The Structure of a Cold-adapted Family 8 Xylanase at
1.3 Å Resolution
STRUCTURAL ADAPTATIONS TO COLD AND INVESTIGATION OF THE ACTIVE SITE*
Received for publication, July 10, 2002, and in revised form, December 2, 2002 Published, JBC Papers in Press, December 9, 2002, DOI 10.1074/jbc.M206862200 Filip Van Petegem‡§¶, Tony Collins§储, Marie-Alice Meuwis储, Charles Gerday储, Georges Feller储, and Jozef Van Beeumen‡**
From the ‡Laboratorium voor Eiwitbiochemie en Eiwitengineering, Ghent University, Ledeganckstraat 35, B-9000 Gent, Belgium and the储Laboratoire de Biochimie, University of Lie`ge, Institute of Chemistry, Sart-Tilman B-4000 Lie`ge, Belgium
Enzymes from psychrophilic organisms differ from their mesophilic counterparts in having a lower thermo-stability and a higher specific activity at low and mod-erate temperatures. The current consensus is that they have an increased flexibility, enhancing accommoda-tion and transformaaccommoda-tion of the substrates at low energy costs. Here we describe the structure of the xylanase from the Antarctic bacterium Pseudoalteromonas
halo-planktis at 1.3 Å resolution. Xylanases are usually
grouped into glycosyl hydrolase families 10 and 11, but this enzyme belongs to family 8. The fold differs from that of other known xylanases and can be described as an (␣/␣)6 barrel. Various parameters that may explain
the cold-adapted properties were examined and indi-cated that the protein has a reduced number of salt bridges and an increased exposure of hydrophobic res-idues. The crystal structures of a complex with xylo-biose and of mutant D144N were obtained at 1.2 and 1.5 Å resolution, respectively. Analysis of the various sub-strate binding sites shows that theⴙ3 and ⴚ3 subsites are rearranged as compared to those of a family 8 hom-olog, while the xylobiose complex suggests the existence of a ⴙ4 subsite. A decreased acidity of the substrate binding cleft and an increased flexibility of aromatic residues lining the subsites may enhance the rate at which substrate is bound.
It has been stated that life can be successful almost every-where on our planet and micro-organisms have indeed been isolated from some of the most extreme environments on earth, including the extremes of pH, pressure, and temperature. Of these extremophilic micro-organisms, the thermophiles and
hyperthermophiles have been extensively studied for several years, while cold-dwelling organisms have only recently at-tracted attention. Cold-adapted, or psychrophilic, organisms have developed adaptation mechanisms to overcome the low temperature challenge, the most important of which is the production of cold-active enzymes. Various adaptational strat-egies have been proposed for these enzymes. The current ac-cepted consensus is that they have an increased flexibility, thus enabling the conformational changes necessary for activity at low temperature (see Refs. 1 and 2 for recent reviews). They are characterized by an increased turnover number and physiolog-ical efficiency (kcat/Km) at low and moderate temperatures, as
well as by a reduced stability. To date, only four crystal struc-tures of cold-adapted enzymes originating from psychrophilic micro-organisms have been reported (3– 6) and no general rules as to how these proteins maintain sufficient flexibility have been deduced. The structures of psychrophilic enzymes can provide clues to how their stability at higher temperatures can be improved while their flexibility in a colder environment is maintained, a trait that makes them useful for many biotech-nological applications (7).
Here we describe the structure of a xylanase from the Ant-arctic bacterium Pseudoalteromonas haloplanktis. Xylanases (EC 3.2.1.8) catalyze the hydrolysis of the -1,4-D-glycosidic bonds in xylan, a major component of plant hemicellulose. A classification system for glycosyl hydrolases has been intro-duced by Henrissat (8) and, at present, at least 89 different families have been identified, with xylanases usually being grouped into families 10 and 11.1Family 10 enzymes have an
(␣/)8 barrel fold (10) while the family 11 members have a
-jelly roll fold (11). The psychrophilic xylanase, on the con-trary, can be classified into glycosyl hydrolase family 8 on the basis of its primary structure (EMBL nucleotide sequence data base AJ427921), sharing 20 –30% sequence identity with its members (12). The latter family mainly contains endoglu-canases (EC 3.2.1.4), but also chitosanases (EC 3.2.1.132) and lichenases (EC 3.2.1.73).
The psychrophilic xylanase hydrolyzes xylan to principally xylotriose (X3) and xylotetraose (X4) and, in contrast to other
currently identified xylanases, it operates with inversion of anomeric configuration. The activity of the enzyme on X5 is extremely low, while the catalytic efficiency on X6 is much
higher, indicating that the enzyme has a large substrate bind-ing cleft, containbind-ing at least six xylose bindbind-ing subsites (12). The structure of the xylanase is analyzed in terms of cold
* This work was supported in part by the European Union (Network Contract No. CT97-0131), the Region Wallone (Contract BIOVAL 981/ 3860), and the Fond National de la Recherche Scientifique (Contract 2.4515.00) (to T. C., M. A. M., C. G., and G. F.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in ac-cordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The atomic coordinates and structure factors (code 1H12, 1H13, and 1H14) have been deposited in the Protein Data Bank, Research Collabo-ratory for Structural Bioinformatics, Rutgers University, New Bruns-wick, NJ (http://www.rcsb.org/).
§ Both authors contributed equally to this work.
¶Research Fellow of the Fund for Scientific Research-Flanders. ** Recipient of Research Grant G006896 from the Fund for Scientific Research-Flanders and Project 12050198 from the Research Council of the University of Gent. To whom correspondence should be addressed. Tel.: 32-0-9-264-51-09; Fax: 32-0-9-264-53-38; E-mail: Jozef.Van-Beeumen@rug.ac.be.
1P. M. Coutinho and B. Henrissat, Carbohydrate-Active Enzymes
server at URL: afmb.cnrs-mrs.fr/⬃cazy/CAZY/index.html.
© 2003 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in U.S.A.
adaptation, and features that may be important for substrate binding and selectivity are described. In addition, the native enzyme is compared with an enzyme/xylobiose complex and a partly inactivated D144N mutant.
EXPERIMENTAL PROCEDURES
Wild Type Psychrophilic Xylanase—The psychrophilic xylanase (pXyl)2was expressed in Escherichia coli and purified as previously
described (12).
D144N Mutant—The psychrophilic xylanase gene, including its signal sequence, was introduced into the Ndel and Xhol sites of the cloning vector pSP 73® (Novagen). The mutation was introduced by PCR with Pwo polymerase, using the sense primer (5⬘-GCCCCGCT-CCCAATGGCGAAGAGTAC-3⬘) containing the Asp to Asn mutation D144N (underlined), and the antisense primer (5⬘-CTCATCCACTT-TATAAACAAAGCCGTTTTGA-3⬘). The PCR product was purified and circularized, used to transform to E. coli DINO RR1威, and double-strand sequenced using an ALF DNA sequencer (Amersham Bio-sciences). The mutated xylanase gene was excised with NdeI and XhoI, ligated into the pET22b(⫹) cloning vector (Novagen) and used to transform to E. coli BL21 (DE3) cells (Stratagene). Production and purification was carried out as described for the wild type recombi-nant xylanase (12). The kcat/Km ratio was determined from initial
rates at substrate concentrations of 2.0, 2.2, and 2.5 mg/ml soluble birchwood xylan, using the following relation: kcat/Km ⫽ v0/S0E0,
which is valid at S0⬍⬍ Km. The pH activity profile and an estimate of
the apparent Kmof the wild type and mutant xylanase were
deter-mined at 25 °C as already described (12).
Crystallization and Data Collection—Crystallization, data collection, and SAD phasing of the native enzyme and the selenomethionine-labeled mutant were performed as described previously (13). Crystals of the D144N mutant were obtained under the same conditions as the wild type enzyme. A complex of the native enzyme with xylobiose was obtained by soaking the native crystals in a solution containing 70% 2-methyl-2,4-pentanediol, 0.1 M sodium phosphate, pH 7.0, and 10 mg/ml xylobiose for 48 h. Data collection statistics for the complexed xylanase and the D144N mutant are shown in Table I. These data were collected at the protein crystallography beamline of the ELETTRA synchrotron (Trieste, Italy). Statistics for the native xylanase and the selenomethionine-labeled mutant used in SAD phasing have already been described (13).
Refinement and Structural Analysis—A FOM of 0.93 was obtained for the selenomethionine mutant after density modification, resulting in an interpretable electron density map for the native enzyme and the autotracing of 400 residues using ARP-wARP (14). The structure was further refined against the native 1.3 Å data using the graphics pro-gram TURBO-FRODO (15) and Refmac5 (16). B-factors were refined anisotropically in the latter refinement rounds. The stereochemistry was checked using PROCHECK (17), which showed that all the resi-dues are in allowed regions of the Ramachandran plot. The structures of the xylobiose complex and mutant D144N were refined using the coordinates of the native enzyme as a starting model. Refinement was as for the native enzyme except that the B-factors were refined isotro-pically for mutant D144N. Refinement statistics are shown in Table II. Side chains with missing electron density were not modeled. For the xylobiose complex, final electron density was good enough to place only one xylosyl residue. The hydroxyl group on the anomeric carbon of this residue was refined with a double conformation corresponding to 65% -anomer and 35% ␣-anomer, typical values for xylose in solution (18). Hydrogen bonds and ligand interactions were calculated with the programs HBPLUS (19) and LIGPLOT (20), respectively. Superposi-tions were performed using PROSUP (21) and LSQKAB (16) while surface features were analyzed with GRASP (22). Unless otherwise stated, figures were prepared using MOLSCRIPT (23) and BOBSCRIPT (24).
RESULTS AND DISCUSSION
Overall Fold—The structure of the psychrophilic xylanase consists of 13␣-helices and 13 -strands. The helices form a barrel with roughly 6 pairs of helices surrounding the central axis, and the structure can be described as a distorted (␣/␣)6
barrel (Fig. 1, A and B). A stereodiagram of the C␣ atom positions is shown in Fig. 2. This protein folding topology is common among inverting glycosidases and is observed in fam-ily 8 and 9 endoglucanases (25, 26), famfam-ily 15 glucoamylases (27), and family 48 cellobiohydrolases (28). The inner helices of the barrel have many hydrophobic residues that constitute the core of the protein, and an extra ␣-helix in comparison to common (␣/␣)6 barrel proteins, comprising residues 19 –25, is
present. One 2-stranded sheet is present at the bottom side in Fig. 1A, while the other-strands occur at the top side, forming either small or irregular sheets. On the top side, an acidic cleft 2
The abbreviations used are: pXyl, cold-adapted xylanase from P. haloplanktis; CelA, endoglucanase from C. thermocellum; FOM, fig-ure-of-merit; SAD, single wavelength anomalous dispersion.
TABLE I
Data collection statistics for the xylanase/xylobiose complex and the xylanase mutant D144N
Values in parentheses indicate the highest resolution shell.
Xylobiose complex D144N mutant
Wavelength (Å) 1.00 1.00
Resolution (Å) 20–1.20 (1.22–1.20) 20–1.50 (1.53–1.50)
Space group P212121 P212121
Unit cell parameters a⫽ 50.972 a⫽ 51.087
b⫽ 90.734 b⫽ 90.891 c⫽ 97.581 c⫽ 98.023 No. reflections Total 1,757,738 479,600 Unique 141,707 74,080 Rmerge a(%) 3.2 (23.5) 6.4 (26.7) I/(I) 43.52 (3.69) 16.21 (2.57) Completeness (%) 99.4 (91.9) 99.7 (95.3) Mosaicity (°) 0.289 0.166 a
Rmerge⫽ ⌺h⌺i兩I(h,i) ⫺ 具I(h)典兩/⌺h⌺i(h,i), where I(h,i) is the intensity of
the ith measurement of reflection h and具I(h)典 is the average value over multiple measurements.
TABLE II
Refinement statistics for the native cold-adapted xylanase, the xylobiose complex, and the D144N mutant
Native Xylobiose complex D144N mutant
Resolution (Å) 20–1.3 20–1.2 20–1.5 R/Rfree(%) 11.87/14.63 10.70/13.02 14.33/17.07 r.m.s.d.abond lengths (Å) 0.022 0.022 0.029 r.m.s.d. angles (°) 1.811 1.586 2.169 No. atoms Protein 3287 3250 3270 Solvent 433 390 388 Ligand 9 Average B-factor (Å2) Main chain 12.169 10.580 9.613 Side chain 13.919 12.399 11.747 Solvent 25.784 25.964 21.120 Ligand 31.577 Ramachandran plot
% most favored regions 93.3 91.9 91.9
% additional allowed 6.7 8.1 8.1
is clearly visible (Fig. 3A), indicative of the catalytic site. Both the N and C terminus of the protein lie at the bottom side and are separated by a distance of 39 Å. Two cysteine residues are
present, forming one disulfide bridge. Cys-324 is at the end of helix 10, while Cys-339 lies before helix 11.
Comparison with Clostridium thermocellum Endoglu-canase—The psychrophilic xylanase belongs to glycosyl hydro-lase family 8, which mainly comprises endoglucanases (9). The structure of the catalytic domain of one of these endoglu-canases, CelA from the thermophilic bacterium C. thermocel-lum, has been reported earlier (25). The (␣/␣)6 fold is also observed in this enzyme; however, there are many differences between the two structures (Fig. 1). CelA has only 12 helices and also fewer-strands. The barrel is less distorted than in pXyl and has a more circular cross-section. Furthermore, 5 cysteine residues are present, none of which occur in disulfide bridges. The acidic cleft is larger than in the case of pXyl and has a greater accessibility (Fig. 3). The full-length CelA also contains a C-terminal dockerin domain, not present in the crystal structure, which serves to anchor the protein to the cellulosome (29). In addition, the N and C terminus of the catalytic domain are separated from each other by only 8.13 Å, in contrast with 39 Å for the pXyl.
General Adaptation to Low Temperature—It is generally ac-cepted that cold-adapted proteins are more flexible than their mesophilic counterparts, with a reduced number of weak inter-actions. This flexibility often coincides with a reduced stability of the psychrophilic protein. In order to identify the cold-adapted properties of pXyl, the structure was analyzed and compared in detail with that of the C. thermocellum CelA, a summary of which is given in Table III. It must be taken into account, however, that the amount of sequence identity be-tween both enzymes is quite low (23%), and differences may therefore be due to this distant relationship. In relation to their adaptation to temperature, it has been found that CelA has an optimum temperature for activity near 80 °C (30), while pXyl has maximum activity near 35 °C (12). Differential scanning calorimetry studies have also demonstrated the lower stability of the psychrophilic enzyme, with an estimated melting tem-perature (Tmm) of 52.6 °C (12) and 83.4 °C3for pXyl and CelA
(minus the C-terminal dockerin domain), respectively. It can be seen from Table III that the most striking difference between the two proteins is in the number of salt bridges, with clearly more stabilizing ionic pairs in CelA. This latter protein also has two extra arginine residues, and as arginines can form 5 different hydrogen bonds, they are therefore expected to be important for stabilization. In addition, pXyl has a higher accessible surface area than CelA and it exposes a larger per-centage of hydrophobic residues, a destabilizing factor due to the ordering of water molecules. An increased exposure of hydrophobic residues has also been reported for the structures of␣-amylase (4) and citrate synthase (3) from Antarctic bacte-ria and for trypsin from Arctic salmon (31).
Previous studies have indicated that, in addition to the fea-tures already mentioned, some psychrophilic enzymes may be characterized by a decreased number of hydrogen and/or disul-fide bonds, a decreased number of proline residues, an in-creased number of glycine residues and/or by insertions in loops, as compared with their mesophilic and thermophilic counterparts (32, 33). From Table III, however, it can be seen that this is not the case for pXyl, indicating that this enzyme does not use these strategies for cold-adaptation.
In addition to the destabilizing adjustments noted above, many psychrophilic enzymes are characterized by alterations in their active site with, in particular, an increased size and accessibility (3, 32) as well as an optimization of the
electro-3T. Collins, M. A. Meuwis, C. Gerday, and G. Feller, manuscript in
preparation. FIG. 1. Overview of the structures of the cold-adapted
xyla-nase and the C. thermocellum endoglucaxyla-nase.␣-Helices are in red and-strands in blue while an extra ␣-helix in the psychrophilic xyla-nase is shown in green. A, side view of the (␣/␣)6barrel of the native
psychrophilic xylanase. B, top view of the barrel in A. C, top view of the (␣/␣)6barrel of C. thermocellum CelA showing that this barrel has a
static potential (6, 32). Fig. 3 gives a surface representation of CelA and pXyl, and it is clear that the substrate binding groove is larger and contains more acidic residues in the case of CelA. The xylanase is known to cleave xylan, which can be substi-tuted with negatively chargedD-glucuronic acid or 4-O-methyl-D-glucuronic acid at the OH-2 position (12, 34, 35). At low
temperatures, both a decreased flexibility of the enzyme and a decreased diffusion rate can influence the overall enzymatic reaction rate. Thus, the decreased negativity of pXyl compared with CelA may be an adaptation of the enzyme to circumvent repulsive interactions between negatively charged residues and substrate, and may enhance the rate at which pXyl binds substituted xylan. Nonetheless, it is necessary to compare this
FIG. 2. Stereo drawing of the C␣
at-oms in the native cold-adapted xyla-nase. The orientation of the molecule is the same as in Fig. 1A.
FIG. 3. Comparison of the molecular surfaces and active site
clefts (GRASP representations) of the cold-adapted xylanase (A) and the C. thermocellum endoglucanase (B). Positive potentials are shown in blue and negative potentials are displayed in red. A decreased negativity and accessibility of the substrate binding cleft is visible for the cold-adapted xylanase.
TABLE III
Parameters affecting stability and flexibility in the cold-adapted xylanase (pXyl) and the C. thermocellum endoglucanase (CelA)
Native pXyl Native CelA
No. salt bridges 13 30
No. hydrogen bonds 418 382
No. disulfide bridges 1 0
Accessible surface (Å) 15,203 13,215
% hydrophobic 16.67 13.98
Hydrophobic surface (Å) 2,534 1,847
No. glycine residues 31 36
No. arginine residues 11 13
No. proline residues 15 11
In loops 12 9
In helices 1 2
In-sheets 2 0
Net surface charge ⫹4 ⫺8
% residues in secondary structure elements
51.36 51.79
TABLE IV
Relative B-factors for catalytically important residues and aromatic residues lining the subsites of the cold-adapted xylanase (pXyl) and
the Clostridium thermocellum endoglucanase (CelA) The relative B-factors were obtained by dividing the mean B-factor of the residue by the overall B-factor value. The native structure was used for CelA.
CelA pXyl
Residue Relative B-factor Residue Relative B-factor
% % Glu-95 83 Glu-78 79 Asp-152 66 Asp-144 74 Asp-278 95 Asp-281 92 Trp-205 (subsite⫺3) 102 Tyr-194 107 Trp-132 (subsite⫺2) 79 Trp-124 99
Tyr-372 (subsite⫹1) 79 Tyr-381 113
Tyr-277 (subsite⫹2) 79 Phe-280 109
situation with the structure of mesophilic family 8 xylanases in order to determine whether or not this is a true factor of cold adaptation. Indeed, the importance of redistribution of charges has also been described for a cold-adapted citrate synthase (3) and a psychrophilic malate dehydrogenase (6) where decreased negative potentials or increased positive potentials are found at the protein surface surrounding the binding site for negatively charged substrate.
A greater accessibility of the active site may enhance the binding of substrate to enzyme and may be an adaptive strat-egy of cold-adapted enzymes (3). Mutagenesis studies have, however, questioned this proposition (36). In pXyl, the accessi-bility of the substrate-binding region is substantially lower than in CelA (Fig. 3), which is mainly the result of loop 263–276 folding over the groove in pXyl. The pXyl structure therefore also questions the importance of substrate accessibility in the cold adaptation of this enzyme.
The flexibility of specific residues in the substrate binding site is another factor suggested to be important for adaptation to low temperatures and, indeed, in pXyl the aromatic residues that line subsites ⫹1 and ⫹2, Tyr-381 and Phe-280 respec-tively, were refined with a double conformation. Both double conformations are dependent on one another: conformation B from Tyr-381 sterically clashes with conformation A from Phe-280 (Fig. 5). In the case of a cold-adapted protease, it has been observed that an active site tyrosine adopts a double conforma-tion as well, corresponding to both a substrate-bound and a substrate-free form (37). The flexibility of residues in the active site was therefore proposed to be a cold-adapted feature. In pXyl it is not clear why extra flexibility of these two residues is necessary because, in CelA, a similar conformation is found in both the native and the substrate-bound enzyme (25, 38). How-ever, small movements of these side chains may be necessary for the accommodation of substrate, and an increased flexibility of these residues may therefore assist in binding the substrate rapidly.
In the structure of a psychrophilic malate dehydrogenase (6), it was observed that the relative B-factors for residues
inter-acting with substrate were higher than for a thermophilic counterpart. This suggests that the relative flexibility of these residues is higher in the psychrophile, which may lead to an increased catalytic efficiency. The relative B-factors for the aromatic residues lining the different subsites in pXyl and CelA are shown in Table IV. Absolute B-values can vary be-cause of differences in data quality or refinement procedures, and the usefulness of relative B-factors resides in the fact that this bias is removed. It is clear that the highest flexibility is observed for Tyr-378 in both enzymes, and compared with CelA, a relatively higher B-factor is observed for Trp-124, Tyr-381, and Phe-280 in pXyl. No significantly increased flexibility in terms of relative B-values is observed for the aromatic res-idues lining subsites⫺3 and ⫹3. In addition, no conclusions can be drawn concerning relative B-factors on comparison of their values for the catalytically important residues Glu-78, Asp-144, and Asp-281 (Table IV).
FIG. 4. Binding of xylobiose to pXyl. A, omit map of a complex of the enzyme with xylobiose, showing the positioning of the reducing end xylose in the xylanase structure. The map is contoured at 2 (pink), 3 (blue), and 4 (green). The refined coordinates for the xylosyl residue and for Tyr-378 are shown. B, LIGPLOT diagram showing the interactions between the xylosyl residue and protein residues/water molecules (dark gray spheres). Hydrogen bonds are represented by dashed lines. The side chain of Tyr-378 is in hydrophobic contact with the C-5 atom of the xylosyl residue.
FIG. 5. Protein-carbohydrate stacking interactions for the C. thermocellum endoglucanase (red) and superposition of the corresponding residues in the cold-adapted xylanase (dark blue). Conformation B of Tyr-381 and Phe-280 in the psychrophilic xylanase are shown in light blue. The glucopentaose substrate, as deduced from an enzyme-substrate complex (35) is shown in yellow to indicate the position of the subsites. The position of a glucosyl residue occupying subsite⫹3 in the same complex is shown in gray, and the xylosyl residue found in the pXyl-xylobiose complex is displayed in green. The xylosyl residue that was not modeled in the latter complex points toward the⫹3 site. Residue numbering is as for the cold-adapted xylanase.
Taking all these features together, we can therefore conclude that the main cold-adaptation features in pXyl are a drastically reduced number of ionic pairs, an increased exposure of hydro-phobic residues, and possibly an optimization of the electro-static potential at the active site and an increased flexibility of the aromatic residues lining the subsites.
Substrate and Product Binding—-1,4-Linked xylobiose was soaked into existing xylanase crystals and an omit map showed the position of the ligand in the structure (Fig. 4A). However, final 2Fo ⫺ Fc electron density maps only allowed a clear
positioning of the xylose at the reducing end, indicating that the other residue is flexible. The positioned xylose adopts a normal4C
1chair conformation, forms various hydrogen bonds
with the enzyme either directly or indirectly through water molecules, and partly stacks against the aromatic side chain of Tyr-378 (Fig. 4).
Recently, a complex was obtained between an inactive family 8 endoglucanase mutant (E95Q) and glucopentaose (38). Six different subsites were identified, but none of these subsites (nomenclature of Davies et al., Ref. 39) corresponds to the positioned xylose residue (Fig. 5). The xylose residue that was not modeled points toward the⫹3 subsite from CelA, but does not coincide with it. Indeed, binding of the xylobiose at the new site may highlight an extra subsite⫹4, which may be impor-tant for directing the leaving product.
We have attempted to obtain a complex of the mutated xylanase with xylohexaose, but as yet no successful results have been obtained. However, on comparing the identified sub-sites from CelA (38) with the corresponding regions in the cold-adapted enzyme, it is clear that many differences occur (Table V). The largest discrepancy occurs at subsite⫺3. The aromatic residue Trp-205 from CelA, which stacks against a
TABLE V
Comparison of the subsites and the protein-carbohydrate interactions of the C. thermocellum endoglucanase (CelA) and the cold-adapted xylanase (pXyl)
The indication (w) shows that the interaction occurs through one or more water molecules in the CelA mutant-product complex. mc indicates that the interaction occurs with the main chain of the appointed residue. All other interactions occur directly with the side chains. The aromatic residues involved in stacking interactions with the (partial) hydrophobic surface of glucose/xylose are noted at the end of each subsite section.
CelA equivalent inProbable pXyl
Comments
Subsite⫺3
O2-Ala-149 mc (w) None identified The corresponding Pro-141 is too distant for a direct, and too close for a water-mediated hydrogen bond
O2-Gly-145 mc None identified
O3-Arg-204 None identified
O4-Arg-204 (w) None identified
Stacking: Trp-205 None identified Side chain of Tyr-194 is oriented in a different direction Subsite⫺2
O2-Asp-278 (w) Asp-281
O2-Tyr-215 (w) Tyr-203
O2-Asp-144 mc (w) None identified The side chain from Asp-138 may take over
O3-Gln-271 (w) None identified
O3-Asp-144 mc (w) None identified The side chain from Asp-138 may take over
O5-Tyr-215 Tyr-203
O6-Ala-149 mc None identified Pro-141 would sterically clash with an O6 hydroxyl group
O6-Trp-205 None identified
Stacking: Trp-132 Trp-124
Subsite⫺1
O2-Arg-281 Arg-284
O2-Asp-152 Asp-144 ? Asp-144 adopts a different conformation, but not in the D144N mutant
O2-Tyr-215 (w) Tyr-203
O3-Asp-152 Asp-144 ? Asp-144 adopts a different conformation, but not in the D144N mutant
O3-Ser-94 Thr-77
O3-Glu-95 Glu-78
O4-Tyr-215 (w) Tyr-203
O5-Asp-278 (w) Asp-281
O5-Tyr-215 (w) Tyr-203
O6-Arg-84 None identified
Stacking: / None identified
Subsite⫹1
O2-Ser-87 (w) None identified
O2-Thr-88 (w) None identified
O3-Glu-95 Glu-78 Glu-78 adopts a different conformation, but not in the D144N mutant
O3-Arg-84 Arg-76
O4-Glu-95 Glu-78 Glu-78 adopts a different conformation, but not in the D144N mutant
O6-Asp-278 Asp-281 No O6 in xylose
Stacking: Tyr-372 Tyr-381 Tyr-381 adopts a double conformation
Subsite⫹2
O2/O3-Lys-276 (w) None identified O2/O3-Ser-329 (w) None identified O2/O3-Tyr-277 mc (w) Phe-280
O6-Gly-373 mc (w) Asn-382 mc No O6 in xylose
O6-Tyr-372 mc (w) Tyr-381 No O6 in xylose
Stacking: Tyr-277 Tyr-280 Tyr-280 adopts a double conformation
Subsite⫹3
O3-Tyr-277 None identified
O3-Ser-335 (w) None identified
O6-Ser-87 (w) None identified
O6-Thr-88 (w) None identified
glucose residue of the substrate, is replaced by Tyr-194, whose side chain points in a different direction (Fig. 5). Furthermore, none of the residues that participate in hydrogen bonding to the substrate are conserved. At subsite⫹3, the aromatic resi-due Tyr-378 (pXyl numbering), which stacks against the glu-cose residue in CelA, adopts approximately the same confor-mation in pXyl, although none of the residues involved in hydrogen bonding are conserved. The fact that the⫹3 subsite is not conserved may explain why one xylose residue is very flexible in the pXyl-xylobiose complex. However, a complex between a larger substrate or product and pXyl is needed to unambiguously describe the⫹3 subsite of the xylanase.
The smallest number of differences occurs at subsite ⫺1 where most of the hydrogen bonds involving the catalytically important residues Asp-144, Glu-78, and Asp-281 (pXyl num-bering, see below) are found.
Comparing the subsites also reveals the structural basis for discrimination between glucose and xylose residues. Xylopy-ranose basically differs from glucopyXylopy-ranose by the absence of a CH2OH group at atom C-5 and it is therefore expected that
amino acid residues interacting with this OH-6 group in CelA are not conserved in pXyl. Indeed, interacting residues in sub-sites⫺2, ⫺1, and ⫹3 of CelA do not occur in the pXyl structure. Furthermore, the main chain of Pro141 in subsite⫺2 would sterically clash against an OH-6 group, showing that the bind-ing of cellulose to pXyl is impossible. On the other hand, resi-dues in subsites⫹1 and ⫹2 that interact with OH-6 are con-served in the two structures.
Hydrolytic studies suggest that pXyl can hydrolyze the-1,4 linkage that precedes (at the non-reducing end) a-1,3 linkage, and that it can only cleave-1,4 bonds positioned at least three linkages after a-1,3 bond (12). The first suggestion indicates that a-1,3 linkage occurs between subsites ⫹1 and ⫹2, re-sulting in the⫹2 sugar being orientated differently within the ⫹2 subsite. Nevertheless, its location within the enzyme can be similar to a normal case where only-1,4 linkages occur and, as
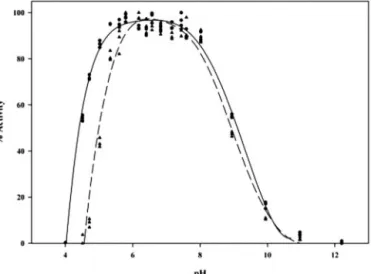
FIG. 7. Profiles of pH versus activity for the wild type cold-adapted xylanase (circles, solid line) and the D144N mutant (triangles, dashed line). Activity was measured at 25 °C with 3% soluble birchwood xylan as substrate.
Fig. 6. Schematic representation of the geometry of the cata-lytic center of the wild type cold-adapted xylanase (A), the psy-chrophilic xylanase mutant D144N (B), and the native C. ther-mocellum endoglucanase CelA (C). Dashed lines indicate hydrogen bonds. Distances between residues not implicated in hydrogen bonding are shown by a double arrow. Hypothetical charges are shown as⫹ or ⫺. All distances shown are in Å. For clarity, water atoms implicated in hydrogen bonds are not included.
a consequence, has a profound effect on the⫹3 xylosyl residue, which now points away from the “normal”⫹3 subsite. As for the second suggestion, a-1,3 linkage occurs just outside of subsite ⫺3. Because of the absence of a ⫺4 subsite, this is structurally tolerated. The first hypothesis also makes sense as subsite ⫹3 is one of the least conserved subsites where no amino acid residues that interact with substrate can be pro-posed. Detailed structures of complexes of an inactivated xyla-nase with various substrates are needed to unambiguously prove this hypothesis.
Catalytic Site—Family 8 glycosyl hydrolases are inverting enzymes that cleave-1,4-glycosidic linkages between consec-utive sugar residues. This results in an inversion of anomeric configuration at C1 and the formation of a hydroxyl group at O-4. The catalytic mechanism normally requires two carboxylic acids, separated by a distance of⬃9.5 Å. One acts as a general base, removing a proton from water, the other acts as a general acid donating a proton to the leaving group. In family 8 en-zymes Glu-78 has been shown to be the catalytic acid, while both Asp-144 and Asp-281 (pXyl numbering) have been pro-posed as the catalytic base (9, 38).
Fig. 6A shows some important interactions and distances between the different residues in the catalytic site of pXyl. The prediction of a hydrogen bond between Asp-144 and Glu-78 depends on the program used, but the two carboxyl groups are very close to each other (2.41 Å), which may mean that at least one of the carboxyl groups is neutral in charge. It can be seen that Asp-144 also forms a 2.93 Å hydrogen bond with Arg-284. As Asp-144 is strictly conserved among the family 8 glycosyl hydrolases, a mutant of pXyl was produced in which this resi-due was replaced by Asn. In agreement with the results for the family 8 endoglucanase K from Bacillus sp. KSM-330 (9), mu-tation of this residue resulted in a reduction of the catalytic efficiency (kcat/Km). However, while a 1820-fold reduction was
obtained in the case of the endoglucanase K mutant (9), the catalytic efficiency of the pXyl mutant (0.051 ⫾ 0.006 ml䡠mg⫺1䡠s⫺1) is only 182 times lower than that of the wild type xylanase (9.3⫾ 0.9 ml䡠mg⫺1䡠s⫺1). Furthermore, while realizing the limitations of the test used, it appears that the apparent Km
of the pXyl mutant (35.6⫾ 7.1 mg/ml) is unchanged (or perhaps very slightly increased) as compared with the wild type enzyme (28 ⫾ 4.5 mg/ml). This is also in good agreement with the results obtained for the endoglucanase K mutant (9) and indi-cates that Asp-144 is not important for substrate affinity. Anal-ysis of the pH activity profile in the presence of substrate shows that the acidic limb is affected in the mutant (Fig. 7), indicating a slight increase in the pKaof the catalytic base and/or
nucleo-phile. These results suggest that Asp-144 may act as the cat-alytic base in pXyl, yet the pKachange observed is small and
the distance (2.41 Å) between Glu-78 and Asp-144 is too short for inverting enzymes.
It has also been suggested that Asp-281 is the catalytic base in family 8 enzymes (38). Structural analysis of CelA has re-cently identified Glu-78 and Asp-281 (pXyl numbering) as the general acid and general base residues in this enzyme, respec-tively. It was also shown that Asp-144 plays a role in hydroly-sis, as it stabilizes the sugar ring in subsite⫺1 in a strained boat conformation (38). The distance between Glu-78 and Asp-281 in CelA is short for an inverting enzyme (6.35 Å for the native enzyme and 6.5 Å for the complexed CelA), and in the pXyl structure this distance is even less (4.5 Å), but may change upon substrate binding. Analysis of the catalytic site of D144N (Fig. 6B) shows that the mutation has an effect on the positions of both Asn-144 and Glu-78. The hydrogen bond be-tween residue 144 and Arg-284 is lost, and the side chain of Glu-78 is turned away from Asn-144. Instead, Glu-78 now forms hydrogen bonds with the side chains of Tyr-380 and Arg-284 (Fig. 6B). Thus, if Asp-281 is indeed the true catalytic base, Asp-144 is also a critical residue, playing a role in the positioning of Glu-78 and perhaps also in facilitating hydrolysis and regulating the pKaof the nucleophile and/or catalytic base.
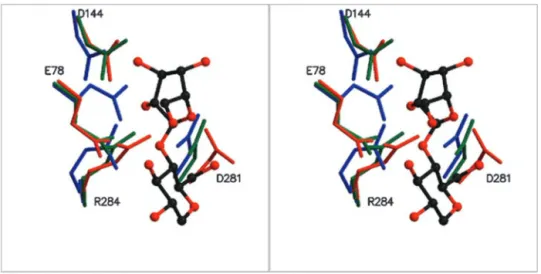
This may lead to a decrease in activity and to the alteration of the pH activity profile as observed in this study. Furthermore, analysis of the local geometry of the catalytic site of pXyl (Fig. 8) indicates that Asp-281 is better positioned than Asp-144, in relation to the general acid and the expected positioning of substrate, to act as the general base. Further studies, including site-directed mutagenesis of Asp-281, as well as structural studies of a complex with substrate, are necessary to clarify this point. In addition, because of the pronounced effects of the D144N replacement, the neutral residue in the Glu-78 –Asp-144 pair is likely to be Glu-78. This corresponds well with the role of this particular glutamate as a general acid.
Curiously, the geometry of the catalytic site residues in CelA resembles more that of the D144N mutant than that of wild type pXyl (Fig. 8). Glu-78 and Asp-144 (pXyl numbering) in CelA are not involved in a hydrogen bond (Fig. 6, B and C), and the two residues adopt the same conformation as in the D144N mutant xylanase. Furthermore, the Asp-281 residue in CelA forms a hydrogen bond with Asn-200, which replaces Asp-200 (pXyl numbering).
Conclusion—The xylanase from the psychrophilic organism P. haloplanktis is a cold-adapted family 8 glycosyl hydrolase displaying an (␣/␣)6fold. Compared with a thermophilic
endo-glucanase from the same family, the structure is destabilized by a drastically reduced number of salt bridges and an in-creased exposure of hydrophobic residues. The rate at which substrate binds may be enhanced by a decreased acidity of the
FIG. 8. Stereo view of the active site region of the wild type cold-adapted xylanase (blue), D144N (green) and the C. thermocellum endoglucanase (red) in complex with glucopentaose. Glucosyl residues in subsites⫺1 (top) and ⫹1 (bottom) are shown in ball-and-stick representation. The general acid (Glu-78) and the putative general bases (Asp-144, Asp-281) of the psychrophilic xylanase are shown, indicating the better position-ing of Asp-281 to act as the general base.
substrate binding site and by an increased flexibility of aro-matic residues lining the different subsites. Contrary to expec-tations, however, is a decreased accessibility of the substrate binding region. Other cold adaptations are not evident, or are more discrete, and this supports the idea that every cold-adapted enzyme has a unique means of dealing with lower environmental temperatures.
Compared with CelA, the substrate binding region differs appreciably. The subsites are not conserved, with large differ-ences occurring at subsites⫹3 and ⫺3, as well as the appear-ance of a potential⫹4 subsite. The selectivity of pXyl for xylan over cellulose is due to the absence of residues that interact with the glucose OH-6 group in CelA and the steric hindrance, which would occur with an OH-6 group in subsite⫺2.
Acknowledgments—We thank access to beamline X13 at the EMBL, Hamburg Outstation, beamline BM14 at the ESRF, Grenoble, and the protein crystallography beamline at ELETTRA, Trieste.
REFERENCES
1. Zecchinon, L., Claverie, P., Collins, T., D’Amico, S., Delille, D., Feller, G., Georlette, D., Gratia, E., Hoyoux, A., Meuwis, M. A., Sonan, G., and Gerday, C. (2001) Extremophiles 5, 313–321
2. Russell, N. J. (2000) Extremophiles 4, 83–90
3. Russell, R. J., Gerike, U., Danson, M. J., Hough, D. W., and Taylor, G. L. (1998)
Structure 6, 351–361
4. Aghajari, N., Feller, G., Gerday, C., and Haser, R. (1998) Structure 6, 1503–1516
5. Alvarez, M., Zeelen, J. P., Mainfroid, V., Rentier-Delrue, F., Martial, J. A., Wyns, L., Wierenga, R. K., and Maes, D. (1998) J. Biol. Chem. 273, 2199 –2206
6. Kim S-Y, Hwang KY, Kim S-H, Sung H-C, Han YS, and Cho Y. (1999) J. Biol.
Chem. 274, 11761–11767
7. Gerday, C., Aittaleb, M., Bentahir, M., Chessa, J. P., Claverie, P., Collins, T., D’Amico, S., Dumont, J., Garsoux, G., Georlette, D., Hoyoux, A., Lonhienne, T., Meuwis, M. A., and Feller, G. (2000) Trends Biotechnol. 18, 103–107 8. Henrissat, B. (1991) Biochem. J. 280, 309 –316
9. Ozaki, K, Sumitomo, N, Hayashi, Y., Kawai, S., and Ito, S. (1994) Biochim.
Biophys. Acta 1207, 159 –164
10. Dominguez, R., Souchon, H., Spinelli, S., Dauter, Z., Wilson, K. S., Chauvaux, S., Beguin, P., and Alzari, P. M. (1995) Nat. Struct. Biol. 2, 569 –576
11. Torronen, A., Harkki, A., and Rouvinen, J. (1994) EMBO J. 13, 2493–2501 12. Collins, T., Meuwis, M. A, Stals, I., Claeyssens, M., Feller, G., and Gerday, C.
(2002) J. Biol. Chem. 277, 35133–35139
13. Van Petegem, F., Collins, T., Meuwis, M. A., Gerday, C., Feller, G., and Van Beeumen, J. (2002) Acta Crystallogr. Sect. D 58, 1494 –1496
14. Perrakis, A., Sixma, T. K., Wilson, K. S., and Lamzin, V. S. (1997) Acta
Crystallogr. Sect. D 53, 448 – 455
15. Roussel, A., and Cambillau, C. (1992) in Silicon Graphics Geometry Directory, Vol. 86, Silicon Graphics, Mountain View, CA
16. The CCP4 Suite: Programs for Protein Crystallography. (1994) Acta
Crystal-logr. Sect. D 50, 760 –763
17. Laskowski, R. A., MacArthur, M. W., Moss, D. S., and Thornton, J. M. (1993)
J. Appl. Crystallogr. 26, 283–291
18. Schmidt, R. K., Karplus, M., and Brady, J. W. (1996) J. Am. Chem. Soc. 118, 541–546
19. McDonald, I. K., and Thornton, J. M. (1994) J. Mol. Biol. 238, 777–793 20. Wallace, A. C., Laskowski, R. A., and Thornton, J. M. (1995) Protein Eng. 8,
127–134
21. Lackner, P., Koppensteiner, W. A., Sippl, M. J., and Domingues, F. S. (2000)
Protein Eng. 13, 745–752
22. Nicholls, A., Sharp, K. A., and Honig, B. (1991) Proteins 11, 281–296 23. Kraulis, P. J. (1991) J. Appl. Crystallogr. 24, 946 –950
24. Esnouf, R. M. (1997) J. Mol. Graphics 15, 132–134
25. Alzari, P. M., Souchon, H., and Dominguez, R. (1996) Structure 4, 265–275 26. Juy, M., Souchon, H., Alzari, P. M., Poljak, R. J., Claeyssens, M., Be´guin, P.,
and Aubert, J. P. (1992) Nature 357, 89 –91
27. Aleshin, A., Golubev, A., Firsov, L. M., and Honzatko, R. B. (1992) J. Biol.
Chem. 267, 19291–19298
28. Parsiegla, G., Juy, M., Reverbel-Leroy, C., Tardif, C., Belaich, J. P., Driguez, H., and Haser, R. (1998) EMBO J. 17, 5551–5562
29. Be´guin, P., Cornet, P., and Aubert, J.-P. (1985) J. Bacteriol. 162, 102–105 30. Petre, J., Longin, R., and Millet, J. (1981) Biochimie (Paris) 63, 629 – 639 31. Smalås, A. O., Heimstad, E. S., Hordvik, A., Willasen, P., and Male, R. (1994)
Proteins 20, 149 –166
32. Smalås, A. O., Leiros, H. K., Os, V., and Willassen, N. P. (2002) Biotechnol.
Annu. Rev. 6, 1–57
33. Feller, G., Arpigny, J., Narinx, E., and Gerday, C. (1997) Comp. Biochem.
Physiol. 118A, 495– 499
34. Sunna, A., and Antranikian, G. (1997) Crit. Rev. Biotechnol. 17, 39 – 67 35. Li, K., Azadi, P., Collins, R., Tolan, J., Kim, J., and Eriksson, K. (2000) Enzyme
Microb. Technol. 27, 89 –94
36. Gerike, U., Danson, M. J., Hough, D. W. (2001) Protein Eng. 14, 655– 661 37. Aghajari, N., Van Petegem, F., Villeret, V., Chessa, J. P., Gerday, C., Haser, R.,
and Van Beeumen, J. (2003) Proteins, in press
38. Gue´rin, D. M., Lascombe, M. B., Costabel, M., Souchon, H., Lamzin, V., Beguin, P., and Alzari, P. M. (2002) J. Mol. Biol. 316, 1061–1069 39. Davies, G. J., Wilson, K. S., and Henrissat, B. (1997) Biochem. J. 321, 557–559