Gene constraint and genotype–phenotype correlations in neurodevelopmental disorders
Texte intégral
Figure

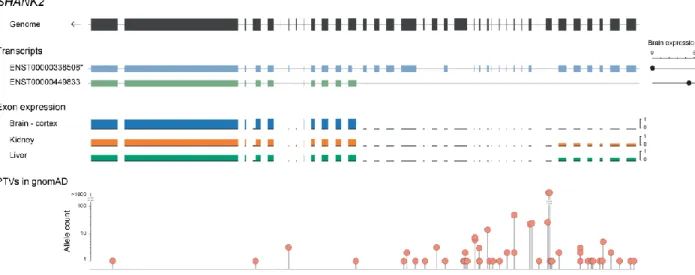
Documents relatifs
Figure 20 shows how our loop detection algorithm and the improved refinement technique are integrated into the traditional abstraction refinement cycle (see Fig. The analyses
Sept patients ont été traités par une combinaison de la technique d’Ilisarov et de péroné libre vascularisé, la consolidation a été atteinte dans tous les cas,
that concern by demonstrating that the robot is capable of machining a full element with a force parameters that are below the safety limit (25 N) and above the
A new k-nearest neighbor based entropy estimator, that is efficient in high dimensions and in the presence of large non-uniformity, is proposed. The proposed idea relies on
Elles lancent au ciel comme en cadeau précieux, Quelques incantations, ou des hymnes joyeux, Dressant à pleine voix les remparts de la foi Pour enfermer chacun dans des textes
Thus, a striking finding of our study is that the highest number of similarly expressed genes was found in the wild type and mutant under drought conditions: only 135 genes with
Ruptured pseudo-aneurysm of a femoral artery in a drug abuser revealed by post-mortem CT angiography.. Katarzyna Michaud & Silke
Tous les deux savaient que leur papa aimait le chocolat, c’est ainsi qu’ils lui ont donné la moitié de leurs chocolats.. Ils ont également donné à leur mama deux chocolats;
