologies for the accelerated development of manufacturing processes to produce new molecules on the system including a strategy for the development and optimization of fully integrated, multi-column processes for straight-through chromatographic purification, and the development of a platform process for the production and purification of single-domain antibodies. We then propose a workflow for the collection of a dataset relating the chromatographic behavior of host-cell proteins to their biophysical characteristics with the goal of building an in silico tool for the prediction of purification processes for any new molecule. Finally, we propose a platform approach, as opposed to a platform process, for the development of manufacturing processes for new
biologics which is based on gaining a deeper understanding of process development challenges with regard to the host and to the molecule itself. Ultimately, we believe that the combination of a small-scale, automated manufacturing platform and accelerated strategies for developing processes to manufacture new products on the platform could enable time- and cost-efficient manufacturing of a wide variety of biologic drugs, increasing access to medicines throughout the world.
Thesis Supervisor: J. Christopher Love
Table of Contents
Abstract ... 2
1. Introduction ... 6
1.1 Motivation ... 6
1.2 Current practices in the manufacturing of biologics ... 8
1.3 Pichia pastoris as an alternative host ... 10
1.4 Development of processes for the purification of biologics ... 11
1.5 Thesis Goals ... 16
1.6 Thesis Structure ... 17
2. On-demand manufacturing of clinical quality biopharmaceuticals ... 20
2.1 Introduction ... 20
2.2 Results and Discussion ... 22
2.3 Conclusions ... 28
2.4 Methods ... 38
3. Manufacturing of a trivalent subunit vaccine in a single campaign ... 57
3.1 Introduction ... 57
3.2 Methods ... 58
3.3 Results and Discussion ... 60
3.4 Conclusions ... 65
4. Rapid optimization of processes for the integrated purification of biopharmaceuticals... 72
4.2 Materials and Methods ... 74
4.3 Results and Discussion ... 80
4.4 Conclusions ... 87
5. Development of a platform process for the production and purification of single-domain antibodies ... 96
5.1 Introduction ... 96
5.2 Materials and Methods ... 98
5.3 Results and Discussion ... 101
5.4 Conclusions ... 108
6. Methods for determining the chromatographic behavior of Pichia pastoris host-cell proteins ... 115
6.1 Introduction ... 115
6.2 Development of a workflow for the collection of host-cell protein chromatographic behavior ... 117
6.3 Potential applications of a dataset of the chromatographic behavior of host-cell proteins ... 123
6.4 Conclusions ... 125
6.5 Methods ... 126
7. Tools for accelerating process development for non-platform biologics ... 133
7.1 Introduction ... 133
7.2 A platform process for the manufacturing of monoclonal antibodies ... 134
7.4 A ‘platform approach’ to process development ... 137
7.5 Selection of a host organism to enable rapid process development ... 142
7.6 Developability ... 143
7.7 Conclusions and outlook ... 144
8. Conclusions ... 150 8.1 Summary ... 150 8.2 Remaining Challenges ... 151 8.3 Implications ... 155 8.4 Personal Contributions ... 155 9. References ... 157 Appendix A ... 167 Appendix B ... 187 Appendix C ... 189 Appendix D ... 196
1. Introduction
1.1 Motivation
Biologics are an important class of therapeutics due on their impact on both global health and financial markets. Conventional biologics are proteins that are manufactured in or derived from living systems, including hormones, cytokines, replacement enzymes, vaccines, blood factors and antibodies. Many biologic drugs provide life-changing therapeutic benefits as vaccines or in areas such as oncology and rare diseases1,2.In 2018 the global biologics market
was valued at $251 billion, and this is expected to increase to $625 billion by 20263.
Unfortunately, global access to these treatments is fairly limited; in 2015 North America and Europe made up approximately 85% of the biologics market, with Asia Pacific contributing about 10% and Latin America, the Middle East, and Africa contributing the remaining 5%4. While
significant growth is predicted in Asia Pacific through 2026 (up to 21%), access in low and middle income countries (LMIC) remains low3. Although some of this difference in market size
could be attributed to differences in sales price, it is still clear that areas such as Latin America, the Middle East, and Africa have severely limited access to these therapeutics.
Limited access to biologics in the developing world is due to challenges regarding both the cost and the logistics of production. The current production strategy for these therapeutics is based on large-volume, centralized manufacturing which provides economical production of a small number of products that can support the overall costs of drug development5. It is
estimated that costs of manufacturing need to come down from ~$50-100/g to ~$1-10/g to enable access in low and middle income countries (LMICs)6. This is especially important for
vaccines, where price targets to reach patients in LMICs can be as low as $0.15 per dose7,8.
Further, in order to reduce the true cost of biologics it is necessary to both reduce the cost of manufacturing and the cost of development.
The current centralized manufacturing paradigm also presents logistical challenges. Most of these products are manufactured in the United States or Western Europe4, and then
they must be shipped to the location of the patient while maintained at a certain temperature. The cold chain infrastructure required for this shipment is lacking in many developing countries. A distributed manufacturing paradigm, utilizing regionalized raw material warehousing and on-site manufacturing with real-time quality assurance and quality control (QA/QC) could provide just-in-time inventory with a reduced cold chain footprint and thus, increase access.
Additional limitations of the current centralized, large-scale manufacturing paradigm for biologics include application to orphan drugs and precision medicine. According to the FDA, orphan drugs are used to prevent or treat a disease which occurs in fewer than 200,000 patients in the United States. Between 2010 and 2016, 42% of newly approved orphan drugs were biologics2. Furthermore, orphan drugs are predicted to contribute 32% of the total growth for
biologics between 2017 and 20221. Thus, orphan drugs represent a growing class of biologics
where a plurality of therapies are used to treat small patient populations. In addition, the
therapeutic use of biologics is becoming increasingly reliant on precise molecular profiles which define diseases of certain, often small, cohorts of patients5. In 2017, the FDA approved the first
cancer treatment based on a common biomarker9.This precision results in a larger number of
small patient populations. As patient populations become more stratified, there is a need for smaller-scale, more agile approaches to biologics manufacturing.
A small-scale, fully integrated and automated, on-demand manufacturing platform for biologics within local hospitals, pharmacies, and health care clinics could address some of the current challenges described above. In addition to a new manufacturing platform, new methods of accelerated process development to produce additional products on the platform are needed to truly enable global access and precision medicine.
1.2 Current practices in the manufacturing of biologics
Many first generation biologics, including granulocyte colony stimulating factor (G-CSF), human growth hormone (hGH), and interferon-α 2b (IFNα-2b), are produced in E. coli, either internally or as inclusion bodies. Following production there are a large number of steps required for the purification of these molecules10–12. Separating the protein of interest from the
cell mass requires some form of cell lysis. Centrifugation is then typically used to separate cell debris from the protein of interest. Often the protein of interest is not in its properly folded form at this point, especially if it was produced in inclusion bodies, and thus a refolding step is
required. A variety of chromatography, precipitation, and/or crystallization steps are then used to get to a final purified product. The manufacturing processes for these first generation products vary significantly in the number, sequence, and method of unit operations10–12. No ‘platform’
process exists, which makes it hard for non-experts to manufacture these proteins at high quality and low cost. Additionally, when similar molecules come along it is difficult to use the current manufacturing methods to rapidly develop a new production process.
In contrast, manufacturing of monoclonal antibodies (mAbs) has become fairly
‘platformized’ over the past few decades13. Most processes use Chinese hamster ovary (CHO)
cells as a host to produce mAbs in a 5-25kL bioreactor operated in fed-batch mode. The cells are then harvested through centrifugation, and the mAb is purified through three
chromatographic separation steps, with protein A affinity chromatography as the first step, followed by anion exchange chromatography and cation exchange chromatography. The purified drug substance is then concentrated and formulated through ultrafiltration / diafiltration. Typically, additional steps concerning virus removal are added both after protein A
chromatography and before the final formulation. Benefits of such a ’platformized’ process include reduced process development times, and low risk of scale-up or process transfer13.
Some argue that the platform process for mAb production does not face any significant challenges13. However, many others believe that the current problems facing the
biomanufacturing industry, including the emergence of competition from biosimilars, the need to gain access to emerging markets, and increasing cost pressures, still require changes from the typical batch processing described above6,14. In order to create more agile and automated
manufacturing processes, there have been numerous efforts to translate the typical batch production platform for mAbs into an integrated and continuous end-to-end manufacturing process15–18. Small-scale experiments have proven the feasibility of this approach, showing
consistent product quality over time with increased process throughput and decreased equipment footprint16.
Few efforts have been made to show end-to-end integrated production of non-mAbs, however, where no ‘platform’ manufacturing exists. Warikoo et al. described the integrated, continuous production and capture of a recombinant human enzyme using a CHO perfusion process and capture with periodic counter-current chromatography (PCC), but few other processes are evident in the literature15. A platform developed for the rapid production of
non-mAbs, such as replacement enzymes, cytokines, and vaccines, could allow for distributed manufacturing of such products and increased access in developing countries.
To enable distributed manufacturing at or near the point-of-care, a manufacturing platform must be small-scale, rapid, and fully automated. Demonstrations of such an on-demand biologics manufacturing platform for rapidly producing limited quantities of
biomolecules on-demand exist, but few have employed any sort of purification process19–21.
Pardee et al. and Adiga et al. attempted purification using affinity tags20,22. Due to the need to
strictly control the presence of such a tag in a therapeutic product, however, this strategy is not ideal for routine biomanufacturing23. While these methods are a good proof-of-concept for the
the quality attributes required by regulatory agencies to actually dose patients, including identity, purity, safety, and potency.
1.3 Pichia pastoris as an alternative host
While many biologics are currently produced in Chinese Hamster Ovary (CHO) cells, there is doubt that the industry will be able to address the need for access to affordable biologics throughout the world using this host6,24. Alternative expression systems, such as the
yeast Pichia pastoris, show numerous advantages over CHO and other widely used hosts such as E. coli with respect to small-scale, rapid and affordable production of biologics24,25. First, P.
pastoris can rapidly grow to high cell densities, reaching >100 g/L wet cell weight (WCW) in less
than 2 days, while a typical CHO system takes about 6 – 8 days to reach this density26. Second,
P. pastoris can secrete recombinant proteins, therefore eliminating the need for cell lysis
common in faster growing hosts such as E. coli27. Third, many classes of proteins, including
FDA-approved therapeutics, have been produced in P. pastoris28. Fourth, there is little to no risk
of viral contamination during production since P. pastoris is not a mammalian organism29. Fifth,
the capability of human-like post-translational modification has previously been demonstrated30.
Sixth, P. pastoris secretes low levels of host-cell proteins, resulting in fermentation broths containing the product of interest at ~80% initial purity24. Finally, a defined media has been
developed for use in cultivations of P. pastoris which contains no animal derived products31.
These advantages enable the rapid production of safe, high-quality biologics with relatively simple purification methods.
In addition to the manufacturing advantages of P. pastoris, there are numerous
advantages with respect to process development. The small genome (9.4 Mbp vs 2450 Mbp in CHO) and ease of transgene insertion enable rapid host strain development24,25. The rapid
growth rate allows for fast upstream process development, and the small secretome and low levels of host-cell protein secretion allow for rapid downstream process development32. To
accelerate the timelines of both production and development on our integrated, on-demand production platform, P. pastoris was selected as the host. Overall, the use of P. pastoris in this work enables rapid cycles of process development for multiple molecules and simplifies the overall structure of the small-scale, integrated system.
1.4 Development of processes for the purification of biologics
As mentioned above, in order to realize the true potential of a small-scale, on-demand manufacturing platform for non-platform molecules, new process development methods are needed to rapidly produce new molecules on the platform. General guidelines exist for the fermentation of P. pastoris which at least offer a starting point for optimization in upstream process development33. No such guidelines exist for purification of non-platform molecules from
P. pastoris, however.
One of the major objectives during purification is to separate the product of interest from host-cell proteins (HCPs) and host-cell DNA. This can be a significant challenge since there can be hundreds to thousands of host-cell proteins with a variety of biophysical characteristics. While some molecules benefit from affinity chromatography, where the product is selectively bound to a resin enabling high purity to be obtained in a single step, this is not true for most non-mAb molecules. Traditionally, separation of the product from HCPs was achieved through trial and error. In the last few decades, there has been a lot of interest in new methods to predict potential purification processes which utilize knowledge of the host-cell proteins and can reduce the number of experiments necessary to develop an optimized process.
As reviewed by Nfor et al., methods for predicting purification processes typically fall within one of four categories: heuristics or knowledge-based systems, algorithmic or
optimization based methods, methods based on high throughput experiments (HTE), or hybrid methods, which are typically a combination of algorithmic and HTE methods34. Expert systems
host-cell proteins to create a promising process flowsheet for purification35–37. Most expert systems
concentrate on properties such as concentration, molecular weight, hydrophobicity, and charge profiles. Unfortunately, most systems are only able to consider a handful of host-cell proteins that are determined to be the most important, based on concentration or similarity to the product protein35. Furthermore, use of expert systems cannot guarantee the selection of the most
optimal process since many processes are never considered34. This is especially true in the
case of multi-step processes, where the optimal conditions at a single step may not be the same as the optimal conditions for that step when the entire process is considered.
Algorithmic methods typically select the best process flowsheet by examining all possible scenarios and utilizing models for each of the unit operations34. With this method all
potential processes are considered, so it is possible to select the most optimal process. These methods can require significant computational power, however, especially when complex mechanistic models are used. Further, although these methods minimize the number of experiments necessary by using models, this requires models to be available for every unit operation and model parameters to be available for every protein important in the process.
Methods based on high throughput experiments take advantage of the strategies developed for high throughput screening of chromatographic separations to predict purification processes based on a limited amount of experimental data38–40. The major advantages of high
throughput experiments, including lower material needs and faster experimental timelines, are based on the ability to reduce the scale of and parallelize chromatographic screening
experiments. Design of experiments (DoE) becomes critical here for obtaining the most useful data with the smallest number of experiments34. A disadvantage of this method for predicting
purification processes is that the potential operating conditions are limited by the extent of the experiments carried out.
Hybrid methods are a combination of algorithmic, or optimization based methods and high throughput experiments. Most of the recent works relevant to predicting processes for purification of a target protein from HCPs are hybrid methods. For example, Nfor et al. implemented HTE utilizing ion exchange chromatography (IEX), hydrophobic interaction chromatography (HIC), and size exclusion chromatography (SEC) to characterize 35 of the major protein impurities present in a hybridoma cell culture supernatant41. The parameters
determined from these experiments were then used as inputs for mechanistic column models to enable in silico prediction and optimization of a purification process. Hanke et al. developed a similar multidimensional fractionation approach for identifying molecular properties relevant to chromatographic behavior for an undisclosed host42. Pirrung et al. further built on these
methods to determine the isotherm parameters of a variety of critical CHO host-cell proteins to allow for mechanistic modelling of their chromatographic behavior43. These methods are limited
in the number of proteins they consider, however, because using mechanistic modelling with a large number of proteins is computationally expensive.
Some strategies have been proposed to reduce the amount of computing power required to examine many potential proteins or many potential resins. Pirrung et al. utilized a combination of mechanistic models and artificial neural networks to optimize a process with up to three different chromatographic steps with respect to yield at a given purity threshold44. Since
evaluating all possible scenarios using mechanistic models would require a significant amount of computing power, artificial neural networks were trained to mimic the detailed mechanistic models based on datasets of inputs and outputs generated from the mechanistic models. Using the artificial neural networks decreased the computational time by ~50% (including the time needed to train the networks). The final model was used to examine a case study with a single product and four major impurities. Liu et al. used an optimization based framework along with data from HTE to identify a sequence of resins to maximize yield and purity of a particular
purification45. A database was created based on a target protein along with its low molecular
weight and high molecular weight variants across 8 different resins. A process was then
predicted utilizing two chromatographic steps to optimize yield and purity of the product by using a multiobjective mixed integer nonlinear programming (MINLP) model. Using this model, the best resins and best sequence were identified in a matter of minutes45. While reducing the
computational power required as compared to mechanistic models, these approached still examine only small numbers of impurities (four and two, respectively). Extending these approached to predict resins for purifying a target protein from a group of host-cell proteins (100s to 1000s of varied proteins) is expected to be difficult.
A different approach is to use alternative methods to predict the chromatographic behavior of proteins instead of using mechanistic models. Methods such as quantitative structure-activity relation (QSAR), or quantitative structure-property relation (QSPR), can be used to predict the chromatographic behavior of a protein based on its structure. Such methods have been used to predict the elution pH or salt required for model proteins on a variety of resins including ion exchange46,47, hydrophobic interaction48, and multimodal resins49. The
calculation of descriptors used for these predictions typically requires the 3D structure of a protein, however, which limits the number of proteins that can be considered50. More recently,
the use of homology modelling has been demonstrated for the prediction of descriptors used in QSAR models51. Buyel et al. used homology modelling to calculate the descriptors of ~55% of
common tobacco plant host-cell proteins and subsequently used those descriptors with
previously developed QSAR models to predict the retention behavior of the tobacco plant HCPs on three different resins52. Swanson et al. determined three main biophysical features
(molecular weight, charge, and surface hydrophobicity) of ~500 host-cell proteins from corn germ and used a multivariate random forest (MVRF) method to predict the elution pattern of individual proteins on a cation exchange and an anion exchange resin based on these
biophysical characteristics50,53. While each of the methods detailed above represent significant
advances over traditional trial-and-error experiments, they are all limited in the percentage of relevant host-cell proteins considered, the number of resins considered, or both. New methods are still required which consider the entire host-cell protein burden of a given host across a wide variety of resins. This could enable the rapid prediction of processes for the purification of new products produced in a given host.
1.5 Thesis Goals
As discussed above, the conventional large-scale, centralized, single-product manufacturing model for biologics does not allow for the economical production of drugs for small patient populations or for the distribution of these drugs in developing countries. A decentralized model featuring small-scale, fully automated, multi-product manufacturing of biologics at the point-of-care could address some of these issues. To truly realize the benefits of such a manufacturing paradigm, it must also be paired with rapid process development methods for the production of new molecules. This is especially important for product classes other than monoclonal
antibodies (mAbs), where platform production processes typically do not exist. To that end, this thesis has two specific goals:
1. Demonstrate on-demand production of high-quality biologic drugs using a fully integrated and automated, pharmacy-scale platform
2. Establish accelerated process development strategies for rapidly producing new biologics, particularly non-mAbs, on the automated, pharmacy-scale platform
a. Demonstrate strategies for developing and optimizing fully integrated purification processes for a new molecule
b. Develop methods to relate protein biophysical properties and/or sequence to chromatographic behavior
1.6 Thesis Structure
• Chapter 2 discusses the development of a bench-scale, automated, multi-product manufacturing system for the end-to-end production of hundreds to thousands of doses of clinical quality protein medicines in about 3 days. The system, called Integrated Scalable Cyto-Technology (InSCyT), includes fully integrated modules for sustained production, efficient purification without the use of affinity tags, and formulation to a final dosage form. We demonstrate that InSCyT can accelerate process development from sequence to purified drug in as few as 12 weeks. The InSCyT system is used to produce human growth hormone, interferon α-2b, and granulocyte colony-stimulating factor with purity and potency comparable to marketed reference products.
• Chapter 3 describes the application of the InSCyT platform to manufacture a trivalent rotavirus vaccine in a single campaign through co-expression and co-purification. We first developed and deployed processes for the production of each individual subunit antigen, while maintaining similarity in process operating conditions. While our initial processes removed process-related variants, product-related variants persisted. In order to maintain process conditions compatible with all three subunits, we addressed the issue of product-related variants at the molecular level by strategically altering specific amino acids. We then created a single Pichia pastoris strain that expressed all three engineered subunits and executed a single manufacturing campaign on the InSCyT system to produce all three subunits. Successful co-expression and co-purification of all three subunits were confirmed by RPLC, SEC, and LC-MS.
• Chapter 4 discusses a method for the development and optimization of fully integrated, multi-column processes for straight-through purification, compatible with the InSCyT system. Resin selection was performed using an in silico tool for the prediction of
straight-through purification processes originally developed alongside the InSCyT system. A two-step optimization was then conducted to maximize yield while minimizing process- and product- related impurities. First, potential operating regions were identified through conventional single-column screening with limited analytics. The identified operating regions were then used as inputs to build a statistical model of the multi-column process, based on design of experiments (DoE), to predict overall process yield and impurity levels, including host-cell protein, DNA, and aggregate. Using these methods, we developed integrated processes for the purification of two different
proteins, a single-domain antibody and a cytokine, with overall purification yields of 88% and 86%, respectively, and process-related impurities reduced below regulatory
guidelines for nonclinical use.
• Chapter 5 describes the development of a platform process for the rapid production and purification of single-domain antibodies (sdAb). We demonstrate this process through the production of eight different single-domain antibody products at phase-appropriate quality. We determined that our platform process could be applied to a variety of sdAbs with the modification of a single parameter, the pH of the bridging buffer. Further, we demonstrated that this parameter can be predicted based only on the biophysical
characteristics of the sdAb. Using these methods, we produced nonclinical quality sdAbs within five weeks of identifying the product sequence. We then further optimized the process for individual sdAbs to maximize yield and minimize impurities in only three weeks using the methodology discussed in Chapter 4.
• Chapter 6 proposes a workflow for the collection of a dataset relating the
chromatographic techniques combined with liquid chromatography mass spectrometry (LC-MS). Further, we propose data analysis methods which could be used to identify relationships between protein sequence and chromatographic behavior using this dataset, as well as a potential algorithm structure for predicting integrated processes for the purification of new products produced in the same host.
• Chapter 7 examines the question of how to get the benefits of a platform manufacturing process, such as that developed for monoclonal antibodies, in the development of non-platform products. We propose a ‘non-platform approach’ to process development, rather than a ‘platform process’. This platform approach includes a toolbox of process development techniques covering strain engineering, upstream, downstream, and formulation, which can be used for the development of any recombinant protein. The proposed methods focus on gaining a deeper understanding of process development challenges with regard to the host and to the molecule itself. We expect that phenomena learned from such understanding will be more predictive across molecular classes.
• Chapter 8 concludes this thesis, discusses remaining challenges, and provides perspective on the implications of the technologies presented herein.
2. On-demand manufacturing of clinical quality
biopharmaceuticals
This work has previously appeared as: Crowell, L. E. et al. On-demand manufacturing of clinical-quality biopharmaceuticals. Nat. Biotechnol. (2018) doi:10.1038/nbt.4262.
2.1 Introduction
Conventional manufacturing of protein biopharmaceuticals in centralized, large-scale, single-product facilities is not well-suited to the agile production of drugs for small patient populations or individuals. Previous solutions for small-scale manufacturing are limited in both process reproducibility and product quality, owing to their complicated means of protein expression and purification19–22. We describe an automated, benchtop, multiproduct
manufacturing system, called Integrated Scalable Cyto-Technology (InSCyT), for the end-to-end production of hundreds to thousands of doses of clinical-quality protein biologics in about three days. Unlike previous systems, InSCyT includes fully integrated modules for sustained
production, efficient purification without the use of affinity tags, and formulation to a final dosage form of recombinant biopharmaceuticals. We demonstrate that InSCyT can accelerate process development from sequence to purified drug in 12 weeks. We used integrated design to
produce human growth hormone, interferon α-2b, and granulocyte colony-stimulating factor with highly similar processes on this system and show that their purity and potency are comparable to those of marketed reference products.
Biologic medicines, such as recombinantly expressed cytokines, hormones, replacement enzymes, blood factors or antibodies, are routinely used to treat cancer, autoimmune disorders and rare diseases. Increasingly, protein biologics are tailored to small groups of patients based on an understanding of the underlying biology of their disease54. The need for small numbers of
in large volumes to achieve economies of scale5. Furthermore, different classes of
biopharmaceuticals (for example, enzymes, hormones, vaccines) generally require unique customized processes for each molecule from expression to purification, constraining commercial facilities to a single class of product.
New technologies to manufacture many different pharmaceutical-quality biologics in small quantities with efficiency and agility are needed to make precision biologic medicines both available and economically feasible55. Technologies such as automated laboratory-scale batch
processes, in vitro transcription and translation, and microfluidics can rapidly produce limited quantities of different biomolecules on-demand19–22. While some of the products generated
show biological activity, they lack sufficient quality attributes for clinical use, including identity, purity, safety and potency as required by regulatory agencies. To address this need, we developed an automated multiproduct manufacturing system capable of rapidly producing clinical-grade recombinant proteins and requiring only minimal reconfiguration to make different biopharmaceuticals. Unlike previous solutions, InSCyT comprises fully integrated and
automated modules for sustained production, efficient purification of the native protein, and final formulation for parenteral use. The yeast-based production module allows both rapid production of tens to hundreds of doses in under 80 h and sustained production for > 100 h of up to
thousands of doses as needed, whereas cell-free solutions19,20,22 provide only short-term
expression. The multistage purification module (and associated methods for rapid process development of suitable purification sequences) enables purification of multiple clinical-grade products using highly similar processes without requiring affinity tags that alter the drug sequence and present risks of immunogenicity. In contrast, previous approaches used affinity tags for purification or had no integrated purification step. Finally, the integrated formulation module prepares protein biopharmaceuticals in a final dosage form. These features, combined
with the use of integrated design principles to create simplified processes, facilitate the production of recombinant proteins with quality attributes sufficient for clinical use.
2.2 Results and Discussion
We selected Pichia pastoris as our expression host because it can grow quickly to high cell densities and efficiently secrete recombinant proteins27. Other advantages of P. pastoris
include low levels of secreted host-cell proteins; little to no risk of viral contamination; validated expression of myriad proteins, including therapeutics approved by the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA); and the capability for human-like post-translational modifications in engineered strains28,56.
InSCyT uses fluidically connected modules for fermentation, multi-stage
chromatography, and ultrafiltration and/or diafiltration, as well as integrated sensors and system controllers for system-wide programmed operations (Fig. 2.1a,b and Fig. A.1). We
implemented continuous fermentation by perfusion to reduce the volume of the bioreactor and enable high space-time yields14. To this end, we adapted a sub-liter benchtop bioreactor for
in-tank perfusion and equipped it with sensors to control input and output flows, pH, temperature, impeller speed, and dissolved oxygen (Fig. 2.1c). The bioreactor was connected to a module for inline pH adjustment of the cell culture fluid prior to chromatographic separations; this module allowed the balancing of flow rates between those for production and purification. An integrated module for purification was designed to enable either two or three stages of chromatographic separation (Fig. 2.1c). This module allowed straight-through processing with no intermediate holding tanks or adjustments between purification steps. Our design simplifies the operation of the module relative to traditional purifications, wherein multiple intermediate procedures are often required to adjust pH, conductivity, concentration, and composition of fluids between steps of purification. The final module in the system is a tangential flow-filtration system for buffer exchange and formulation to a final liquid dosage form of the product (Fig. 2.1c). A custom
integrated software architecture unified operation of all three modules with appropriate controls as a fully automated single system.
We built and used three independent InSCyT systems to demonstrate consistent operational performance for both production and purification processes (Fig. 2.1d,e). We first produced and purified the common biologic medicine human growth hormone (hGH), used to treat growth deficiencies57. The extensive knowledge available about this drug’s quality, safety,
and potency aided our assessment of InSCyT and the novel process we developed for production of hGH. Our production process used two straight-through stages of
chromatographic purification and required fewer than half as many operations as the innovator process (Fig. A.2). We implemented our process on three separate InSCyT systems. After inoculation, each process ran “hands-free” and produced > 100 doses of formulated product in less than one week, with initial yields for the process ranging from 27%-31% on each system (Fig. 2.2a).
We compared the biophysical and biochemical attributes of our purified product from multiple time points on each system to a marketed drug substance using multiple analytics commonly used to establish identity, potency, safety, and purity of therapeutic proteins58 (Fig.
2.2b,c and Fig. A.3). We confirmed the protein sequence (100% coverage) by mass
spectrometry (Fig. A.4). Potency of InSCyT hGH was comparable to the National Institute for Biological Standards and Control (NIBSC) World Health Organization (WHO) 98/574 reference standard (96%-104%) using a cell-based proliferation assay. Key contributors to product safety include the levels of potentially immunogenic product-related impurities, such as aggregates, and the presence of process-related impurities, including host-cell proteins (HCPs) and host-cell DNA. Minimal high molecular weight species were present in the InSCyT hGH product (<0.5%), and levels of process-related impurities were each below typical values for clinical-stage
consider limits for host-cell proteins on a case-by-case basis, although in vitro studies using peripheral blood mononuclear cells from both healthy and diseased individuals have shown that HCP levels up to 4,000 p.p.m. from Chinese hamster ovary (CHO) cells do not pose a higher immunogenicity risk than a highly purified monoclonal antibody (<50 p.p.m.)60. A small
percentage (average: 6%) of a proteolytically cleaved form of hGH was also observed (two-chain variant); this natural form is both highly potent and previously has been determined as clinically irrelvant62. Further assessment for product-specific impurities by tandem liquid
chromatography-mass spectrometry (LC-MS), however, showed that InSCyT hGH was not comparable to marketed products because of increased levels of deamidation and oxidation (Fig 2.2c and Fig. A.5).
Given the consistency of both the operation of individual InSCyT systems and the hGH products produced by each system, we attributed the deamidation and oxidation observed in our product to process parameters, rather than the InSCyT system specifically. We therefore modified our process for hGH without any significant hardware changes by changing to a defined cultivation medium, adjusting the set point for dissolved oxygen and eliminating agitation in the surge tank. Using this adjusted process, a single InSCyT system produced nearly 50 maximum weight-based doses of hGH in 75 h (Fig. A.6). Oxidation and deamidation were reduced to below 1% at each residue, while all other quality attributes were maintained (Fig. 2.2c,d and Fig. A.6). Process yield also increased to nearly 80%. Together, these data demonstrate the capability of our manufacturing system to rapidly and reproducibly produce tens of doses of a potent and pure form of a biologic drug in an automated, short production cycle.
We next sought to demonstrate on-demand production of hGH. The upstream perfusion process was operated fully automated for 240 h. In addition to the initial purification and
were performed on-demand during days 6 and 10 (Fig. A.6). Each on-demand cycle produced between 50 and 75 doses of hGH within 12 h, with product quality and yields similar to those of the batch produced on day 3 (Fig. 2.2c,d and Fig. A.6). (We attributed the reduced yield (50%) and increased host-cell protein levels observed during the day 6 cycle to overloading the
capture column, which we adjusted before the day 10 cycle.) Further optimization of the process and column sizing could improve the consistency from batch to batch. Nonetheless, the ability to produce small lots of this product on demand shows the potential for manufacturing medicines as needed and highlights the stability of yeast-based bioprocesses in continuous operations.
Biopharmaceuticals typically require custom manufacturing processes that vary widely, especially for proteins other than monoclonal antibodies, and require unique facility designs14.
This constraint limits facility flexibility to provide additional products, which would be essential for on-demand supply. We therefore assessed whether our modular manufacturing system and choice of host could readily produce other molecules with no substantial hardware alterations. We selected interferon-α (IFN-α)-2b as a second example. This potent 19.2-kDa cytokine is used in both monotherapies and drug combinations to treat cancer and hepatitis, and is
produced commercially in a specialized 13-step process using E. coli63 (Fig. A.2). Owing to the
ease of targeted transgene insertion and simplicity of upstream process development in P.
pastoris, we developed a draft fermentation process for producing secreted IFNα-2b less than 4
weeks after identifying the product sequence (Fig. 2.3a). We have found that P. pastoris routinely secretes a consistent set of host-cell proteins along with the heterologous product during fermentation to yield a high level of initially pure product (>80%)24. This feature made it
possible to develop an in silico tool to predict draft multistage purification processes32. With this
in silico tool, we selected a process to purify IFNα-2b within another 4 weeks. The procedures
developed for both production and purification did not require modifications to the InSCyT system itself.
Initial biophysical analyses of IFNα-2b produced in our first run on the InSCyT system indicated minimal high molecular weight species (0.34%), and process-related impurities below typical values for clinical development (Fig. 2.3c). A cell-based viral replication assay
demonstrated that the potency of our IFNα-2b was the same or greater than that of a reference drug substance. InSCyT-generated IFNα-2b was highly potent (134%) in part owing to a
naturally-occurring C-terminal truncation known to increase potency64. Assessment for purity by
LC-MS and reversed-phase liquid chromatography (RPLC), however, showed that our product quality was not sufficient due to the presence of oxidized forms that could potentially promote aggregation and immunogenicity65 (Fig. 2.3c). These data show that rapid production of
biomolecules with acceptable bioactivity is necessary, but not sufficient, to define a clinical-quality biologic product.
To address these attributes, we optimized process conditions on InSCyT, performing experiments on individual modules simultaneously with fully integrated experiments comprising connected modules (Fig. 2.3a and Fig. A.7). After 27 d of process development, a final run showed that oxidation was reduced to < 1.5% at all residues (Fig. 2.3b,c). During this run the system produced nearly 8,000 formulated doses of IFNα-2b in less than one week (Fig. A.8). The identity and purity of the product was confirmed by multiple analytical methods at four time points during the campaign (Fig. 2.3c and Fig. A.8). RPLC, size-exclusion chromatography, and LC-MS showed levels of purity within targeted specifications; other analytics showed the specifications for safety and potency of our product were also achieved. Differences in chromatographic behavior of our product were confirmed by matrix-assisted laser desorption and ionization mass spectrometry as the naturally-occurring C-terminal truncation mentioned previously64 (Fig. A.8). The low overall process yield (~11%) is attributed to removal of an
sequence during expression by our host. Further engineering of the expression vector with alternative signal sequences could alleviate the expression of this variant66.
Next, we produced granulocyte colony stimulating factor (G-CSF), used to stimulate blood cell proliferation and reduce infections in cancer patients treated with myelosuppressive chemotherapy67. This drug has manufacturing challenges due to complex folding and a
propensity for aggregation and oxidation at specific amino acid residues68,69. We designed a
new process for production of G-CSF and implemented it on three InSCyT systems (Fig. A.2). After inoculation, each automated process yielded more than 165 doses of formulated drug in less than 100 h (Fig. 2.4a and Fig. A.9). Typical process yields for each cycle ranged from 70 to 90% (average 77%). We assessed biophysical and biochemical attributes of the product using multiple analytics to establish its identity, purity, safety, and potency (Fig. 2.4a and Fig. A.9). InSCyT G-CSF was comparable to a drug substance from a licensed product. We confirmed the protein sequence (100% coverage) by mass spectrometry (Fig. A.10). Minimal high molecular weight species were present in the product (0.33%-0.65%). Levels of process-related
contaminants in our formulated G-CSF were each below values typical for early-stage clinical development (1,000 p.p.m. for HCPs and 100 pg/dose for DNA)59–61. Potency of the InSCyT
G-CSF was comparable to the NIBSC WHO 09/136 reference standard (89.6%-141.1%) in a cell-based proliferation assay. Our product contained a minor variant comprising an N-terminal truncationand was a mixture of aglycosylated and glycosylated forms (Fig. A.9). Neither of these variants are likely to be clinically meaningful, as both truncations and glycosylation have been observed in licensed products without impact on product activity or safety70,71. Overall,
these data demonstrate that InSCyT can rapidly and consistently produce therapeutic proteins that are comparable to currently marketed products.
We performed further nonclinical studies with InSCyT-produced G-CSF to provide a framework for future clinical development. We assessed the pharmacokinetics,
pharmacodynamics, and toxicology of the InSCyT-produced G-CSF by comparing our product to a licensed product (Neupogen) in a rat model. We found InSCyT-produced G-CSF to be comparable to Neupogen in neutrophil activation during a single-dose administration study (Fig.
2.4b). InSCyT G-CSF and Neupogen showed no statistically significant difference in
pharmacokinetic profile when administered at the same dose (Kolmogorov-Smirnov test, P = 0.9963) (Fig. 2.4b). A 5-d repeat-dosing study also showed our product was comparable to Neupogen in toxicity as based on survival, clinical signs, body weight, quantitative food consumption, hematology, serum chemistry, organ weights, and macroscopic findings (Fig.
2.4b and Fig. A.11). In all of these studies, no abnormal clinical signs of toxicity, including
injection site inflammation, were observed in any animals dosed with InSCyT G-CSF (58 in total). Together, these data suggest that the InSCyT-produced G-CSF has potency in vivo and that potentially immunogenic process-related impurities are appropriately minimized.
2.3 Conclusions
InSCyT can produce a variety of clinical-quality recombinant therapeutic proteins in a liquid dosage form through integrated production, purification and formulation under a single control architecture. The efficient secretion of proteins by P. pastoris, combined with a holistic design of purification sequences, enabled new and intensified processes for hGH, IFNα-2b, and G-CSF that reduced the total number of processing steps by 45% or more and did not require refolding, excursions in pH or other substantial changes to the protein itself during processing (Fig. A.2). We demonstrated fast cycles of process development to reach clinically relevant target specifications in 12 weeks, aided by testing production at scale in a modular and integrated manner on InSCyT. The combination of the manufacturing system with the demonstrated strategy for process development could facilitate the rapid transition of lead molecules into the clinic for translational studies, and reduce subsequent iterations in process development and technology transfer for late-stage and commercial manufacturing.
Further engineering of InSCyT to comply with current good-manufacturing practices (cGMP) and concurrent development of an appropriate control strategy would enable its use for chemistry, manufacturing and controls of new drugs. A fill and finish module would enable product vial dispensing for simple administration to patients. Several relevant solutions have emerged, including systems from MedInstill and Vanrx. Modular facilities for housing
manufacturing equipment, such as G-Con PODs and Germfree BioGO™ Modules, also are becoming widely available for aseptic containment of small-scale manufacturing facilities.
InSCyT could be used in its current form to produce many other products, such as monoclonal antibodies, vaccine components, nanobodies and other antibody-like proteins (for example, bispecific T-cell engagers, Fabs), blood products (such as erythropoietin), and therapeutic enzymes (for example, β-glucocerebrosidase). Other products, such as insulin or modified products such as antibody-drug conjugates or PEGylated versions of products, would require additional modules for enzymatic processing, chemical ligation or crystallization; such systems could include de novo synthesis of the key starting materials or active pharmaceutical ingredient as well72. Further integration of multiple units may also facilitate blended products of
multicomponent vaccines or unique drug combinations tailored for applications for regional use or precision medicine.
DSP USP g TFF
e
c
b
a
d
Cycle #1 Column 1 UV Column 2 UV Cycle #2 Cycle #3 Cycle #4 A280 (mAU) 0 100 200 300 0 100 200 300 0 100 200 300 200 300System 1 System 2 System 3
Purification (DSP) 0.1 0.2 0.3 Stirrer (1e 4 RPM) Sparge (SPLM) 0 0.05 0.1 (ml/min) Addition rate
0 100 Level (% contact) 0 50 20 40 60 80 100 120 140 Time (hrs) 0 20 40 60 80 100 120 140 Time (hrs) 0 20 40 60 80 100 120 140 Time (hrs) 0
System 1 System 2 System 3
DO Stirrer O sparge Reactor Block pH Acid addition Base addition Level Feed B Feed A 0 200 400 DO (%) 15 20 25 30 6.0 6.5 7.0 pH Temperature ( oC) 0.0 0.5 1.0 Flow (g/min) Production (USP) Diverter Valve Pressure Regulator Pump Hollow Fiber Filter Product Retentate Reservoir Formulation Buffer To Waste From DSP To Waste Buffer Inputs Mixing Valve Capture
Column Column(s)Polish
To TFF Dilution Buffer Surge Tank pH UV UV UV UV From USP Production Media Inoculum Off Gas (O2,CO2) Air To DSP Perfusion Vessel Growth
Media Acid Base
pH DO T RPM Final Product Production (USP) Purification (DSP) Formulation (TFF) Production (USP) Purification (DSP) Formulation (TFF) MEDIA WASTE BUFFER MEDIA BUFFER 10 cm
Figure 2.1 Schematic of the InSCyT system for on-demand biomanufacturing and
demonstration of consistent operation across three distinct InSCyT systems. (a) To-scale rendering of the InSCyT system. Human figure is approximately 5’7”. (b) Photograph of an operational InSCyT system. (c) Detailed schematic of the InSCyT system including interactions between modules and key control points for the production (upstream processing, USP), purification (downstream processing, DSP) and formulation (tangential flow filtration, TFF) modules. Process parameter profiles collected by the control software from (d) the production (USP) module and (e) the purification (DSP) module of three separate InSCyT systems during hGH fermentation.
0
a
b
c
d
Circular Dichroism WHO Intl Std InSCyT hGH 0 1 2 3 Day 10 Day 6 0 1 2 3 Response 0 1 2 3 Day 3 Activity Marketed hGH InSCyT hGH -12000 -10000 -8000 -6000 -4000 -2000 2000 4000 ) MRW 200 210 220 230 240 Wavelength (nm) 250 M Std 0 3 6 10 3 6 10 MP Std M PerfusateSample Day #* Sample Day #**Formulated
2x diluted * **
MP - Master Pool 20x diluted
Rx Std - 0.5 and 0.1 mg/mL 180 130 100 70 55 40 35 25 15 10 kDa Aggregate M14 M125 M170 Oxidation 0 5 10 15 20 Percent V ariant (%) Marketed hGH InSCyT hGH 0 5 10 15 20 Percent V ariant (%) Pre-optimization Post-optimization Product-Related Variants InSCyT hGH
FDA Guideline (DNA) EMA Guideline (DNA) Target Range (HCP) 1 10 100 1000 10000
Host Cell DNA
(PPB) Host Cell DNA 0 500 1000
Host Cell Protien (PPM)
Host Cell Protein Process-Related Variants WHO Intl Std InSCyT hGH 0.1 1 0 1000 2000 3000 0.1 1 0 1000 2000 3000 hGH Dose 0.1 1 0 1000 2000 3000 Response Activity System #1 #2 #3 M Std 0 3 4 5 3 4 5 6 M Perfusate
Sample Day # Sample Day #Formulated* 6 180 130 100 70 55 40 35 25 15 10 M Std 0 3 4 5 3 4 5 6 M Perfusate
Sample Day # Sample Day #Formulated* 6 #3 #2 *4x diluted Rx Std - 0.27, 0.13, and 0.07 mg/mL M Std 0 3 4 5 3 4 5 6 M Perfusate
Sample Day # Sample Day #Formulated* 6 180 130 100 70 55 40 35 25 15 10 kDa System #1 Formulated doses System 1 System 2 System 3 Unpurified doses System 1 System 2 System 3 Wet Cell Weight (WCW) System 1 System 2 System 3 0 50 100 150 0 50 100 150 200 250 0 50 100 150 Time (h)
Wet Cell Weight (g/L)
hGH (doses)
Inoculation Induction Production
USP DSPTFF DSPTFF DSPTFF DSPTFF Production (USP) 1) Inoculate with hGH expressing strain 2) Accumulate biomass (32 h) 3) Induce expression 4) Production with perfusion
Purification (DSP)
1) Adjust supernatant (pH) 2) Multimodal cation exchange (bind and elute)
3) Multimodal anion exchange (flow-through)
Formulation (TFF)
1) Concentration (as needed) 2) Buffer exchange hGH
Figure 2.2 Production of hGH on the InSCyT system. Dose size used was 1.75 mg57. Center
values and error bars represent the mean and range, respectively, of technical triplicates unless otherwise indicated. (a) Process flow chart (left), timeline and yields (right) for production of hGH using InSCyT. Wet cell weight (WCW) (black), unpurified (orange) and formulated (blue) doses of hGH produced are shown. Grey circles represent individual data points. (b) Product quality analyses for InSCyT-produced hGH pre-optimization alongside a reference drug substance from a licensed hGH product produced in E. coli. SDS-PAGE (12% tris-glycine) analysis of samples from the USP during biomass accumulation and production (perfusate samples), final, formulated samples (formulated samples) and the reference (Std). Activity of InSCyT hGH alongside the WHO international standard (NIBSC 98/574). The final formulated sample (day 6) was analyzed from each system. Quantification of host-cell protein and host-cell DNA impurities in formulated InSCyT hGH. Host-cell protein limits are shown as a target
range59,60. Host-cell DNA guidelines are based on 100 pg/dose (FDA) and 10 ng/dose
(EMA)61,73. For host-cell protein data, each point represents a unique sample (12 points total; 4
time points from each of three InSCyT systems). For host-cell DNA, data each point represents a single pooled sample from each system comprising equal volumes of samples from each time point (3 points total; 1 per system). (c) Analysis of product-related variants in formulated InSCyT hGH pre-optimization (top) and post-optimization (bottom) alongside levels typically found in marketed products (Fig. A.5). Each data point represents a unique sample; there are 12 data points for pre-optimization runs (four time points from each of three InSCyT systems) and 3 data points for post-optimization runs (three time points from a single InSCyT system). Black boxes represent the range of InSCyT hGH samples with an additional line at the mean. (d) Product quality analyses for InSCyT produced hGH post-optimization alongside reference drug substance from a licensed hGH product. SDS-PAGE (12% tris-glycine) analysis of samples from the USP during biomass accumulation and production (perfusate samples), final, formulated samples (formulated samples) and the reference (Std). Activity of InSCyT hGH alongside the WHO international standard (NIBSC 98/574). Secondary structure analysis of InSCyT hGH (individual formulated samples from days 3, 6, and 10) and the reference hGH standard using circular dichroism (CD).
1 - 4 weeks 4 - 8 weeks 4 - 8 weeks
a
b
c
200 220 240 260 Wavelength (nm) -25000 -20000 -15000 -10000 -5000 0 5000 ) MRW InSCyT IFND-2b Reference IFND-2b Drug Substance Circular DichroismFirst At-scale Process Development Run Qualification Run Reference IFND-2b Drug Substance
Oxidation N-Terminal VariantAggregation
M148 M111 M59 M21 M16 0 5 10 15 20 25 % Impurity Product-Related Variants
Host Cell DNA (PPB)
Typically Accepted Range (EMA) Typically Accepted Range (FDA) InSCyT IFND-2b Target Range InSCyT IFND-2b First At-scale Process Development Run Qualification Run 1 10 100 1000 10000 100000 1000000 First At-scale Process Development Run Qualification Run
Host Cell Protein (PPM)
0 500 1000
Process-Related Variants
Host Cell Protein Host Cell DNA
180 130 100 70 55 40 35 25 15 10 180 130 100 70 55 40 35 25 15 10 Rx Std - 0.5 and 0.1 mg/mL P - USP sample during production F - Formulated InSCyT IFND-2b (2x diluted)
Rx Std - 0.5 and 0.1 mg/mL
P - USP sample during production (2x diluted) F - Formulated InSCyT IFND-2b (2x diluted)
F
M Std P
M Std P F
First At-scale
Process Development Run Qualification Run
IFND-2b USP DSP TFF Qualification Run Time (days) 0 7 9 10 17 1920 27 30
At-Scale Process Development (IFND-2b)
USP
~3 days ~1 dayDSP ~1 dayTFF manufacturing campaign(s)End-to-end
Complete facility use (connected modules);
7-10 days Independent use of modules
Identify product sequence Production strain High throughput batch cultivation Chromatographic screening of product In-silico synthesis of purification processes Characterization of host related impurities Strain development Purification development
Cell culture fluid from batch cultivation
One-time database collection (product independent)
8 -12 weeks 4 - 8 weeks 1 - 4 weeks
Figure 2.3 Accelerated process development using the InSCyT system and production of
IFNα-2b. Dose size was 12 µg63. Center values and error bars represent the mean and range,
respectively, of technical triplicates unless otherwise noted. (a) Process development timeline for new manufacturing processes using the InSCyT system, including simultaneous unit operation development (comprised of strain development and purification development), at-scale process development (comprised of simultaneous experiments on individual modules and on the fully integrated system), and process qualification. (b) Timeline for at-scale process development for IFNα-2b. Horizontal colored bars represent the modules that were used in each experiment (USP – orange, DSP – purple, TFF – blue). Each new bar represents a new set of experimental conditions on that module. (c) Product quality for InSCyT-produced IFNα-2b from the first at-scale run after initial unit operation development (first at-scale process development run) and the final qualification run alongside a reference drug substance produced in E. coli. SDS-PAGE (12% tris-glycine) analysis of samples from the USP during production (P), a final, formulated sample (F) and a reference drug substance (Std). Analysis of process-related variants in formulated InSCyT IFNα-2b (per Fig. 2.2b). Each data point represents a unique sample, there is 1 data point from the first at-scale process development run and 4 data points from the qualification run (four time points from a single InSCyT system). Product-related variants detected in formulated InSCyT IFNα-2b alongside levels typically found in a reference drug substance (Fig. A.5). Black boxes represent the range of InSCyT IFNα-2b samples with an additional line at the mean. Secondary structure analysis of InSCyT IFNα-2b (triplicate analyses of an individual sample from the qualification run) and reference drug substance (duplicate analyses of an individual sample) using circular dichroism (CD).
Circular Dichroism
a
b
Vehicle
ControlInSCyTG-CSF Neup-ogen
Hematology 9.15* 32.65** 39.61** Neutrophils 86.15* 59.78ං 52.74 1.93* 4.42** 4.17** Clinical Chemistry (M) 155.2* 289.4** 384.6** 65.6* 193.8** 217.8** 102.10* 98.96** 99.42ං 0.458* 0.420 0.364** Lymphocytes Monocytes ALP Clinical Chemistry (F) ALP Chloride Creatinine Toxicology 0 1 2 3 0 10 20 30 40 80 120 InSCyT High Dose (575 Pg/kg)
InSCyT Low Dose (115 Pg/kg) Neupogen Low Dose (115 Pg/kg)
0 10 20 30 40 80 120 Time (Hrs) 0.0 0.2 0.4 0.6 0.8 G-CSF Concentration ( Pg/mL) Vehicle
Control NeupogenLow Dose (115 Pg/kg) InSCyT Low Dose (115 Pg/kg) InSCyT High Dose (575 Pg/kg) 0 10 20 30 40 50
Relative Neutrophil Counts
(10
3 cells/
PL)
Neutrophil Activation Pharmacokinetics
0.001 0.01 0.1 1 0 1 2 3 4 0.001 0.01 0.1 1 0 1 2 3 4 0.001 0.01 0.1 1 0 1 2 3 4 0.001 0.01 0.10 1 1 2 3 4 0.001 0.01 0.10 1 1 2 3 4
G-CSF Dose G-CSF Dose G-CSF Dose G-CSF Dose G-CSF Dose G-CSF Dose 0.001 0.01 0.10 1 1 2 3 4 WHO Intl Std InSCyT G-CSF Response Batch #1 #2 #3 #4 #5 #6 Activity Marketed G-CSF InSCyT G-CSF Neupogen Wavelength (nm) 200 210 220 230 240 250 ) MRW -30000 -20000 -10000 0 10000 20000 Target Range (HCP)
InSCyT G-CSF FDA Guideline (DNA) EMA Guideline (DNA)
Host Cell DNA 1 10 100 1000 10000 100000
Host Cell DNA (PPB)
Host Cell Protein
0 500 1000
Host Cell Protein (PPM)
Marketed G-CSF InSCyT G-CSF Neupogen Aggregate Q12 Deamidation M1 M122 M127 M138 Oxidation 0 1 2 3 4 5 Percent V ariant (%)
Product-Related Variants Process-Related Variants
8.2 8.0 7.8 7.5 7.1 7.0 6.5 6.0 5.1 4.65 M Std G P F M Std InSCyT 180 130 100 70 55 40 35 25 15 10 Rx Std - 0.5 and 0.1 mg/mL
G - USP sample during biomass accumulation P - USP sample during production F - Formulated InSCyT G-CSF (5x diluted)
Perfusate Formulated
Vials SDS-PAGE IEF
Wet Cell Weight (WCW) Unpurified doses Formulated doses 0 50 100 150 200 G-CSF (doses) 0 20 40 60 80 Time (h) 0 50 100 150 200 250
Wet Cell Weight (g/L)
Inoculation Induction Production
G-CSF
Figure 2.4 Production of G-CSF on three identical InSCyT systems. Dose size 300 µg67. Center
values and error bars represent the mean and range, respectively, of technical triplicates unless otherwise noted. (a) Timeline and yields for production of G-CSF using the InSCyT system for a single representative sample (Batch #1). Wet cell weight (WCW) (black circles) and cumulative unpurified (orange) and formulated (blue) doses of G-CSF are shown. Grey circles represent individual data points. Product quality for InSCyT-produced G-CSF alongside drug substance from a licensed product produced in E. coli and Neupogen (produced by Amgen in E. coli). A photo of vials comparing material sampled from the USP (perfusate) to final formulated material (formulated). SDS-PAGE (12% tris-glycine) analysis of Batch #1 from the USP during biomass accumulation (G) and production (P), and a final, formulated InSCyT sample (F) alongside drug substance from a licensed product (Std). Analysis of product purity by isoelectric focusing (IEF) for formulated Batch #1. Gel analyses of Batch #1 are representative of all six batches (Fig.
A.9). Analysis of product-related variants and process-related variants. Each data point
represents a unique batch (2 time points from each of 3 distinct systems). Paired data points indicate analyses from a single batch. Product-related variants are shown alongside levels typically found in marketed products (Fig. A.5). Black boxes represent the range of InSCyT G-CSF samples with an additional line at the mean. Process-related variants are shown alongside common guidelines (per Fig. 2.2b). Analysis of the secondary structures of InSCyT G-CSF (Batches 1-6) and a reference drug substance from a licensed product using circular dichroism (CD). Activity of InSCyT G-CSF alongside that of the WHO International standard (NIBSC 09/136). (b) Analysis of pharmacokinetics (PK), pharmacodynamics (PD), and toxicology of InSCyT-produced G-CSF and a licensed product (Neupogen) in a rat model. Neutrophil
activation and pharmacokinetic profile of low dose (115 µg/kg, n=3 animals, t1/2 = 2.1 h) and high
dose (575 µg/kg, n=3 animals, t1/2 = 4.6 h) InSCyT G-CSF in rats compared to Neupogen (115
µg/kg, n=3 animals, t1/2 = 1.4 h) (PK: p=0.9963, Kolmogorov-Smirnov test). For neutrophil
activation, grey boxes represent the range of three individual animals with an additional line at the mean. For PK center points and error bars represent the mean and range, respectively, of three individual animals. Summary of statistically significant results comparing the toxicology of InSCyT G-CSF and Neupogen to a vehicle control. Values represent the mean; standard deviation and sample size can be found in Fig. A.11. Statistical significance was determined by one-way ANOVA. ALP – alkaline phosphatase

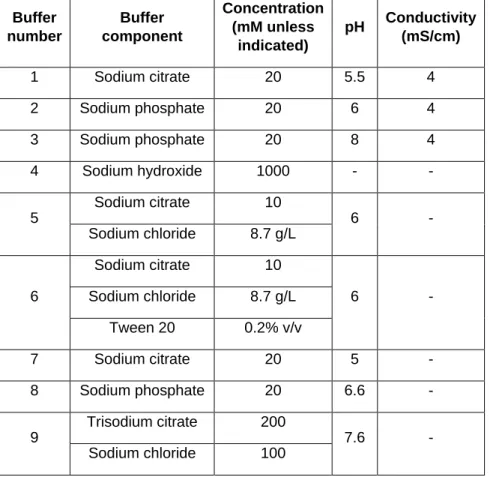
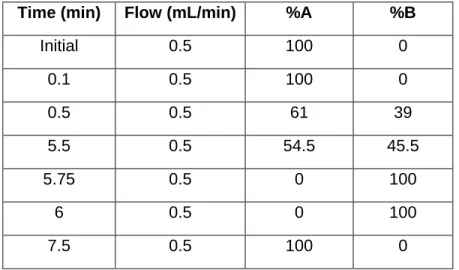
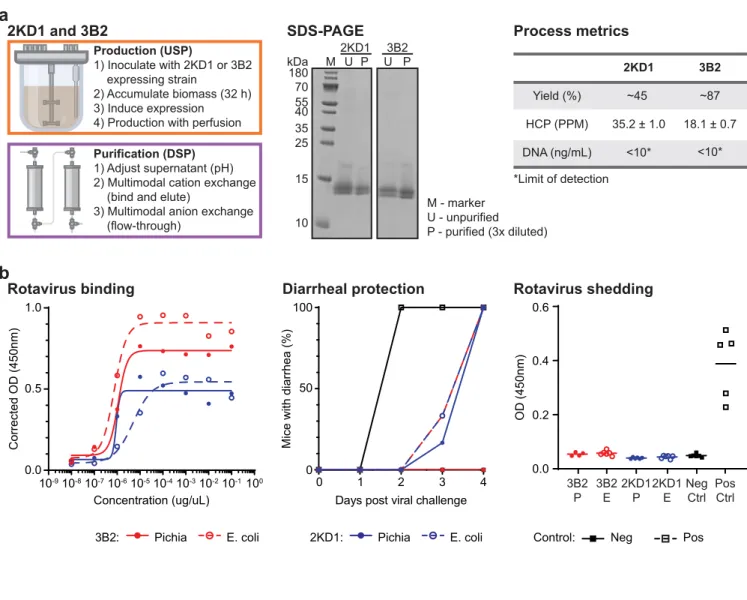
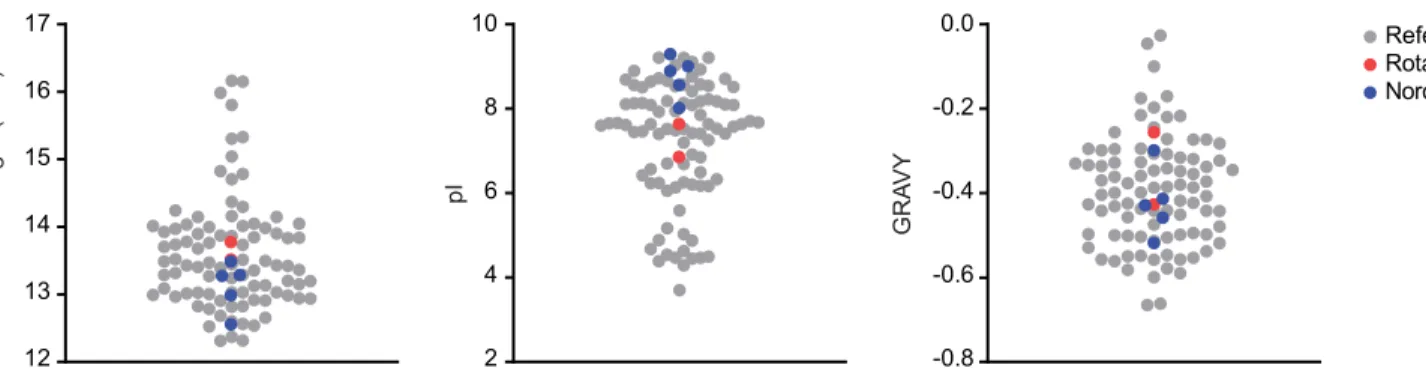
![Figure 3.1. Production and purification of P2-VP8-P[4] (original) on the InSCyT system](https://thumb-eu.123doks.com/thumbv2/123doknet/13819841.442514/67.918.74.834.83.545/figure-production-purification-p-vp-p-original-inscyt.webp)
![Figure 3.2. Production and purification of engineered P2-VP8-P[4] on the InSCyT system](https://thumb-eu.123doks.com/thumbv2/123doknet/13819841.442514/68.918.75.506.88.444/figure-production-purification-engineered-p-vp-p-inscyt.webp)
![Figure 3.3. Production and purification of engineered P2-VP8-P[8] on the InSCyT system](https://thumb-eu.123doks.com/thumbv2/123doknet/13819841.442514/69.918.81.839.82.596/figure-production-purification-engineered-p-vp-p-inscyt.webp)
![Figure 3. 4. Production and purification of engineered P2-VP8-P[6] on the InSCyT system](https://thumb-eu.123doks.com/thumbv2/123doknet/13819841.442514/70.918.76.513.87.452/figure-production-purification-engineered-p-vp-p-inscyt.webp)
![Figure 3. 5. Production and purification of P[4], P[6], and P[8] subunits in a single manufacturing campaign on the InSCyT system](https://thumb-eu.123doks.com/thumbv2/123doknet/13819841.442514/71.918.82.844.80.828/figure-production-purification-subunits-single-manufacturing-campaign-inscyt.webp)