Digest: Demographic inferences accounting for selection at linked sites
Texte intégral
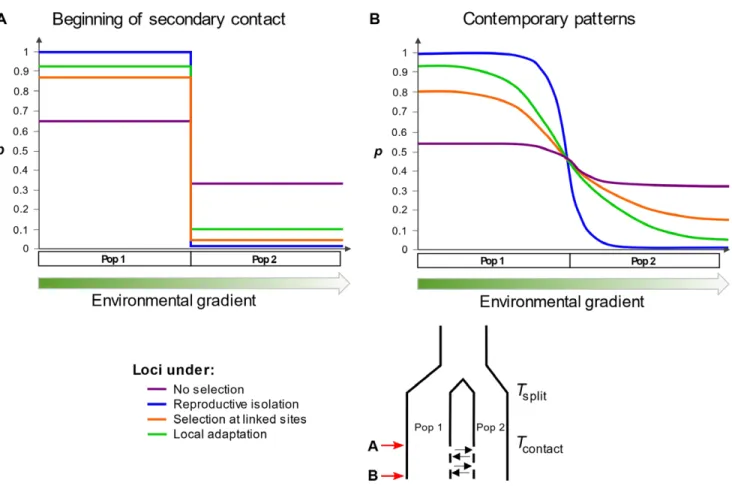
Figure
Documents relatifs
kernelPSI: a Post-Selection Inference Framework for Nonlinear Variable Selection that estimate the association between a given kernel and an.. outcome of interest, that can
quo n’est donc pas une solution. Doter l’Alsace d’une collectivité à l’instar de la Corse peut sembler une solution souhaitable. Mais entre le vote des textes législatifs et
(scénarios démographiques et/ ou évolutifs trop complexes) 1-Tirer une valeur du paramètre dans le prior. 2-Simuler un jeu de données avec cette valeur de paramètre Ex : taux
We show that our algorithm, applied to a toy exam- ple and to an individual-based social model, requires significantly less simulations to reach a given quality level of the
The aim of applying these additional criteria is to build an evaluation tree that minimizes the total computational cost of compiling (building the corresponding probability table)
As a toy model we inferred the population–mutation parameter u ¼ 4Nm of a panmictic population model from the number of segregating sites S of a sample of sequences with 10,000 bp
We used posterior predictive checks to study the entropy of relative synonymous codon usage (RSCU, fig. 6) and of rela- tive codon frequencies (RFC, supplementary fig.
Our Bayesian variable selection method for probit mixed models is developed to select a few important probesets, among tens of thousands, which are indicative of the activity of
