Modélisation cinétique de la réduction de minerais par le monoxyde de carbone utilisés dans le procédé de combustion en boucle chimique
179
0
0
Texte intégral
Figure
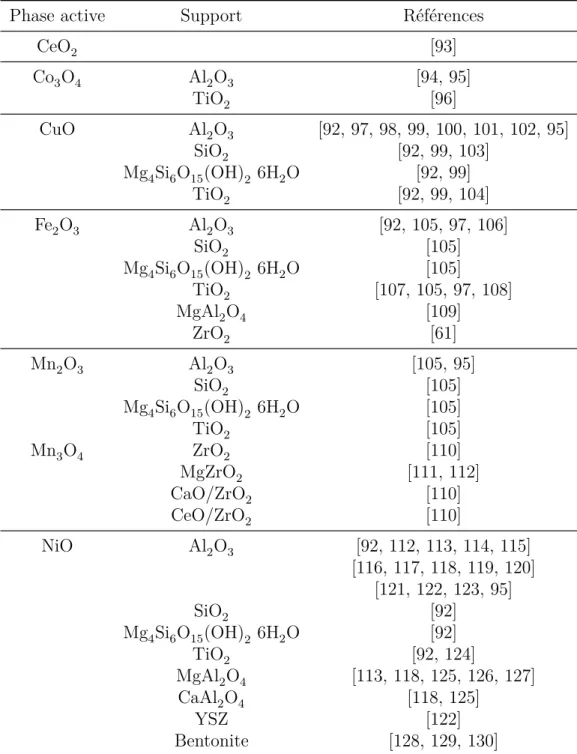


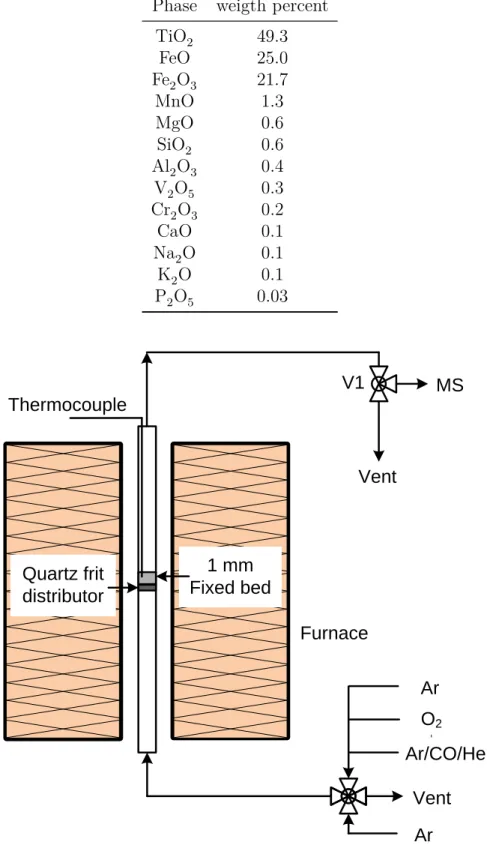
+7
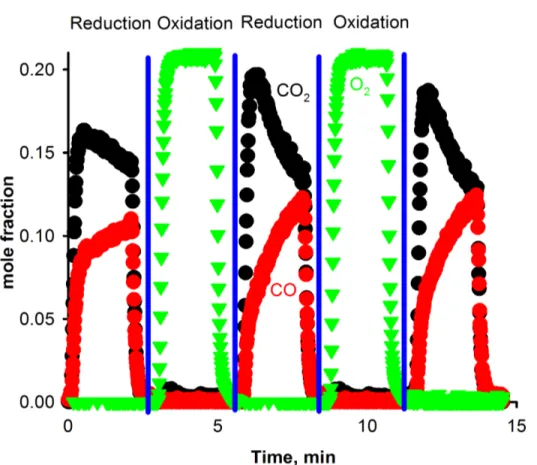
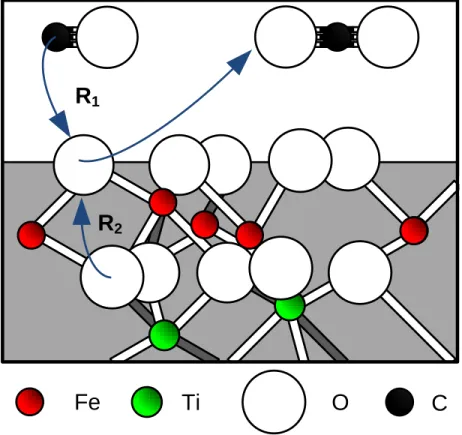
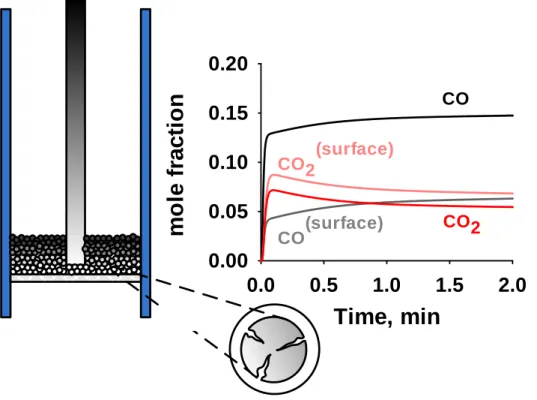
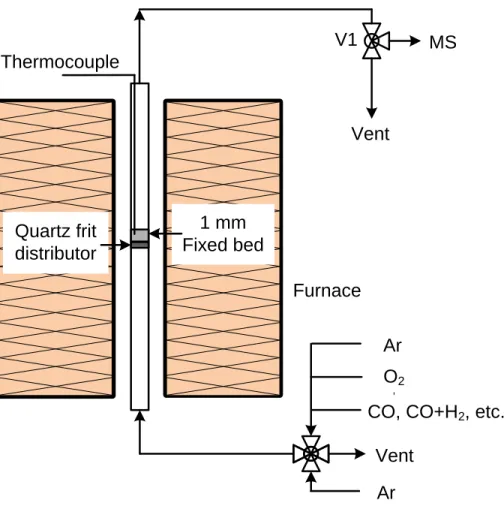
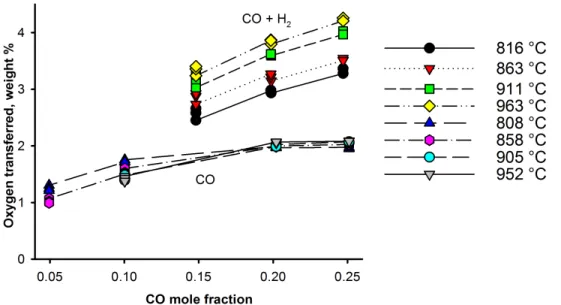
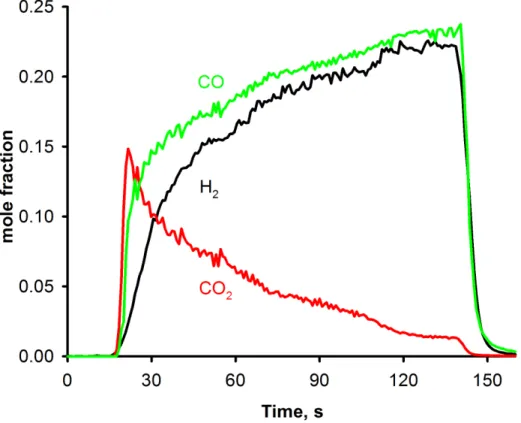
Documents relatifs