Molecular Modeling of Interfacial Phenomena
Texte intégral
Figure
![Figure 1. The place of computer simulations in the hierarchy of scientific research. (Image taken from [1])](https://thumb-eu.123doks.com/thumbv2/123doknet/14612118.545717/11.892.296.589.152.526/figure-place-computer-simulations-hierarchy-scientific-research-image.webp)
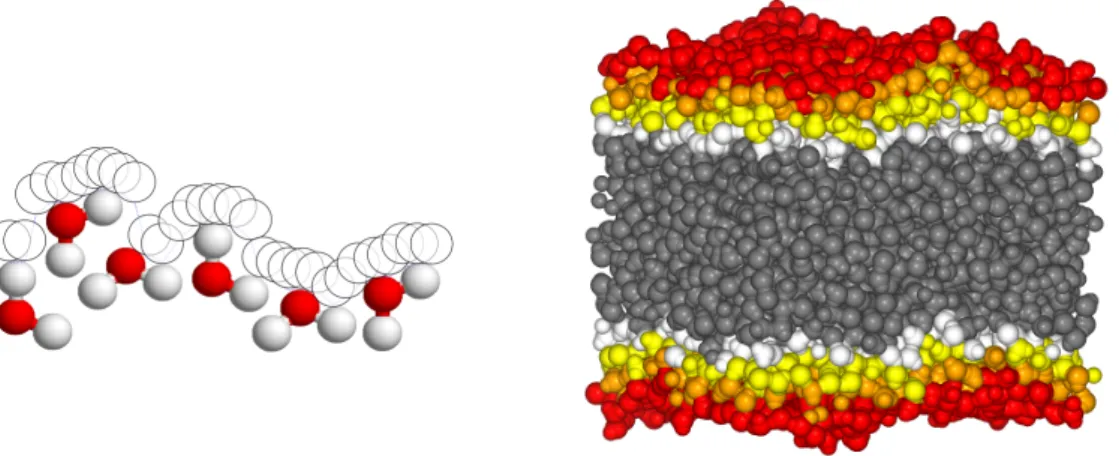

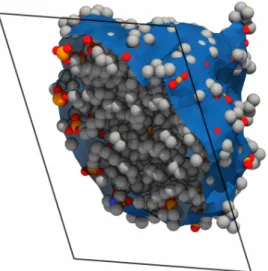


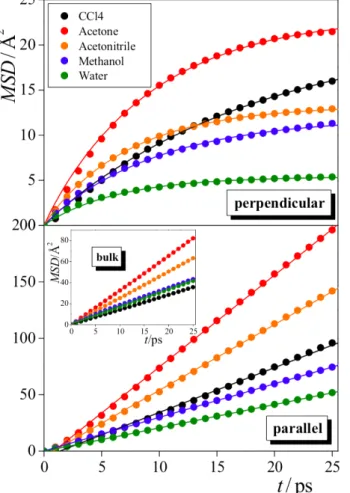
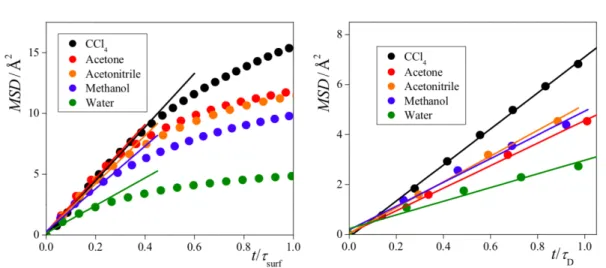

Documents relatifs
The synthetic case was used in order to test four data types for model calibration: one water content profile at a single sampling time (case 1); one water content profile and one
This simple example illustrates the fact that the use of a weighted or a non weighted graph for the join count statistics can have a strong influence on the result, depending on
Für Kinder ohne Sprachentwicklungsstörung ist eine eindeutige Schlussfolgerung nicht mög- lich – zwar finden sich in der SU-Gruppe einzelne Probanden, die sehr schwache Ergebnisse
Parmi les patients non diabétiques, les résultats obte- nus à l’aide de la collecte urinaire étaient comparables à ceux calculés par les différentes formules, contraire- ment
Ainsi, nous avons cherché à constituer deux sous-corpus de publications déjà référencées dans HAL en fonction de termes se trouvant mentionnés soit dans leur résumé, soit
La première (M1, publiée dans le n° 1) a consisté à rechercher les thèses d’université (excluant donc les thèses dites d’exercice, en médecine, pharmacie ou de
La présente étude concerne l’évaluation de la qualité physico-chimique et organique des eaux souterraines dans le périmètre irrigué. Une campagne d’échantillonnage a
tem is a positive and monotonically decreasing function of the thickness of the ice layer. It follows that an arbitrarily thick layer of ice should form at the water vapor interface
