Learning a Markov Logic network for supervised gene regulatory network inference
Texte intégral
Figure

Documents relatifs
The main objective of this research is to improve estimates of biomass by combining ALOS-2 SAR, Landsat 8, and field plots data in a tropical forest with high biomass values.. First,
Type C vessels - The database contains only two damage events for Type C vessels, both caused by the contact with MY ice.. Damage to Type D vessels was caused in ice regimes with
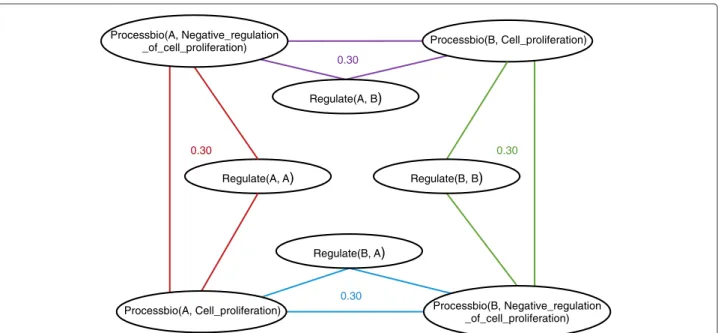
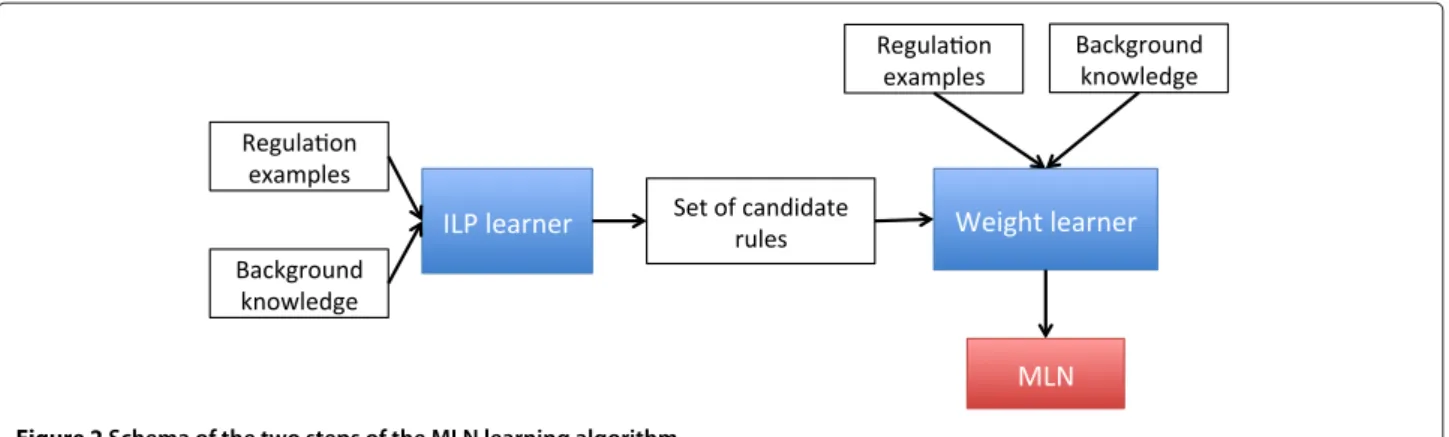
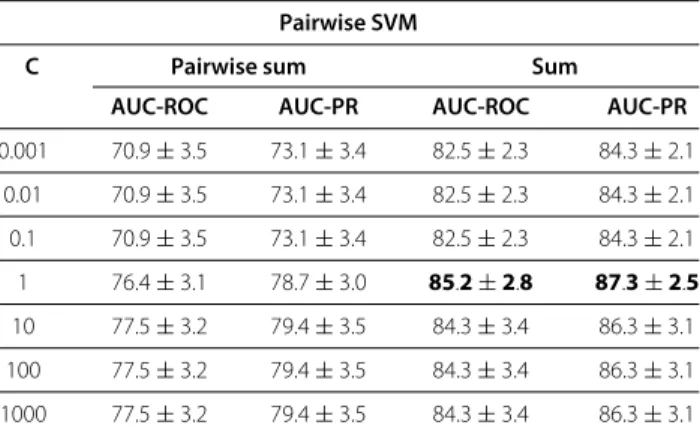
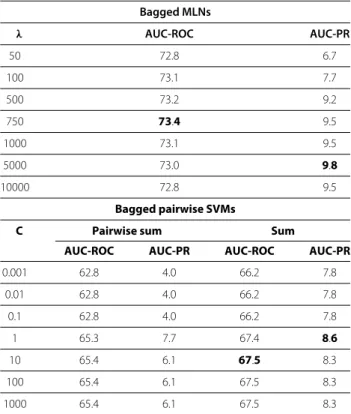
Despite the large number of available Gene Regulatory Network inference methods, the problem remains challenging: the underdetermination in the space of possible solutions
Durante i negoziati internazionali riguardanti il finan- ziamento dello sviluppo e l’agenda post-2015 sono emerse visioni assai diverse in merito alla responsa- bilità che i Paesi
• As we believe that floating-point on FPGA should exploit the flexibility of the target and therefore not be limited to IEEE single and double precision, the algorithm
/ La version de cette publication peut être l’une des suivantes : la version prépublication de l’auteur, la version acceptée du manuscrit ou la version de l’éditeur.. Access
L’objectif est ici d’apporter des éléments de réponse à la question du devenir des éléments traces dans un sol contaminé en comprenant mieux les interactions
The reactor performance was predicted to lie between the convection model where only the laminar flow profile is considered and the dispersion model which includes
