One-Pot Cascade Synthesis of (3S)-Hydroxyketones Catalyzed by
Transketolase via Hydroxypyruvate Generated in Situ from
d-Serine by d-Amino Acid Oxidase
Mélanie L’enfant,
aFelipe Bruna,
aMarion Lorillière,
aNazim Ocal,
aWolf-Dieter Fessner,
bLoredano Pollegioni,
cFranck Charmantray,
a,* and
Laurence Hecquet
a,*
aUniversité Clermont Auvergne CNRS
SIGMA Clermont, Institut de Chimie de Clermont-Ferrand (ICCF) F-63000 Clermont-Ferrand (France)
E-mail: franck.charmantray@uca.fr; laurence.hecquet@uca.fr
b
Institut für Organische Chemie und Biochemie Technische Universität Darmstadt
64287 Darmstadt (Germany)
c
Department of Biotechnology and Life Sciences Università degli Studi dell’Insubria
Varese (Italy)
Manuscript received: January 24, 2019; Revised manuscript received: March 14, 2019;
Version of record online: April 9, 2019
Supporting information for this article is available on the WWW under https://doi.org/10.1002/adsc.201900109
Abstract:
We described an efficient in situ generation of hydroxypyruvate from serine catalyzed by a
d-amino acid oxidase from Rhodotorula gracilis. This strategy revealed an interesting alternative to the
conventional chemical synthesis of hydroxypyruvate starting from toxic bromopyruvate or to the enzymatic
transamination from l-serine requiring an additional substrate as amino acceptor. Hydroxypyruvate thus
produced was used as donor substrate of transketolases from Escherichia coli or from Geobacillus
stearothermophilus catalyzing the stereoselective formation of a carbon carbon bond. The enzymatic cascade
reaction was performed in one-pot in the presence of d-serine and appropriate aldehydes for the synthesis of
valuable (3S)-hydroxyketones, which were obtained with high enantio- and diastereoselectivity and in good
yield. The efficiency of the process was based on the irreversibility of both reactions allowing complete
conversion of d-serine and aldehydes.
Keywords: biocatalysis; transketolase; d-amino acid oxidase; hydroxypyruvate; ketoses; stereoselectivity
Introduction
Hydroxypyruvate (HPA) is a key substrate for several
HPA-dependent enzymes including aldolases
[1]and
transketolases (TK)
[2]used in biocatalysis for the
synthesis of valuable monosaccharides and analogues
by stereoselective carbon carbon bond formation. For
synthetic purposes conducted at gram-scale, the main
problem is the cost of the commercial HPA. This
compound can also be synthetized by hydrolysis of
toxic bromopyruvic acid in the presence lithium
hydroxide.
[3]Enzymatic in situ generation of HPA has
been previously described, in particular from l-serine
using transaminases (TA) (Scheme 1A).
[4–6]The
rever-sibility of the TA-reaction requires the simultaneous
coupling to an irreversible enzyme-catalyzed reaction
to shift the overall equilibrium toward HPA formation
and to avoid its accumulation and degradation,
partic-ularly
at
high
temperature,
and/or
in
buffered
solution.
[5]The irreversible TK-catalyzed
decarboxyla-tion of HPA is a powerful tool for such an approach.
Indeed, TK, a thiamine dependent enzyme, catalyzes
the subsequent transfer of the ketol group of HPA to an
aldehyde acceptor leading to a (3S)-ketose by the
stereoselective formation of the C2 C3 bond.
[1]The
coupling of the TA and the TK reactions in a one pot
has been developed producing the expected ketose
products.
[4–6]The major drawback of this approach is
the requirement of an α-keto acid as an amino group
acceptor, which is a second substrate generating the
corresponding amino acid as an additional by-product
if not recycled. In addition, the equilibrium shift
toward the formation of the product needs an excess of
one of the two TA substrates, i. e., d-serine or α-keto
acid, and in the latter case inhibition of TA was
observed.
[5,6]To circumvent all these limitations, another strategy
consists in using an aminoacid oxidase (AAO) to
catalyze the oxidative deamination of serine into HPA
(Scheme 1B). Substrate specificity studies of AAOs
have shown that the best substrates for lAAOs are
hydrophobic aminoacids
[7]while dAAOs display a
broad aminoacid spectrum including d-serine. Hence,
we have selected the latter enzyme family for the
approach developed in this study.
dAAOs (EC 1.4.3.3) are flavin adenine
dinucleo-tide (FAD)-containing flavoenzymes that catalyze the
deamination of d-amino acids to their imino acid
counterparts with concomitant reduction of FAD which
is subsequently re-oxidized by molecular oxygen with
generation of hydrogen peroxide. The released imino
acid spontaneously hydrolyses to the corresponding
α-keto acid and ammonia (Scheme 2). dAAO activity
has been identified in many organisms, ranging from
fungi to humans.
[8]The well-known pig kidney dAAO
has been first reported for use in biocatalytic
applications,
[9–11]but the enzyme from the yeast
Rhodotorula gracilis (DAAO
Rg) showed much higher
turnover number, better stability under a wide range of
reaction conditions, and a larger active site to
accom-modate various substrates.
[12,13]Due to these properties,
the yeast enzyme has been used in a variety of
biotechnological processes at industrial scale such as
the production of glutaryl-7-aminocephalosporanic
acid from cephalosporin C,
[14]the resolution of racemic
mixtures of amino acids,
[15]and the production of
α-keto acids from natural and synthetic d-amino acids.
[16]DAAO
Rgaccepts the polar amino acid d-serine with
higher specific activity compared to dAAO from pig
kidney.
[9]However, this enzyme has never been
investigated to produce the α-keto acid HPA from
d-serine, especially at preparative scale.
The aim of this work is to study the reaction
catalyzed by DAAO
Rgin the presence of d-serine as
the substrate to evaluate the in situ synthesis of HPA at
preparative scale. It should be noted that d-serine is
commercially available at approximatively the same
price as l-serine and can be produced at industrial
scale by a microbial process.
[17]To validate the
strategy, the DAAO
Rg-catalyzed reaction will be first
studied independently and then coupled with TK,
requiring and consuming HPA as donor substrate. The
irreversibility of TK-catalyzed reaction and the
stereo-selectivity of this enzyme are major advantages. On
this side, wild-type or evolved TKs from mesophilic
[18]or thermophilic
[5,6b,19]sources have been reported as
efficient biocatalysts for the conversion of a wide
range of aldehydes leading to the synthesis of various
(3S)-hydroxyketones. In this study, we have selected
the mesophilic TK from Escherichia coli (TK
eco)
[18d–h]
and the thermostable TK from Geobacillus
stearother-mophilus (TK
gst)
[19,20]to be coupled with DAAO
Rgin a
one-pot two step procedure, starting only from the two
substrates d-serine and an appropriated aldehyde
acceptor for obtaining valuable (3S)-hydroxyketones.
Results and Discussion
Properties of dAAO from R. Gracilis
DAAO
Rgwas overexpressed in E. coli and then
extracted and purified by Ni
2 +chelating affinity
column chromatography as reported earlier.
[10]In order
to use DAAO
Rgunder the best conditions for the
synthesis of HPA in situ from d-serine, we first
determined at analytical scale the stability of the
recombinant flavoenzyme over time at different
tem-peratures and its kinetic parameters using d-serine as
substrate in the presence of oxygen. During turnover
the tightly bound FAD cofactor is reduced by the
d-amino acid to FADH
2and then reoxidized by O
2generating hydrogen peroxide. Since the accumulation
of hydrogen peroxide can lead to an enzyme inhibition,
addition of catalase is required for the dismutation of
hydrogen peroxide to water and oxygen (Scheme 2).
Scheme 1. In situ generation of HPA from TA ordAAO-catalyzed reaction coupled with TK-dAAO-catalyzed reaction.
Scheme 2. Reaction of DAAORg coupled with l-lactate
DAAO
Rgactivity was determined by a coupled
reaction with l-lactate-dehydrogenase (l-LDH)
allow-ing the reduction of HPA in the presence of NADH
(Scheme 2). The disappearance of NADH was
fol-lowed over time by spectrophotometry at 340 nm
(LDH assay).
An optimum DAAO
Rgactivity was previously
described in a 37–45
°C temperature range and a 8.0–
8.5 pH range.
[10]Under those conditions, we observed
that DAAO
Rgfully maintained its activity during
25 hours at 25
°C and at pH 7.5 while at higher
temperature the enzymatic activity decreases
drasti-cally (Figure 1).
The DAAO
Rgkinetic parameters were determined
using the LDH assay described above. The kinetic
parameters toward d-serine were approximatively the
same than those reported in the literature using a
polarographic assay method.
[11]This latter assay has
been used for the determination of kinetic parameters
of other amino acids (Table 1).
DAAO
Rgoffers more substrate flexibility compared
to other dAAOs.
[8,11]Indeed, based on the K
M
values,
the best affinities were apparent for methionine,
d-phenylalanine, d-leucine, and d-alanine. Further amino
acids are also accepted with lower affinities such as
d-serine, d-valine, and d-cysteine. The V
maxobserved for
d-serine is approximately half of the value obtained
with the reference substrate D-alanine. Even if d-serine
is not the best substrate for DAAO
Rg, the K
Mand V
maxvalues are more suitable for biocatalytic applications
compared to those obtained toward l-serine converted
into HPA by TA-catalyzed reaction (K
M22.4 mM; V
max3.3 U. mg
1; V
max/K
M0.14 U. mg
1
. mM
1).
[5]In
addi-tion, the DAAO
Rgcatalyzed reaction is irreversible
offering the possibility to convert d-serine completely
into HPA and making this strategy an efficient
synthetic way for obtaining HPA at preparative scale.
Conversion of d-Serine into HPA Catalyzed by
DAAO
RgFor HPA synthesis from d-serine at preparative scale,
catalase was added dropwise to the reaction mixture,
because addition of a specific quantity of catalase as a
single dose at the beginning of the reaction was found
to be insufficient to ensure the total dismutation of
hydrogen peroxide during the entire reaction period,
thus causing a decrease of dAAO activity. Oxygen was
bubbled into the reactor at a rate of 10 mL. min
1at
atmospheric pressure. The reaction was followed by
quantification of HPA and d-serine over time by
spectrophotometry in aliquots from the reaction
mix-ture. After the completion of the reaction, HPA and
d-serine quantification was performed by in situ
1H
NMR analysis of the reaction mixture.
d-Serine at 50 mM led to almost complete
con-version (90%) in 24 hours while HPA quantification
corresponded to 78% of initial d-serine concentration
(Figure 2). This result may be explained by a slight
HPA degradation over time as previously observed.
[19]In addition, we noted that DAAO
Rgactivity decreased
during the reaction (SI) due to an inhibition by HPA
Figure 1. DAAORgthermostability. DAAORgactivity wasdeter-mined by the LDH assay in the presence of d-serine (50 mM) and O2 at ♦ 25°C, ~ 30°C, * 40°C, & 60°C. 100%
corresponds to initial activity (37.5 U. mg 1 at 25°C and
pH 7.5).
Table 1. Apparent kinetic parameters of DAAORgtoward different d-amino acids (at air saturation).
d-amino acids KM mM Vmax U. mg 1 Vmax/KM U. mg1. mM 1 kcat/KM mM. 1s 1 d-methionine[10] 0.18 115 639 416 d-phenylalanine[10] 0.29 144 497 323 d-leucine[10] 0.50 120 240 156 d-alanine[10] 0.83 140 169 110 d-serine 13.7[10] 61[10] 4.4[10] 3.0
14.0 � 1[a] 48 � 7[a] 3.5 � 0.8[a] 2.3 � 0.5
d-valine[10] 18.9 195 10.3 6.7
d-cysteine[10] 21.4 130 6.1 4.0
already described with 2-oxo-4-methylthiobutyric acid,
the α-keto acid produced from d-methionine
[21a]and
other α-keto acids analogs of amino acids.
[21b]Indeed,
conversion of d-serine into HPA was 66 and 90% at
250 mM and 50 mM, respectively, clearly showing an
inhibition of DAAO
Rgby the ketoacid. To avoid HPA
accumulation and to limit DAAO
Rginhibition, the
DAAO
Rg-catalyzed reaction was therefore coupled
with an irreversible reaction that consumes HPA as
substrate such as a TK-catalyzed reaction.
Hydroxyketone Synthesis from d-Serine and
Vari-ous Aldehydes by DAAO
Rg/TK
gstcoupling
The one-pot, two-step cascade reaction catalyzed by
DAAO
Rgand TK was performed using two substrates:
d-serine and an appropriated aldehyde as TK acceptor
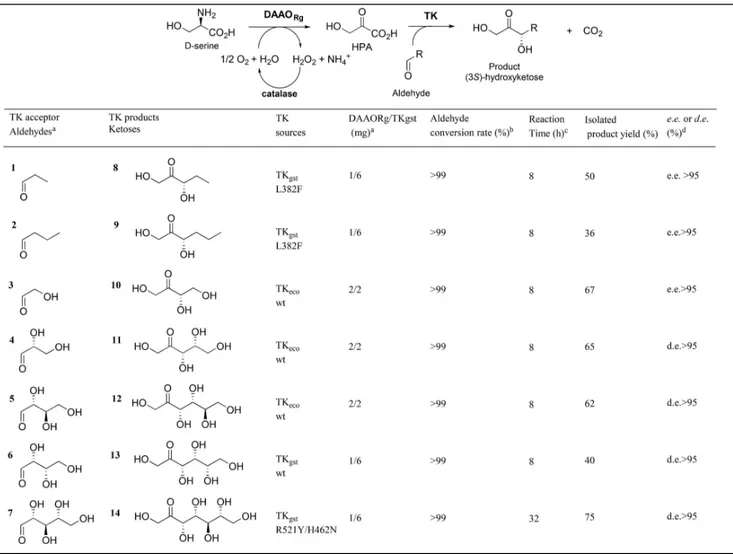
substrate. To validate the strategy, various aldehydes
were chosen, such as non-hydroxylated (1, 2) and
hydroxylated (3–7) aldehydes (Table 2). For better
efficiency, TKs from different sources were used
depending on the aldehyde. Previous substrate
specif-icity studies showed that for wild-type TKs best
substrates are (2R)-configured hydroxylated aldehydes
with hydroxylated short carbon chain length (C
2C
4).
Hence, for hydroxylated aldehydes 3–5 and 6, we
employed wild-type TK
eco[2]and wild-type TK
gst,[19]respectively, reported as the best TK sources for these
substrates, respectively. For non-hydroxylated
alde-hydes 1/2 and for long-chain hydroxylated aldehyde 7,
the reactions were performed using previously
identi-fied, specially designed TK
gstvariants L382F
[20b]and
R521Y/H462N,
[22]respectively.
To study the influence of different concentrations of
both enzymes and substrate, the reaction mixtures were
monitored by in situ
1H NMR of the reaction mixture
for the determination of aldehyde conversion rate and
by enzymatic analysis from aliquots for HPA detection
using the LDH assay described above.
For all reactions, the same concentration (50 mM)
of both substrates d-serine and aldehyde was used. The
ratio DAAO
Rg/TK was adjusted to obtain a total
conversion of d-serine considering that a minimum
amount of both enzymes was used and that HPA was
gradually consumed by TK-catalyzed reaction. As an
example, Figure 2 shows the simultaneous
quantifica-tions of d-serine and HPA in the reaction mixture
containing both DAAO
Rgand TK
ecoenzymes in the
presence of glycolaldehyde 3 as TK
ecoacceptor
substrate. The conversion of d-serine into HPA was
achieved in 8 hours. In parallel, a complete
disappear-ance of glycolaldehyde 3 was observed by in situ
1H
NMR. Hence, the bi-enzymatic cascade avoids HPA
accumulation and DAAO
Rginhibition as shown by the
very rapid conversion of d-serine and 3 (8 hours) as
compared to the longer reaction time required for
d-serine conversion (24 hours) when DAAO
Rgwas not
coupled with TK. This procedure also allowed a rapid
and total conversion (8 hours) of other hydroxylated
(4–6) aldehydes as well as non hydroxylated (1, 2)
aldehydes using the appropriate TKs (Table 2).
All the ketose products 8–13 were purified and
characterized as the only products arising with high
enantio- and diastereoselectivity (> 95%).
Enantiomer-ic excess of products 8, 9, 10 were determined by
chiral GC after peracetylation.
[20c]Moreover, the
absolute configurations of these compounds have been
confirmed by comparison with reference samples as
previously described.
[20c]These results show that DAAO
Rg/TK coupling
allows greatly increasing the d-serine conversion rate
as compared to the dAAO
Rgalone, avoiding the
inhibition of HPA by its gradual consumption in the
TK-catalyzed reaction.
The conversion of aldehyde 7 into product 14 was
performed with the ad hoc designed TK
gstvariant
R521Y/H462N.
[22]We showed that this synthesis must
be carried out at 60
°C for improved conversion rate of
aldehyde 7.
[22]Since DAAO
Rgis not stable at this
temperature for longer times, the reaction was first
performed at 25
°C for HPA synthesis from d-serine
(8 hours) and then TK and aldehyde 7 were added with
increasing the temperature to 60
°C for 24 hours. This
procedure allowed to obtain ketose product 13 in
reasonable reaction time (32 hours) with good isolated
yield (72%) and excellent diastereoselectivity (> 95%).
Conclusions
We developed the first application of DAAO
Rgfor
rapid in situ generation of HPA from d-serine. For
improvement of the HPA synthesis, the DAAO
Rgreaction was coupled with TK
gstin a one-pot, two-step
cascade sequence for ketose synthesis from two
substrates, d-serine and an appropriate aldehyde. The
aldehyde was used as TK acceptor substrate and it was
Figure 2. Comparison of d-serine conversion and HPAforma-tion overtime obtained with DAAORgalone (& d-serine and ♦
HPA) or with DAAORg and TKgst in the presence of
introduced in the reaction mixture at the same
concentration as D-serine. dAAO and TK-catalyzed
reactions were performed simultaneously and applied
to the synthesis of ketose products 8–13 from
aldehydes 1–6 with complete conversion of all
sub-strates in 8 hours. All the targeted compounds were
recovered with high enantio- and diastereoselectivity.
For the conversion of long carbon chain aldehyde 7,
TK-catalyzed reaction requiring elevated temperature
(60
°C) and longer reaction time (32 hours), the two
steps were performed sequentially. The produced
polyhydroxylated products 10–14 are valuable
com-pounds in various fields: l-erythrulose 10 is used in
self-tanning formula,
[23]d-xylulose 11 is an important
intermediate for d-xylose fermentation to ethanol,
[24]d-fructose 12 is a well-known sweetener,
[25]l-sorbose
13 is a precursor of ascorbic acid
[26]and
d-altro-heptulose 14 is a marker of sugar metabolism disorders
such as cystinose.
[27]This environmentally friendly procedure represents
an interesting alternative to the conventional chemical
synthesis of HPA from toxic bromopyruvate and to the
in situ generation of HPA by reversible enzymatic
TA-catalyzed reaction from l-serine requiring an
addi-tional α-keto acid as an amino acceptor and excess of
one substrate to shift the equilibrium. This
straightfor-ward approach could be applied to other aldehydes
using wild-type or variants TK
gstand could be used in
additional cascade reactions involving further
HPA-dependent enzymes such as aldolases.
Table 2. Reaction conditions of the one-pot multi-enzymatic cascade synthesis of ketose products.
[a]Reactions were carried out with DAAO
Rg(1 or 2 mg) and TKgst(2 or 6 mg), ThDP (0.1 mM), MgCl2(1 mM), aldose (50 mM),
D-serine (50 mM) at pH 7 and 25°C.
[b]Aldose acceptor conversion determined by in situIH NMR analysis. [c] 1H NMR of reaction mixture (SI).
[d]Enantiomeric excess (e.e.) determined by chiral GC after peracetylation[20c]and diastereoisomeric excess (d.e.) determined by1H
Experimental Section
General. All chemicals were purchased from Sigma-Aldrich, Alfa-Aesar and CarboSynth. Bradford reagent was from Bio-Rad. Ni-NTA resine was obtained from QIAGEN. Proteins and enzymes were acquired from Sigma-Aldrich. Lyophilisation was carried out with Triad LABCONCO dryer. UV-visible absorbance was measured using a Spark control 10 microplate reader from TECAN and an Agilent Technologies, Cary 300 UV-Vis spectrophotometer enabling Peltier temperature control. MARCHEREY-NAGEL GmbH & Co KG 60/40-63 mesh silica gel for Liquid Flash Chromatography and MARCHEREY-NAGEL GmbH & Co KG 60 F254 silica gel TLC plates with anisaldehyde stain for detection were used. Reaction pH for preparative synthesis was maintained using a TitroLine®7000 autotitrator. NMR spectra were recorded in D2O or DMSO on a
400 MHz Bruker Avance III HD spectrometer. Chemical shifts are referenced to the residual solvent peak. The following multiplicity abbreviations are used: (s) singlet, (d) doublet, (t) triplet, (m) multiplet.
Expression of transketolases (TKecoand TKgst) and d-amino acid oxidase (DAAORg). Escherichia coli strain BL21(DE3) pLysS was used for wild-type TKgst,[19] TKgst L382F,[20c] TKgst
R521Y/H462N[22]and TK eco
[28]overexpression with the plasmid
pET47b and for DAAORg overexpression[11b] with the plasmid
pT7. These strains were stored at 80°C in glycerol (10%). One colony of each recombinant E. coli strain, grown on selective LB agar plates, was transferred into 30 mL liquid LB medium containing ampicillin (100 μg. mL 1) or
kanamy-cin (30 μg. mL 1) and grown at 37°C, 130 rpm for 12 h.
20 mL of the pre-culture was used to inoculate 1 liter of culture medium containing ampicillin (100 μg. mL 1) or
kanamycin (30 μg. mL 1) and grown at 37°C, 200 rpm.
Isopropyl β-D-1-thiogalactopyranoside (IPTG) at 0.5 mM was added when the OD600nm range was 0.7–0.8. The cells
were then grown overnight at 30°C, under stirring at 130 rpm, and harvested by centrifugation at 8000 rpm at 4°C for 15 min. Bacterial pellets were washed twice with phosphate buffer (NaH2PO4· 2H2O, 50 mM), NaCl (300 mM),
pH 8.0) and harvested ( � 5 g/L for TKs and � 2 g/L for DAAORg).
Purification of transketolases (TKeco and TKgst) and D-amino acid oxidase (DAAORg). Harvested recombinant cells from 1 liter of culture were resuspended in 35 mL of phosphate buffer (50 mM) containing NaCl (300 mM) at pH 8.0 for TKs and 14 mL of phosphate buffer (50 mM) containing 2-mercap-toethanol (5 mM) and FAD (0.1 mM) at pH 7.2 for DAAORg.
The cells were disrupted by sonication on ice for 30 min and the insoluble pellets were discarded after centrifugation at 8000 rpm for 15 min at 4°C. Crude extracts were applied to a Ni NTA column equilibrated with phosphate buffer for TKs and with phosphate buffer (50 mM) containing NaCl (1 M), imidazole (20 mM) and glycerol 5% at pH 7.2 for DAAORg.
After washing each column with the same buffer respectively for TKs and DAAORg, the His6-tagged TKs or DAAORg were
finally eluted with phosphate buffer (50 mM) containing NaCl (300 mM) and imidazole (500 mM) at pH 8.0 for TKs and phosphate buffer (50 mM) containing glycerol (5%) imidazole (50 mM) at pH 7.2 for DAAORg. The fractions containing the
eluted proteins were collected and dialyzed against
triethanol-amine buffer (2 mM, pH 7.5) and then against water (pH 7.5) through dialysis tubing (cut-off 14 kDa) at 4°C for TKs and twice against water (pH 7.5) though dialysis tubing at 4°C for DAAORg. Then, these protein solutions were lyophilized.
Protein concentration was determined by the Bradford method and bovine serum albumin (BSA) was used as the standard. The specific activities of lyophilized TKecoTKgst, and DAAORgwere
21 U, 2 U and 37 U per mg of total protein respectively at 25°
C. The purity and molecular mass of these samples were analyzed by SDS-PAGE[29]using Precision Plus Protein™ All
Blue Standards#161-0373 (10–250 kDa, BioRad) as standard. The proteins were revealed with Coomassie Blue G-250. Determination of wild-type TKs (TKeco and TKgst) or variant TKgstactivity.
[5]
One unit of TKgstactivity was defined
as the amount of enzyme that catalyzes the formation of 1 μmol of ketose product per minute at 25°C in glycylglycine buffer (100 mM, pH 7.5). TKgst enzymatic assay was
per-formed in the presence of l-erythrulose and d-ribose-5-phosphate (dR5P) leading to d-sedoheptulose-7-d-ribose-5-phosphate (dS7P) and glycolaldehyde. The glycolaldehyde formed was reduced by yeast alcohol dehydrogenase (ADH) to ethylene glycol in the presence of nicotine adenine dinucleotide reduced form (NADH). l-erythrulose (100 mM), d-R5P (9.1 mM), ThDP (0.1 mM), MgCl2 (0.5 mM), NADH (0.2 mg. mL 1),
5 μL of ADH (25 U. mL 1) and the TK
gst suspension (10 μL)
were added to disposable plastic cuvettes and completed to 1 mL with glycylglycine buffer. The disappearance of NADH was followed by spectrophotometry at 340 nm (value of ɛNADH
at 340 nm is 6220 M. 1cm 1). All measurements were
per-formed in triplicate.
Determination of DAAORg activity. One unit of DAAORg
activity was defined as the amount of enzyme that catalyzes the formation of 1 μmol of hydroxypyruvic acid (HPA) product per minute at 25°C in Tris-HCl buffer (50 mM, pH 7.5). The assay was performed in the presence of d-serine leading to HPA, ammonia and hydrogen peroxide. Catalase, which is essential for the protection of the enzyme, allows dismutation of H2O2
into H2O and O2. The HPA formed was reduced by rabbit
muscle L-lactate dehydrogenase (LDH) to L-lactate in the presence of nicotine adenine dinucleotide reduced form (NADH). d-serine (50 mM), catalase from bovine liver (528– 1320 U. mL 1), LDH (11 U. mL 1), NADH (0.2 mg. mL 1) and
the DAAORg suspension (50 μL) were added to disposable
plastic cuvettes and completed to 1 mL with Tris-HCl buffer. The disappearance of NADH was followed by spectrophotom-etry at 340 nm. All measurements were performed in triplicate. Determination of HPA concentration using LDH assay. The HPA concentration was determined by an enzymatic assay using LDH. A sample of HPA (20 μL) was introduced in a cuvette containing 25 μL of NADH (0.2 mg. mL 1), 20 μL LDH
(187 U. mL 1) and 935 μL of TEA buffer (100 mM) at pH 7.5.
The disappearance of NADH was followed by spectrophotom-etry at 340 nm and difference between initial and final absorbance was used to calculate the HPA concentration using the Beer-Lambert law. All measurements were performed in triplicate.
Determination of d-serine concentration using DAAORg/ LDH assay. The d-serine concentration was determined by a coupled enzymatic assay using a combination of DAAORg,
catalase and LDH. In the spectrophotometric continuous assay d-serine is converted into HPA by DAAORgin the presence of
O2 releasing hydrogen peroxide which is then dismutated in
water and O2 by catalase. HPA produced in situ is reduced by
LDH in the presence of NADH. The assay cuvette (1 mL) contained
25 μL of NADH (10 mg. mL 1), 20 μL of LDH (187 U. mL 1),
50 μL of catalase (17000–44000 U. mL 1), 15 μL of DAAO Rg
(40 U. mL 1) in 855 μL of Tris buffer (50 mM) at pH 7.5. The
reaction was initiated with d-serine or an aliquot of the reaction mixture containing d-serine (20 μL). The disappearance of NADH was followed by spectrophotometry at 340 nm and difference between initial and final absorbance gave the d-serine concentration using the Beer-Lambert law. When assaying the d-serine concentration for DAAORg or DAAORg/
TKgst reaction monitoring, two aliquots (containing both
d-serine and HPA) were removed overtime from the mixture. The HPA concentration was determined using LDH assay from the first aliquot. The second aliquot was assayed with DAAORgand
catalase (DAAORg/LDH assay) in order to quantify the total
concentration of d-serine and HPA at once. Indeed d-serine concentration is not directly accessible since at first the enzyme DAAO converts serine to HPA, which is readily assayed by the enzyme LDH. Thus, the d-serine concentration was obtained by substracting HPA concentration (LDH assay) to the total concentration of d-serine and HPA (DAAORg/LDH assay). All
measurements were performed in triplicate.
In situ1
H NMR measurements. Progress of preparative scale enzymatic synthesis were monitored by using quantitative
in situ 1H NMR relative to
3-trimethylsilyl-2,2,3,3-tetradeuter-opropionate (TSP-d4) as internal standard. Aliquots of the
reaction mixture were removed overtime (450 μL) and mixed with 50 μL of TSP-d4(50 mM in D2O).
Procedure of HPA synthesis catalyzed by DAAORg. d-serine (50 mM) was dissolved in H2O and the pH was adjusted to 7
with 0.1 M NaOH. To this stirred solution were added catalase (1 mg, i. e. 1998 U–4995 U/2 h), and DAAORg (2 mg, 86 U)
giving a final volume of 20 mL. Oxygen was bubbled into the reactor (10 mL. min 1 at atmospheric pressure). The reaction
mixture was stirred (100 rpm) at 25°C, and the pH was automatically maintained at 7 by adding HCl (0.1 M) using a pH stat. Oxygen was bubbled into the reactor (10 mL. min 1at
atmospheric pressure). The reaction was monitored by measur-ing HPA appearance usmeasur-ing LDH assay and d-serine consump-tion using dAAORg/LDH assay. The completion of the reaction
was evidenced by in situ1H NMR.
Procedure of simultaneous or sequential cascade synthesis catalyzed by TK and dAAORg. For simultaneous cascade synthesis, ThDP (0.1 mM), MgCl2· 6H2O (1 mM), d-serine
(50 mM) and acceptor aldehyde (50 mM) were dissolved in H2O and the pH was adjusted to 7 with 0.1 M NaOH. To this
stirred solution were added catalase (1 mg/2 h, 1998–4995 U/ 2 h), TKgst (6 mg, 14 U) and dAAORg (2 mg, 86 U) giving a
final volume of 20 mL. Oxygen was bubbled into the reactor (10 mL. min 1 at atmospheric pressure). The reaction mixture
was stirred (100 rpm) at 25°C and the pH was automatically maintained at 7 by adding 0.1 M HCl using a pH stat.
For sequential cascade synthesis, d-serine (50 mM), DAAORg
(2 mg, 86 U) and catalase (1 mg/2 h, 1998–4995 U/2 h) were dissolved in 20 mL H2O and the pH of the solution was adjusted
to 7 with 0.1 M NaOH. The reaction proceeded at 25°C under stirring (100 rpm) until d-serine was totally converted. After-wards, TKgst (6 mg, 14 U) and its cofactors ThDP (0.1 mM),
MgCl2· 6H2O (1 mM) were added to this solution and the
reaction mixture was heated at 60°C under stirring (100 rpm), while the pH was automatically maintained at 7 by adding 0.1 M HCl, using a pH stat. Oxygen was bubbled into the reactor (10 mL. min 1at atmospheric pressure).
Sequential and simultaneous cascades were monitored by measuring d-serine and aldehydes consumption by in situ 1H
NMR and TLC. After total conversion of d-serine and/or aldehydes (8–32 hours), silica was added to the supernatant and the suspension was concentrated to dryness under reduced pressure before dry loading onto a flash silica column. After silica gel chromatography using eluent CH2Cl2/CH3OH (9:1–
8:2) for products 11–14, ethyl acetate/CH3OH (9:1) for product
10 and cyclohexane/ethyl acetate (1:1) for products 8–9, products 8–14 were isolated.
(3S)-1,3-dihydroxypentan-2-one 8. 8 was isolated as colorless
oil (59 mg, 50% yield with TKgstvariant L382F). TLC: Rf 0.38
(cyclohexane/ethyl acetate, 1/1 v:v). NMR data for 8 were identical to those previously described.[20c] 1H NMR (400 MHz,
D2O): 4,57 (d, 1H, J = 19,3 Hz, H1a); 4,48 (d, 1H, J = 19,3 Hz, H1b); 4,33 (dd, 1H, J = 7,7 Hz, J = 4,4 Hz, H3); 1,82 (dqd, 1H, J = 15,0 Hz, J = 7,5 Hz, J = 4,4 Hz, H4a); 1,66 (dq, 1H, J = 14,5 Hz, J = 7,4 Hz, H4b); 0,93 (t, 3H, J = 7,5 Hz, H5). 13C NMR (101 MHz, D2O): 214,2 (C2), 75,80 (C3), 65,04 (C1), 26,25 (C4), 8,47 (C5). m/z HRMS found [M + HCOO] 163.0612, C6H11O5 requires 163.0601 (obtained with TKgst
variant L382F).
(3S)-1, 3-dihydroxyhexan-2-one 9. 9 was isolated as colorless
oil (42 mg, 36% yield with TKgstvariant L382F). TLC: Rf 0.36
(cyclohexane/ethyl acetate, 1/1 v:v). NMR data for 9 obtained with either TK were identical to those previously described.[20c] 1H NMR (400 MHz, D 2O): 4,56 (d, 1H, J = 19,3 Hz, H1a); 4,47 (d, 1H, J = 19,3 Hz, H1b); 4,36 (dd, 1H, J = 8,3 Hz, J = 4,1 Hz, H3); 1,80–1,64 (m, 1H, H4a); 1,64–1,51 (m, 1H, H4b); 1,48– 1,30 (m, 2H, H5a/b); 0,90 (t, 3H, J = 7,3 Hz, H6). 13C NMR (101 MHz, DMSO): 213,54 (C2), 74,32(C3), 65,13 (C1), 35,45 (C4), 17,92 (C5), 13,75 (C6). m/z HRMS found [M + HCOO] 177.0768, C7H13O5 requires 177.0760 (obtained with TKgst
variant L382F).
(3S)-1,3,4-trihydroxy-butan-2-one 10. 10 was isolated as colorless oil (81 mg,: 67% yield with wt-TKeco). TLC: Rf 0.37
(ethyl acetate/CH3OH; 9:1). NMR data for 10 obtained with
either TK were identical to those previously described.[20b] 1H
NMR (400 MHz, D2O): δ (ppm) 4.55 (dd, J = 19.4 Hz, 2H,
H1a/b); 4.44 (t, J = 4.1 Hz, 1H, H3); 3.89 (dd, J = 4.1 Hz, J = 2.0 Hz, 2H, H4a/b).13C NMR (101 MHz, D
2O): δ (ppm) 212.24
(C2); 75.8 (C3); 65.77 (C1); 62.81 (C4). m/z HRMS found [M + HCOO] 165.0405, C5H9O6requires 165.0396 (obtained with
wt-TKeco).
(3S, 4R)-1,3,4,5-tetrahydroxy-pentan-2-one 11. 11 was iso-lated as colorless oil (92.8 mg, 62% yield with wt-TKeco). TLC:
either TK were identical to those previously described[30](ratio: β-d-xylulo-2,5-furanose/Acyclic form/α-d-xylulo-2,5-furanose: 62/21/17; lit. Ratio: 62/20/18).[31] 1H NMR (400 MHz, D 2O): δ (ppm) β-d-xylulo-2,5-furanose 4.43–4.37 (m, 1H, H4); 4.21 (dd, J = 9.5 Hz, J = 6.5 Hz, 1H, H5a); 4.07 (d, J = 5.6 Hz, 1H, H3); 3.66 (d, 1H, H5b); 3.65–3.56 (m, 2H, H1a/b); acyclic form 4.71–4.51 (m, 2H, H1a/b); 4.48 (d, J = 2.2 Hz, 1H, H3); 4.10 (d, J = 2.8 Hz, 1H, H4); 3.71 (t, 1H, H5b); 3.68 (m, 1H, H5b); α-d-xylulo-2,5-furanose 4.33–4.28 (m, 1H, H4); 4.26 (m, 1H, H5a); 4.10 (q, 1H, H3); 3.89–3.95 (dd, 1H, J = 9.4 Hz, J = 4.1 Hz, H5b); 3.74 (m, 1H, H1a); 3.68 (m, 1H, H1b).13C NMR (101 MHz, D2O): δ (ppm) β-d-xylulo-2,5-furanose 104.39 (C2); 77.69 (C3); 76.34 (C4); 71.29 (C5); 64.48 (C1), acyclic form 214.4 (C2); 76.76 (C3); 73.42 (C4); 67.49 (C1); 63.33 (C5), α-d-xylulo-2,5-furanose 107.18 (C2); 81.96 (C3); 77.30 (C4); 73.37 (C5); 63.88 (C1). m/z HRMS found [M + Cl] 185.0222, C5H10O5Cl requires 185.0217 (obtained with wt-TKgst);
C5H10O5Cl requires (obtained with wt-TKeco).
(3S, 4R, 5S)-1,3,4,5,6-pentahydroxy-hexan-2-one 12. 12 was isolated as colorless oil (72 mg, 40% yield with wt-TKgst). TLC:
Rf 0.40 (CH2Cl2/CH3OH; 8:2). NMR data for 12 obtained with
either TK were identical to those previously described (ratio: 96/4; lit. Ratio: 98/2).[32] 1H NMR (400 MHz, D 2O): δ (ppm) α-l-sorbo-2,6-pyranose 3.74 (d, J = 4.8 Hz, 1H, H6a); 3.71 (d, J = 11.8 Hz, 1H, H1a) 3.69 (d, J = 1.9 Hz, 1H, H4); 3.65 (d, J = 10.1 Hz, 1H, H6b); 3.64 (m, 1H, H5); 3.52 (d, J = 18 Hz, 1H, H1b); 3.51 (d, J = 2.9 Hz, 1H, H3); α-l-sorbo-2,5-furanose 4.43 (t, J = 6.3 Hz, 1H, H4); 4.31 (tt, J = 6.6 Hz, J = 3.3 Hz, 1H, H5); 4.12 (d, J = 6.1 Hz, 1H, H3); 3.81 (d, J = 3.7 Hz, 1H, H6a); 3.78 (d, J = 4.0 Hz, 1H, H6b); 3.62 (d, J = 3.5 Hz, 1H, H1a); 3.59 (d, 1H, J = 5.3 Hz, H1b). 13C NMR (101 MHz, D2O): δ (ppm) α-l-sorbo-2,6-pyranose 98.48 (C2); 74.59 (C4); 71.12 (C3); 70.16 (C5); 64.23 (C1); 62.52 (C6), α-l-sorbo-2,5-furanose 102.5 (C2); 78.45 (C5); 76.70 (C3); 76.05 (C4); 63.82 (C1); 61.41 (C6). m/z HRMS found [M + Cl] 215.0328, C6H12O6Cl requires 215.0322 (obtained with wt-TKgst).
(3S, 4R, 5R)-1,3 4 5,6-pentahydroxy-hexan-2-one 13. 13 was isolated as colorless oil (118 mg, 65% yield with wt-TKeco).
TLC: Rf 0.19 (CH2Cl2/CH3OH; 8:2). NMR data for 13 obtained
with either TK were identical to those previously described (ratio: 73/22/5; lit. Ratio: 75/21/4).[32,33] 1H NMR (400 MHz,
D2O): δ (ppm) β-d-fructo-2,6-pyranose 4.09–4.04 (m, 1H, H6a); 4.04–4.02 (m, 1H, H5); 3.93 (dd, J = 10.0 Hz, J = 3.5 Hz, 1H, H4); 3.83 (d, J = 10.0 Hz, 1H, H3); 3.76–3.72 (m, 2H, H6b, H1a); 3.60 (d, J = 11.7 Hz, 1H, H1b), β-d-fructo-2,5-furanose 4.17–4.13 (m, 2H, H3, H4); 3.84 (m, 1H, H5); 3.83 (m, 1H, H6a); 3.69 (m, 1H, H6b); 3.63 (d, J = 12.1 Hz, 1H, H1a); 3.58 (d, J = 12.1 Hz, 1H, H1b), α-d-fructo-2,5-furanose 4.17–4.13 (m, 2H, H3, H5); 4.05–4.01 (m, 1H, H4); 3.86–3.82 (m, 1H, H6a); 3.74–3.70 (m, 1H, H6b); 3.72–3.68 (m, 2H,H1a/b). 13C NMR (101 MHz, D2O): δ (ppm) β-d-fructo-2,6-pyranose 99.12 (C2); 70.72 (C4); 70.25 (C5); 68.61 (C3); 64.94 (C1); 64.42 (C6), β-d-fructo-2,5-furanose 102.54 (C2); 81.72 (C5); 76.44 (C3); 75.49 (C4); 63.74 (C1); 63.43 (C6), α-d-fructo-2,5-furanose 105.49 (C2); 83.0 (C3); 82.37 (C5); 77.09 (C4); 63.96 (C1); 62.15 (C6). m/z HRMS found [M + Cl] 215.0328, C6H12O6Cl requires 215.0321 (obtained with wt-TKeco).
(3S,4R,5R,6R)-1,3,4,5,6,7-hexahydroxy-heptan-2-one 14. 14 was isolated as colorless oil (157 mg, yield: 75% with TKgst
variant R521Y/H462N). TLC: Rf 0.22 (CH2Cl2: CH3OH, 80:20
v:v). NMR data for 14 obtained with either TK were identical to those previously described[22] (ratio:
β-D-altro-heptulo-2,5- furanose/α-D-altro-heptulo-2,6-pyranose/α-D-altro-heptulo-2,5-furanose: 67/19/14; lit. ratio: β-D-altro-heptulo-2,5-fura-nose/α-D-altro-heptulo-2,6-pyranose/α-D-altro-heptulo-2,5 fur-anose: 64/20/16.[20d,22] 1H NMR (400 MHz, D 2O): δ (ppm) = (β-D-altro-heptulo-2,5-furanose): 4.30-4.23 (m, 1H, H4), 4.05 (d, J = 7.9 Hz, 1H, H3), 3.80 (d, J = 3.3 Hz, 1H, H6), 3.73–3.72 (m, 1H, H5), 3.63–3.57 (m, 2H, H7a/b), 3.53 (d, J = 12.1 Hz, 1H, H1a), 3.54 (d, J = 12.1 Hz; H1b); (α-D-altro-heptulo-2,6-pyranose): 4.04 (d, J = 7.4 Hz, 1H, H4), 3.98 (m, 1H, H6), 3.91 (d, J = 3.7 Hz, 1H, H3), 3.83 (dd, J = 4.1 Hz, J = 1.9 Hz, 2H, H7a/b), 3.81 (m, 1H, H5), 3.65 (d, J = 11.7 Hz, 1H, H1a), 3.46 (d, J = 11.7 Hz, 1H, H1b); (α-d-altro-heptulo-2,5-furanose): 4.14 (dd, J = 6.1 Hz, J = 4.5 Hz, 1H, H4), 3.94 (d, J = 2.6 Hz, 1H, H3), 3.88–3.84 (m, 2H, H5, H6a), 3.79–3.75 (m, 1H, H6b), 3.69 (d, J = 9.3 Hz, 1H, H1a), 3.51 (d, J = 11.7 Hz, 1H, H1b). 13C NMR (101 MHz, D 2O): δ (ppm) = (β-d-altro-heptulo-2,5-furanose): 101.6 (C-2), 80.1 (C-5), 75.6 (C-3), 75,3 (C-4), 72.6 (C-6), 62.4 (C-1), 62.2 (C-7); (α-d-altro-heptulo-2,6-pyranose): 97.4 (C-2), 70.8 (C-4), 68.7 (C-6), 68.2 (C-3), 64.0 (C-1), 63.4 5), 61.2 7); (α-d-altro-heptulo-2,5-furanose): 104.6 (C-2), 81.7 (C-3), 81.6 (C-5), 76.4 (C-4), 71.5 (C-6), 62.7 (C-1), 62.3 (C-7). m/z HRMS found [M + HCOO] 245.0431, C8H15O9requires 245.0431 (obtained with TKgstvariant R521Y/
H462 N).
Acknowledgements
This work was funded by the Fonds Régional Innovation Laboratoire (grant DOS00494484/00, to L.H.), by the Agence Nationale de la Recherche (grant ANR-09-BLAN-0424-CSD3 to L.H.) and by ERA CoBioTech TRALAMINOL – ID: 64 (grant to W.D.F and L.H.).
References
[1] P. Clapes, W.-D. Fessner, G. Sprenger, A. K. Samland,
Curr. Opin. Chem. Biol. 2010, 14, 154–167.
[2] N. J. Turner, Curr. Opin. Biotechnol. 2000, 11, 527–531. [3] F. Dickens, D. H. Williamson, Biochem. J. 1958, 68, 74–
81.
[4] L. Hecquet, J. Bolte, C. Demuynck, Tetrahedron 1996,
52, 8223–823.
[5] M. Lorillière, M. De Sousa, F. Bruna, E. Heuson, F. Gefflaut, V. de Berardinis, T. Saravanan, D. Yi, W.-D. Fessner, F. Charmantray, L. Hecquet, Green Chem. 2017,
19, 425–435.
[6] a) F. Subrizi, M. Cárdenas-Fernández, G. J. Lye, J. M. Ward, P. A. Dalby, T. D. Sheppard, H. C. Hailes, Green
Chem. 2016, 18, 3158–3165; b) M. Bawn, F. Subrizi,
G. J. Lye, T. D. Sheppard, H. C. Hailes, J. M. Ward,
Enzyme Microb. Technol. 2018, 116, 16–22.
[7] a) L. Pollegioni, G. Molla, S. Sacchi, E. Rosini, R. Verga, M. S. Pilone, Appl. Microbiol. Biotechnol. 2008,
78, 1–16; b) L. Pollegioni, P. Motta, G. Molla, Appl. Microbiol. Biotechnol. 2013, 97, 9323–9341.
[8] L Pollegioni, L Piubelli, S Sacchi, M. S. Pilone, G. Molla, Cell. Mol. Life Sci. 2007, 64, 1373–1394.
[9] a) B. Curti, S. Ronchi, S. M. Pilone, D- and L-amino
acid oxidases, Chemistry and Biochemistry of
Flavoen-zymes, 1991, 3, CRC Press, Boca Raton, pp. 69–96; b) M. S. Pilone, L. Pollegioni, Biocatal. Biotransform.
2002, 20, 145–159.
[10] C. Demuynck, J. Bolte, L. Hecquet, H. Samaki,
Carbohydr. Res. 1990, 206, 79–86.
[11] a) L. Pollegioni, A. Falbo, M. S. Pilone, Biochim.
Biophys. Acta 1992, 120, 1–16; b) S. Fantinato, L.
Pollegioni, S. M. Pilone, Enzyme Microb. Technol. 2001,
29, 407–412.
[12] L. Pollegioni, K. Diederichs, G. Molla, S. Umhau, W. Welte, S. Ghisla, M. S. Pilone, J. Mol. Biol. 2002, 324, 535–546.
[13] L Pollegioni, S Iametti, D Fessas, L Caldinelli, L Piubelli, A Barbiroli, M. S. Pilone, F. Bonomi, Protein
Sci. 2003, 12, 1018–1029.
[14] a) T. A. Savidge, Enzymatic conversion used in the
production of penicillins and cephalosporins. In: E. J.
Vandamme (ed.), Biotechnology of industrial antibiotics. Dekker, New York. 1984, 171–224; b) M. S. Pilone, S. Butò, L. Pollegioni, Biotechnol. Lett. 1995, 17, 199–204; c) L. Pollegioni, L. Caldinelli, G. Molla, S. Sacchi, M. S. Pilone, Biotechnol. Prog. 2004, 20, 467–473.
[15] a) T. Tosa, R. Sano, I. Chibata, Agric. Biol. Chem. 1974,
38, 1529–1534; b) A. Caligiuri, P. D’Arrigo, E. Rosini,
D. Tessaro, G. Molla, S. Servi, L. Pollegioni, Adv. Synth.
Catal. 2006, 348, 2183–2190.
[16] a) S. Tu, D. B. McCormick, Sep. Sci. 1972, 7, 403–408; b) L. Pollegioni, G. Molla, Trends Biotechnol. 2011, 29, 276–283.
[17] H. Ikeda, Y. Yonetani, S. I. Hashimoto, Process for producing D-serine, US 7,186,532 B2, Kyowa Hakko Kogyo Co Ltd.
[18] a) C. Wikner, L. Meshalkina, U. Nilsson, M. Nikkola, Y. Lindqvist, M. Sundstrom, G. Schneider, J. Biol. Chem.
1994, 269, 32144–32150; b) F. Charmantray, V. Helaine,
B. Legeret, L. Hecquet, J. Mol. Catal. B: Enzym. 2009,
57, 6–9; c) A. Ranoux, S. K. Karmee J. Jin, A. Bhaduri,
A. Caiazzo, I. W. Arendsand U. Hanefeld,
ChemBio-Chem 2012, 13, 1921–1931; d) G. A. Sprenger, U.
Schörken, G. Sprenger, H. Sahm, Eur. J. Biochem. 1995,
230, 525–532; e) G. R. Hobbs, R. K. Mitra, R. P.
Chauhan, J. M. Woodley, M. D. Lilly, J. Biotechnol.
1996, 45, 173–179; f) T. U. Zimmermann, G. A. Sprenger, W.-D. Fessner, Tetrahedron: Asymmetry 1999,
10, 1643–1646; g) M. E. B. Smith, E. G. Hibbert, A. B.
Jones, P. A. Dalby, H. C. Hailes, Adv. Synth. Catal. 2008,
350, 2631–2638; h) A. Cázares, J. L. Galman, L. G.
Crago, M. E. Smith J. Straford, L. Ríos-Solís, G. L. Lye, P. A. Dalby, H. C. Hailes, Org. Biomol. Chem. 2010, 8, 1301–1130.
[19] J. Abdoul Zabar, I. Sorel, V. Hélaine, F. Charmantray, T. Devamani, D. Yi V. de Berardinis, D. Louis, P. Marlière, W.-D. Fessner, L. Hecquet, Adv. Synth. Catal. 2013, 355, 116–128.
[20] a) J. Abdoul Zabar, M. Lorillière, D. Yi, L. Nauton, F. Charmantray, V. Hélaine, W.-D. Fessner, L. Hecquet,
Adv. Synth. Catal. 2015, 357, 1715–1720; b) K. Benaissi,
V. Hélaine, V. Prévot, C. Forano, L. Hecquet, Adv. Synth.
Catal. 2011, 353, 1497–1509; c) D. Yi, T. Saravanan, T.
Devamani, F. Charmantray, L. Hecquet, W.-D. Fessner,
Chem. Commun. 2015, 51, 480–483; d) T. Saravanan,
M. L. Reif, D. Yi, M. Lorillière, F. Charmantray, L. Hecquet, W.-D. Fessner, Green Chem. 2017, 19, 481– 489; e) T. Saravanan, S. Junker, M. Kickstein, J. Hegen, S. Hein, M. K. Link, S. Witt, M. Lorillière, L. Hecquet, W.-D. Fessner, Angew. Chem. Int. Ed. 2017, 56, 5358– 5362.
[21] a) J. A. Moreno, J. Catalán, M. A. Galán, Biotechnol.
Lett. 1994, 16, 8, 843–848; b) J. A. Moreno, F. J.
Montes, J. Catalán, M. A. Galán, Enzyme Microb.
Technol. 1996, 18, 379–382.
[22] M. Lorillière, R. Dumoulin, M. L’enfant, L. Nauton, A. Rambourdin, V. Thery, W.-D. Fessner, F. Charmantray, L. Hecquet, 2018, submitted.
[23] K. Jung, M. Seifert, T. Herrling, J. Fuchs, Spectrochim.
Acta A Mol Biomol. Spectrosc. 2008, 69, 1423–1428.
[24] K. Karhumaa, R. G. Sanchez, B. Hahn-Hägerdal, M.-F. Gorwa-Grauslund, Microb. Cell. Fact. 2007, 6:5. [25] H. D. Belitz, W. Grosch, P. Schieberle, in Food
Chemistry 2009, 248–270, Heidelberg: Springer-Verlag.
[26] L. L. Gao, G. C. Du, J. W. Zhou, J. Chen, J. Liu,
Biotechnol. Prog. 2013, 29, 1398–1404.
[27] a) M. M. Wamelink, E. A. Struys, E. E. W. Jansen, H. Blom, T. Vilboux, W. A. Gahl, M. Komhoff, C. Jakobs, E. N. Elevtchenko, Mol. Genet. Metab. 2011, 3, 339– 342; b) M. M. C. Wamelink, E. A. Struys, V. Valayanno-poulos, M. Gonzales, J.-M. Saudubray, C. Jakobs,
Prenatal Diagn. 2008, 5, 460–462.
[28] Y. Dong, T. Devamani, J. Abdoul-Zabar, F. Charmantray, V. Helaine, L. Hecquet, W.-D. Fessner, ChemBioChem
2012, 13, 2290–2300.
[29] U. K. Laemmli, Nature 1970, 227, 680–685.
[30] T. Vuorinen, A. S. Serianni, Carbohydr. Res. 1991 209, 13–31.
[31] J. Wu, A. S. Serianni, T. Vuorinen, Carbohydr. Res.
1990, 206, 1–12.
[32] T. Barclay, M. Ginic-Markovic, M. R. Johnston, P. Cooper, N. Petrovsky, Carbohydr. Res. 2012, 347, 136– 141.