Analyse transcriptomique de l’interaction
tripartite Pseudozyma flocculosa‐Blumeria
graminis f.sp. hordei‐Hordeum vulgare
Mémoire
Gowsica Bojarajan Ramakrishnan
Maîtrise en biologie végétale
Maître ès sciences (M.Sc.)
Québec, Canada
© Gowsica Bojarajan Ramakrishnan, 2016iii RÉSUMÉ Afin d’améliorer nos pratiques agricoles dans le contexte d’une agriculture durable, plusieurs agents de lutte biologique (ALB) ont été développés, testés et sont maintenant utilisés dans le monde pour combattre les pertes de rendements causées par les maladies. Blumeria graminis f. sp. hordei ( Bgh) est l’agent pathogène responsable du blanc de l’orge et peut réduire les rendements de cette culture jusqu’à 40%. Un champignon épiphyte, Pseudozyma flocculosa, a été découvert et identifié en 1987 en association étroite avec le blanc du trèfle. Les chercheurs ont alors remarqué que ce champignon exhibait une forte activité antagoniste contre le blanc en détruisant les structures de l’agent pathogène. Suite à d’autres travaux, il est apparu que ce comportement antagoniste était dirigé contre tous les membres des Erysiphales et semblait lié à la synthèse d’un glycolipide antifongique soit la flocculosine. Toutefois, on n’est toujours pas parvenus à associer l’efficacité de l’ALB avec la production de ce glycolipide. Ces observations suggèrent que d’autres facteurs seraient impliqués lorsque les deux protagonistes, l’ALB et le blanc, sont en contact. L’objectif principal de ce projet était donc de chercher d’autres mécanismes moléculaires pouvant expliquer l’interaction P. flocculosa‐blanc et orge, en faisant une analyse transcriptomique complète des trois protagonistes en même temps.
L’interaction tripartite a été échantillonnée à différents temps suivant l’inoculation de P. flocculosa sur des feuilles d’orge présentant déjà une intensité de blanc d’environ 50%. Les échantillons de feuilles prélevés ont ensuite été utilisés pour l’extraction de l’ARN qui ont été ensuite transformés en ADNc pour la préparation des librairies. Cinq répliquats ont été effectués pour chaque temps et le tout a été séquencé à l’aide de séquençage par synthèse Illumina HiSeq.
Les séquences obtenues (reads) ont ensuite été analysées à l’aide du logiciel CLC Genomics Workbench. Brièvement, les séquences obtenues ont été cartographiées sur les trois génomes de référence. Suite à la cartographie, les analyses d’expression ont été conduites et les gènes exprimés de façon différentielle ont été recherchés. Cette étape a été conduite en portant une attention particulière aux gènes codant pour un groupe de
iv
protéines appelées CSEP pour “candidate secreted effector proteins” qui seraient possiblement impliquées dans l’interaction tripartite.
Parmi les protéines exprimées de façon différentielle en présence du blanc ou en absence de ce dernier, nous avons pu constater que certaines CSEP étaient fortement exprimées en présence du blanc. Ces résultats sont prometteurs et nous offrent une piste certaine pour l’élucidation des mécanismes impliqués dans cette interaction tripartite.
v TABLE OF CONTENTS RÉSUMÉ ... iii TABLE OF CONTENTS ... v LIST OF TABLES ... vii LIST OF FIGURES ... ix ACKNOWLEDGEMENTS ... xi FOREWARD ... xiii CHAPTER 1 ... 1 LITERATURE REVIEW ... 1 1. INTRODUCTION ... 1 2. PSEUDOZYMA FLOCCULOSA ... 1 2.1 Biological control agents ... 1 2.2 Classification and ecology ... 2 2.3 Mode of action of P. flocculosa ... 2 2.4 Genetics of P. flocculosa ... 3 2.5 Ustilago maydis and Pseudozyma flocculosa: A tug of war ... 3 3. Transcriptomic analysis – A powerful tool of Next Generation Sequencing ... 4 4. Powdery mildew fungi – The pathogen of interest ... 5 5. Hordeum vulgare – The host ... 6 6. Effector biology – A path to be unraveled ... 7 7. Hypotheses ... 9 8. OBJECTIVES ... 10 CHAPTER 2 ... 11 MANUSCRIPT ... 11 Transcriptomic analysis of the tripartite interaction Pseudozyma flocculosa‐ Blumeria graminis f.sp. hordei‐Hordeum vulgare ... 13 RÉSUMÉ ... 15 INTRODUCTION ... 17 MATERIALS AND METHODS ... 19 Plant material ... 19 Fungal material of Pseudozyma flocculosa ... 19 Inoculation with Pseudozyma flocculosa fungus ... 19 RNA isolation ... 19 cDNA library construction ... 20 RNA sequencing ... 20 Mapping reads to reference genome ... 21 Genome annotation ... 21 Identification of differentially expressed genes ... 22 RESULTS ... 23 Characteristic growth of Pseudozyma flocculosa in culture conditions. ... 23
vi Calibration of P. flocculosa in response to powdery mildew fungus ... 23 Transcriptional dynamics of powdery mildew fungus in response to P. flocculosa... 26 RNA integrity, cDNA library validation and sequencing ... 26 Differential gene expression pattern in P. flocculosa ... 31 Differential gene expression pattern of genes involved in flocculosin production ... 32 P. flocculosa effector candidates are differentially expressed during infection with B. graminis ... 33 Discussion ... 35 ACKNOWLEDGEMENTS ... 37 REFERENCES ... 37 CHAPTER 3 ... 41 GENERAL CONCLUSIONS ... 41 BIBLIOGRAPHY ... 45
vii LIST OF TABLES
Table 1. Sample collection pre and post inoculation of barley leaves with P. flocculosa. ... 30
ix LIST OF FIGURES Figure 1: Characterization of Pseudozyma flocculosa in culture condition. ... 23 Figure 2: Development of powdery mildew disease over time in a healthy plant. ... 24 Figure 3: Antagonism of Pseudozyma flocculosa on barley powdery mildew colonies over time. 25 Figure 4: Scanning electron microscopy observation of barley powdery mildew fungus before and after treatment with Pseudozyma flocculosa. ... 26 Figure 5. Total RNA isolation from the leaf samples and its integrity check using bioanalyser. .. 27 Figure 6 a Principle Component Analysis (PCA) of P. flocculosa samples grown in vitro and in vivo. ... 28 Figure 6 b Principle Component Analysis (PCA) of the samples collected at various conditions. 29 Figure 7. Tripartite interaction mapping. ... 30 Figure 8. Differential gene expression pattern of P. flocculosa genes. ... 31 Figure 9. Differential gene expression pattern of flocculosin producing genes. ... 32 Figure 10. Differential gene expression pattern of effector candidates at different time points .. 33 Figure 11. Differential gene expression pattern of effector candidates ... 34
xi ACKNOWLEDGEMENTS
First and foremost, I would like to thank god for providing me the opportunity to come to Canada for my graduate studies.
I like to thank my supervisor Prof. Richard Bélanger for providing me the opportunity to pursue my graduate studies in his esteemed laboratory. He has been a great teacher for me and guided me to be a better scientist. Every scientific interaction with him makes me more delightful and enthusiastic about science. I admire his passion for science, which has been a great encouragement and inspiration for me during my stay in lab. During all my years of hard work in the laboratory he supported during the tough time and appreciated when I had success. His advice on both research as well as on my career have been priceless.
I would like to thank Dr. François Belzile for his inputs on my project at lab meetings on every Friday and being a part of my thesis committee. I would like to extend my thanks to Prof. Daniel Dostaler for being a committee member of my thesis and for his valuable comments and suggestions to improve my project. My grateful thanks go to Caroline Labbé for all her efforts to teach and train me in this project. A big thank goes to Huma for helping me in crucial bioinformatics steps. I thank my lab members Julien, Rupesh, Julie Anne, Amandine, François, Geneviève, Bastien, Sarah, Marc‐Olivier, Stéphanie, Marie‐Hélène Samuel and Joan for their constant support and encouragement. Special thanks to Aliyeh for being a very good friend and for sharing lots of fun‐filled moments, discussions and ideas. I am lucky enough to have such excellent people as colleagues. Special and loads of thanks go to all my family members. Words cannot express how grateful I am to my family. My beloved thanks to my mother Mrs. Pushpam Ramakrishnan and my father Mr. Ramakrishnan for all of the sacrifices they have made throughout their life to shape up me and my career. Their prayers and blessings for me were what sustained me so far. I would also like to thank my brother Mr. Arun Ramakrishnan for his unconditional love and care towards me. At the end I would like express appreciation to my beloved husband Mr. Girishwaran who spent long lonely days,
xii
sleepless nights without me and stood by me always as my support in each and every moment of my life. His love, care, encouragement and support made me to achieve this degree. I have a special mention to my dog “Dhivan” who left his foot prints in my heart forever. My family is my backbone and strength.
I would like to extend my heartfelt thanks to my friend Senthil Krishnasamy who supported me and guided me in lot of things in my life. I hugely appreciate my friend Preyesh, Jina and my little angel Ishitha for their kindness and support towards me.
I like to thank Wajid Bhat and Ambreen for their kind help during my initial days in Canada. I thank Ranjan, Hemanta, Prakash, Pallavi, Priyanka, Dinesh, Prenitha, Ramesh and all Indian buddies in Quebec City for making my stay pleasant and memorable in Quebec.
Last but not least I would like to dedicate my thesis to two most inspiring women in my life Mrs. Natchiyar Shanmugam and Mrs. Seeniyammal Bojararajan. They are my grand‐mothers who always wanted me to achieve great heights in my life and stood beside me during my hardships. A special mention goes to living legend Mrs. Natchiyar Shanmugam for her unconditional love, care and support, encouragement towards me in my life.
xiii FOREWORD
This thesis includes a literature review that outlines the current knowledge related to the advancement in the field of biological control agents in plant protection. It discusses the importance of effector proteins and reveals their role in fungal‐fungal interactions, a big milestone in effector biology. The first part of the first chapter includes the classification, etiology and genetics of the biocontrol agent Pseudozyma flocculosa. The second part of the first chapter reveals the relationship between P. flocculosa and Ustilago maydis and the structural similarity between flocculosin and ustilagic acid. The hypothesis of this thesis was based on the comparative analysis of P. flocculosa with closely related organisms that revealed features unique to P. flocculosa.
The second chapter of this thesis is presented in the form of a research manuscript where I am the principal author. The manuscript deals with the identification of mechanisms of action of P. flocculosa defining its antagonistic activity against powdery mildew fungi using a novel next generation sequencing technique of transcriptomic analysis. The differential gene expression analysis of RNA‐sequencing data highlights the role of effectors in the interaction P. flocculosa‐powdery mildew fungi, the first such report for fungal‐fungal interactions.
The third and final chapter of this thesis concludes the present results in a broader context.
CHAPTER 1
1 1. Introduction
Plants are a main source of life on earth but their production is constantly threatened by pathogens such as fungi, oomycetes, bacteria, viruses and nematodes. These pathogens need to be controlled to maintain the quality and abundance of food, feed, and fiber produced by growers around the world. Crop protection is a constant concern for human communities since the beginning of agriculture. Different approaches may be used to prevent, mitigate or control plant diseases. Beyond good agronomic and horticultural practices, growers often rely heavily on chemical pesticides. However, the intensive usage of pesticides compromises sustainability and environmental health. In recent decades, the search for new approaches for crop protection that are both more effective and less damaging to the environment and human health has led to the development of promising new tools inspired by biological and ecological processes. Indeed, many microorganisms have the natural ability to inhibit the growth or even kill other species in order to protect their ecological niche or have a source of nutrients. The development of novel alternatives to control plant diseases, based on the exploitation of beneficial organisms, has been at the forefront of many research endeavors around the world. The use of living organisms to combat other living organisms presupposes a thorough knowledge of their ecology. However, many technical and scientific challenges remain to be resolved before a widespread commercial use of these beneficial organisms can be envisioned.
2. PSEUDOZYMA FLOCCULOSA 2.1 Biological control agents
In agriculture, the microbial flora in the environment of cultivated plants has become the object of great interest. With the advent of sophisticated molecular techniques, new microbial species are discovered at an unprecedented pace, screened and described for their beneficial or harmful properties. Exploiting these microorganisms for their beneficial properties is an approach called "biological control". Biological control is defined as the reduction of insects, mites, weeds and plant diseases by natural enemies and also sometimes with the help of an active human role. It is a component of integrated pest management strategies and aims to control any insects or plant diseases by the use
2
of other species that are their natural antagonist or that promote plant defense reactions (Flint et al., 1998). Biological control will try to favor the growth and the dispersal of natural antagonists in agricultural systems to fight plant diseases, as an ecological or natural alternative to fungicides. Most biological control agents (BCAs) identified to date have been categorized as exerting their activity through the manifestation of one or more of four modes of action: competition, parasitism, antibiosis and/or induced resistance (Bélanger and Avis et al., 2001; Whipps et al., 2001). Understanding precisely how BCAs act on their targets will increase their efficacy at reducing various plant disease. For instance, in cases where parasitism appeared to be the predominant mode of action, several attempts have been made to increase production of lytic enzymes such as chitinases and glucanases (Kubicek et al., 2001; Lorito et al., 2001). The approach targeted either the selection of BCA strains with superior ability to produce such enzymes or the direct cloning and over‐expression of relevant genes conferring greater degrading properties. In order to control these plant diseases, the broad spectrum of mechanisms that filamentous fungal pathogens use to colonize host plants needs to be elucidated. 2.2 Classification and ecology
The 1980s were particularly fertile in the search for potential BCAs against various plant diseases. The discovery and characterization of the BCA P. flocculosa was part of this trend. Pseudozyma flocculosa was discovered in 1987 and originally identified as Sporothrix flocculosa, an ascomycetous yeast. It was first discovered as an epiphytic yeast on powdery mildew‐infected clover leaves (Traquair et al., 1988). Since then, it has been extensively studied for the development of an efficient biofungicide to control powdery mildews in many crops. P. flocculosa is an anamorphic fungi which lacks sexual development and were found to be morphologically and phylogenetically related to the Ustilaginales. The phylogenetic analysis and comparison of diagnostic ribosomal DNA sequences made it to reclassify under Ustilaginales family (Begerow et al., 2000).
2.3 Mode of action of P. flocculosa
Initial studies with P. flocculosa showed that it was neither a strong competitor in the phyllosphere nor a direct hyperparasite, since no direct contact between the
3 biocontrol agent and target powdery mildew was required to observe an antagonistic activity. Powdery mildew cells exposed to P. flocculosa suffered rapid plasmolysis, which led to the conclusion that antibiosis was its mode of action (Hajlaoui et al., 1993). This was further supported by the discovery of an unusual glycolipid produced by P. flocculosa, called flocculosin that exhibited strong antifungal activity (Mimee et al., 2005). Shortly thereafter, it was discovered that the molecule was nearly identical to ustilagic acid, found in the culture filtrates of U. maydis in 1951. This finding brought a direct link to the reclassification of P. flocculosa among the Ustilaginales (Begerow et al., 2000). 2.4 Genetics of P. flocculosa Molecular signalling between a plant pathogen and its host plays a fundamental role in pathogenesis and in the establishment of the interaction. These interactions have a profound effect for designing new strategies to combat diseases. To understand genes and their role in the biology and the genetics of an organism, it is necessary to understand the genome sequences. As such, sequencing and assembly of the P. flocculosa genome became a necessity for understanding the implication of flocculosin in the biocontrol activity of P. flocculosa. Using Roche 454 Titanium technology, 525 Mb of shotgun data and 167 Mb of 2.6 and 4.5 kb mate‐pair sequences for a ca. 30X coverage of the genome was generated for genome sequencing of P. flocculosa. The P. flocculosa genome is 23Mb and includes 6877 predicted proteins. The assembly yielded 1583 contigs from which 1187 were oriented and ordered into 37 scaffolds to which three contigs larger than 2Kb were added. The main difference observed between the genome structures of P. flocculosa and other Ustilaginales were found in the proportion of guanine and cytosine (GC) residues and in the structure of genes. The genes identified in P. flocculosa contained an average of four times more introns than U. maydis but the number of transposable elements and simple repeats found in P. flocculosa genome is similar to that of U. maydis (Lefebvre et al., 2013). 2.5 Ustilago maydis and Pseudozyma flocculosa: A tug of war Although the phylogenetic proximity between U. maydis and P. flocculosa has been demonstrated several years ago, details about the content and distribution of genes in P. flocculosa remained the subject of speculation. Indeed, on one hand, the genome of the
4
pathogen U. maydis has been available since 2006, and has led to important discoveries and generated a lot of useful information for different research aspects (Kämper et al., 2006). For instance, using the U. maydis cyp1 cDNA as a probe against all known species of Pseudozyma, it was possible to show that it hybridized specifically with P. flocculosa (Marchand et al., 2007), the only other strain producing flocculosin. This indicated that cyp1 had to be involved in flocculosin production. The presence of cyp1 in P. flocculosa raised the obvious possibility of the existence of a cluster similar to the one found in U. maydis regulating the production of flocculosin. On the basis of sequence homology with genes found in U. maydis, a gene cluster comprising 10 genes that were necessary for the biosynthesis of flocculosin as identified (Teichmann et al., 2010). In contrast to the cluster of U. maydis, the flocculosin biosynthesis cluster contains an additional gene encoding an acetyl‐transferase and is lacking a gene homologous to the α‐hydroxylase Ahd1 necessary for UA hydroxylation. The functions of three acyl/acetyl‐transferase genes (Fat1, Fat2 and Fat3) including the additional acetyl‐transferase were studied by complementing the corresponding U. maydis mutants (Teichmann et al., 2010). This showed that the additional acetyl‐transferase is necessary for acetylation of the glucose moiety, explaining the differences between the two molecules.
3. Transcriptomic analysis – A powerful tool of Next Generation Sequencing
Since the start of genomics research, genome‐wide expression studies have been used as a tool to improve our understanding of the involvement of genes in various biological processes. Measuring gene expression patterns simultaneously across all genes in the genome, i.e. transcriptomics, is a uniquely powerful technology to explore potential novel candidate genes for a particular process. Identifying the full set of transcripts including large and small RNAs, novel transcripts from unannotated genes, splicing isoforms and gene‐fusion transcripts serves as a foundation for the comprehensive study of the transcriptome. Whole‐transcriptome analysis is of growing importance in understanding how altered expression of genetic variants contributes to complex plant diseases. The analysis of genome‐wide differential RNA expression provides greater insights into biological pathways and molecular mechanisms that regulate cell fate, development and disease progression (De Vos et al., 2005). Transcriptomic approaches
5 are a revolutionary functional genomic tool for deciphering plant‐pathogen interactions in the pathogenomics era. The technique is extremely powerful as a first step to implicate novel genes and pathways that may be involved or associated with a particular condition. Transcriptomic data from next generation sequencing technology give us information about the activity of genes that change their expression pattern in response to a signal originating from the host plant or in the host tissue and may reveal mechanisms of pathogenesis and biocontrol activity as initiated by fungal pathogens and fungal BCAs. 4. Powdery mildew fungi – The pathogen of interest
Powdery mildews are amongst the most common, widespread and recognizable of all plant diseases. They are aptly named, for the infection produces a white lawn of fungal mycelium that covers the plant surface, while chains of aerial conidia give the characteristic powdery appearance. Powdery mildews can infect a wide range of hosts, including over 9000 dicotyledonous and over 650 monocotyledonous plant species. The cereals, particularly wheat and barley, are among the most important agricultural crops that suffer from powdery mildew diseases. Indeed, in temperate regions, barley powdery mildew can cause yield losses of some 5‐20% and occasionally as much as 40%. Taken collectively, powdery mildews cause greater losses in terms of crop yield than any other single “type” of plant disease. The powdery mildew diseases are caused by many species of Ascomycete fungi, grouped into several genera. They are true obligate biotrophs, which means that growth and reproduction of these fungi depend on their parasitizing living host plants. Despite the lack of robust and reliable DNA‐mediated transformation method and mutational analysis, significant progress has been made over the past decade towards understanding powdery mildew‐host interactions at both the cellular and molecular level (Hacquard et al., 2013).
Blumeria graminis f. sp. hordei (E.O. Speer DC) is a powdery mildew fungus that infects barley and it can reduce crop yield as much as 40% (Wiese et al., 1987). It was earlier included in order Erysiphales but later its molecular studies placed these into a new taxon Blumeria. The genome of B. graminis f. sp. hordei (Bgh) has recently been sequenced (Spanu et al., 2010). This genome is of size 120Mb and extremely rich in repetitive elements derived mostly from retrotransposons with 90% transposable
6 elements. Additionally, 6540 genes were annotated, from which 437 encoded candidate effector proteins and 165 for non‐secreted candidate effector proteins. The ability to infect tetraploid as well as domesticated hexaploid wheat, was seen to be the result of mildew genomes being mosaics of ancient haplogroups that existed before wheat domestication. This has allowed wheat powdery mildew to maintain genetic flexibility, variability and thus a great potential for pathogen variation. 5. Hordeum vulgare – The host Hordeum vulgare L. (barley) is the world's fourth most important cereal crop and an important model for ecological adaptation. It was one of the first domesticated cereal grains originating in the Fertile Crescent over 10,000 years ago. About two‐thirds of the global barley crop is used for animal feed, while the remaining third underpins the malting, brewing, and distilling industries. Although the human diet is not a primary use, barley has potential health benefits, and is still the major calorie source in several parts of the world. Barley is a member of the grass family. It is a self‐pollinating, diploid species with 14 chromosomes. Genome of barley was sequenced in 2012 by the International Barley Genome Sequencing Consortium (IBSC) and also the UK Barley Sequencing Consortium. The genome is composed of seven pairs of nuclear chromosomes, with a total of 5000 Mbp. In the IBSC assembly, ~2.6 million sequenced contigs were generated using whole‐ genome shotgun sequencing (WGS). Of these, ~723,000 are assigned to specific chromosomal positions (Klaus et al., 2012). It is one of the largest diploid genomes sequenced to date.
7 6. Effector biology – A path to be unraveled
Interactions between organisms are controlled by exchanges of signals between partners. Plants can be colonized by fungi that have adopted highly diverse lifestyles, ranging from symbiotic to necrotrophic. Fungi have adopted diverse strategies to interact with host plants and to overcome a complex network of plant defense mechanisms. Colonization is governed in all systems by hundreds of secreted fungal effector molecules (Sonah et al., 2016). These effectors suppress plant defense responses and modulate plant physiology to accommodate fungal invaders and provide them with nutrients. Fungal effectors either function in the interaction zone between the fungal hyphae and host or are transferred into plant cells.
Effector proteins are mostly secretory proteins that alter host cells to suppress host defense mechanisms and facilitate the interaction by the pathogen so it can derive nutrients from the host. Effectors will also activate resistance mechanisms in the resistant plant genotypes. Candidate secreted effector proteins (CSEPs) are defined as fungal proteins with a signal peptide for secretion, no trans‐membrane domains and no similarity with other obvious protein domains are fairly small in size and usually species‐specific (Jones and Dangl, 2006; Djamei et al., 2011). The centrality of effector proteins in the biology of plant pathogenic microbes is demonstrated by the presence of vast arrays of effector‐like genes that are found in practically all pathogen genomes. This is particularly striking in the genomes of the obligate biotrophic fungi that cause powdery mildews (Spanu et al., 2010; Wicker et al., 2013). In these fungi, many commonly large gene families are reduced to very few members, and some genes are lost altogether. In sharp contrast, the effector‐like gene superfamilies described in cereal powdery mildews comprise over 7% of the conventional protein‐coding gene capacity of the genome. From the pathogens' point of view, these effectors are essential tools to gain entry and switch off the hosts' defense mechanisms. In general, effector proteins interfere with recognition of microbes at the surface of cells and intercellular spaces; they can also target the intracellular immune‐signaling pathways all the way up to and including the activation of the transcription of genes involved in resistance and the defense response. Indeed, the study of protein effectors is a useful instrument to investigate and define the mechanisms
8
of the immune response itself. Recent developments and advances in computational tools and in development of various pipelines make it easier to perform genome‐wide identification of CSEPs (Sonah et al., 2016). This gives us more information on the distribution and organization of CSEPs within a given species. In U. maydis, 426 secretory proteins were identified, 70% of which were with unknown function based on their homology search. It was also found that effector proteins exist as clusters of 3‐26 genes per cluster. Knockout studies of specific genes or clusters identified about 50 effector proteins that were involved in pathogenesis (Kamper et al., 2006). More recently the generation and analysis, including annotation, of the complete genome of P. flocculosa with a comparative analysis to the genomes of U. maydis, S. reilianum and U. hordei highlighted similarities and differences with respect to effector proteins (Lefebvre et al., 2013). The comparative genomic analysis of phylogenetically closely related species revealed a higher conservation of virulent secreted proteins in the three pathogens and a near complete loss in P. flocculosa. These results highlighted that the main difference between phytopathogenic Ustilaginales and P. flocculosa could be attributed to a few specific effector proteins, thus confirming the important role of such proteins in defining plant‐pathogen interactions. Focusing on potential effectors revealed that, in comparison to U. maydis, the genome of P. flocculosa has nearly the same number of predicted secreted proteins. For its part, P. flocculosa possesses 200 specific CSEPs where no orthologs are found in closely related species. P. flocculosa also possess two NPP1 containing proteins, absent in all pathogenic Ustilaginales. Understanding the role of effector proteins will lead us to new paths of defining the potential factors involved in the biocontrol properties of P. flocculosa. (Lefebvre et al., 2013).
9 7. Hypotheses
Extensive studies have characterized many infection‐responsive genes in the pathogen and host plant, separately. To understand the plant‐pathogen interaction and pathogen‐BCA interaction comprehensively, it is valuable to monitor the gene expression profile of all the interacting organisms simultaneously in the same infected plant tissue. The research on P. flocculosa made many assumptions about the genetic basis related to its molecular biology. The close phylogenetic link between P. flocculosa and U. maydis has offered unexpected opportunities to define the factors inherent to specific lifestyles that characterize fungi. More specifically, it allowed the study of genetic determinants that conferred the phytopathogenic nature of some Ustilaginales. On the other hand, the biocontrol process appears to be mediated by an interaction involving nutrients produced by the plant, harvested by the phytopathogen and exploited by P. flocculosa. It would thus constitute a tripartite interaction. Our hypotheses are as follows: 1. P. flocculosa will only develop on powdery mildew colonies/spores present on a living plant; it will not antagonize powdery mildew spores separated from their host 2. The development of P. flocculosa is stopped as soon as the powdery mildew spores are ruptured thus interrupting the flow of nutrients from the plant. This shows that Pseudozyma flocculosa is somehow dependent on the plant and taking the nutrients from the plant through the powdery mildew fungus. These hypotheses lead to the following questions: 1. What are the factors that could be responsible for this kind of interaction between P. flocculosa and B. graminis? 2. How does P. flocculosa recognize B. graminis as a host?
3. What are the genes involved in this process, i.e., that are activated or suppressed during this process?
10
8. Objectives
Based on the available literature, few studies have been carried out to determine the virulence factors of a BCA against another fungus, mainly in the context of a tripartite interaction. Our major objective is to understand the biocontrol mechanisms of Pseudozyma flocculosa against B. graminis.
The present work aims to unravel the genetic determinants associated with the process of suppression of Bgh and to better understand the mode of action of P. flocculosa to improve its effectiveness as a biological control agent. In short, the objectives are designed to reveal the most interesting properties associated with the biology of the biocontrol agent.
The specific objectives are:
1) To optimize the methodology of the sampling time and technique of Pseudozyma flocculosa in culture conditions and in biological control condition 2) To acquire a fundamental understanding of the genetic principles that regulate the biocontrol activity of P. flocculosa against B. graminis. 3) To identify the gene (s) responsible for the tripartite interaction P. flocculosa‐ Bgh – Hordeum vulgare
CHAPTER 2
MANUSCRIPT
13 Transcriptomic analysis of the tripartite interaction Pseudozyma flocculosa‐ Blumeria graminis f.sp. hordei‐Hordeum vulgare
ABSTRACT
Blumeria graminis f. sp. hordei (Bgh) is a powdery mildew fungus that infects barley and can reduce crop yield by as much as 40%. The epiphytic fungus Pseudozyma flocculosa, is often found in close association with powdery mildew of clover leaves and exhibits a strong antagonistic activity by rapidly destroying the invasive structures of the pathogen. The objective of this work was to understand the molecular mechanisms dictating the interaction between a biological control agents (BCA) P. flocculosa and a plant pathogen (Bgh). In the present study, to understand gene expression dynamics during a host‐pathogen‐BCA interaction, a complete RNA‐seq transcriptome profiling was performed on the tripartite interaction. The transcriptome profiling strategy was used to understand the genetic determinants of the interaction involving P. flocculosa, the pathogen Bgh, and the host plant, barley. Our results determined the subtle changes in P. flocculosa gene expression under in vitro and biocontrol conditions. The analysis of differentially expressed genes (DEGs) as performed with an initial emphasis on 200 unique candidate secretory effector proteins (CSEPs) of P. flocculosa in an attempt to determine their role in influencing its interaction with barley powdery mildew. Over 30 CSEPs were upregulated during P. flocculosa interaction with Bgh, including pf02826 that had a near 1000‐fold change. These results suggest strongly that CSEPs are involved in the biocontrol activity of P. flocculosa, and represent, to our knowledge, the first such report for a fungal‐fungal interaction.
15 RÉSUMÉ
Blumeria graminis f. sp. hordei (Bgh) est l’agent pathogène causant le blanc de l’orge. Il peut à lui seul, causer des pertes allant jusqu’à 40% dans cette culture. Un champignon épiphyte, Pseudozyma flocculosa, a été découvert et identifié en 1987 en association étroite avec le blanc du trèfle. Les chercheurs ont alors remarqué que ce champignon exprimait une forte activité antagoniste contre le blanc en détruisant les structures de l’agent pathogène. L’objectif de ce travail était de comprendre les mécanismes moléculaires sous‐ jacents de l’interaction de lutte biologique entre P. flocculosa et l’agent pathogène B. graminis f.sp. hordei. Pour ce faire, une analyse transcriptomique complète par séquençage des cDNA (RNA‐Seq) des trois protagonistes de l’interaction tripartite a été effectuée. Une analyse des gènes exprimés de façon différentielle a été effectuée avec une attention toute particulière à la classe des protéines candidates effectrices ou plus simplement, CSEPs. Il en est ressorti que 30 CSEPs, sur les 200 étudiées, présentaient de grandes différences d’expression de la part de P. flocculosa lorsqu’il se trouvait en absence ou en présence de B. graminis. Par exemple, la protéine CSEP pf02826, avait une expression près de 1000 fois supérieure lorsque le P. flocculosa était en présence de blanc. Ce résultat met en évidence que les CSEPs sont impliquées dans l’activité de lutte biologique de P. flocculosa. De même, pour une première fois, nous mettons en lumière l’implication de protéines effectrices dans une interaction champignon‐champignon.
17 INTRODUCTION
Barley is one of the most widely grown crops in the world. The barley powdery mildew fungus, Blumeria graminis f. sp. hordei (E.O. Speer DC) (Bgh) is an obligate biotrophic pathogen defined as a serious barley disease worldwide where it can reduce yield by as much as 40%. This plant pathogen is able to overcome host defense mechanisms and subsequent immunity manifested during formation of the intracellular feeding structure of the fungus, the haustorium.
Because of their ectotrophic growth, powdery mildews are readily exposed to natural enemies and a few fungal species have been tested for their potential as biocontrol agents (Kiss 2003). Among them, Pseudozyma flocculosa (Traquair, Shaw and Jarvis) has a significant antagonistic activity against powdery mildews (Avis et al., and Paultiz et al., 2001). Pseudozyma flocculosa was discovered in 1987 and originally identified as Sporothrix flocculosa, an ascomycetous yeast (Traquair et al., 1988) and later reclassified as a basidiomycete related to the anamorphs of the Ustilaginales (Begerow et al., 2000). While an effective antagonist of powdery mildews, its specific activity toward this particular group of plant pathogens appears to be a lot more intricate and complex than what was hypothesized in the literature. Earlier studies with P. flocculosa showed that it was neither a strong competitor in the phyllosphere nor a direct hyperparasite, since no direct contact between the biocontrol agent and target powdery mildew was required to observe an antagonistic activity. Powdery mildew cells exposed to P. flocculosa suffered rapid plasmolysis, which led to the conclusion that antibiosis was its mode of action. This was further supported by the discovery of an unusual glycolipid produced by P. flocculosa, called flocculosin that exhibited strong antifungal activity (Mimee et al., 2005).
The structure of flocculosin is highly similar to the structure of ustilagic acid produced by the plant pathogen Ustilago maydis. This finding brought a direct link to the reclassification of P. flocculosa with the Ustilaginales. With the availability of U. maydis genome in 2006 (Kämper et al., 2006) and P. flocculosa genome, the gene clusters responsible for the synthesis of ustilagic acid and flocculosin were identified (Teichmann et al., 2010). More recently, in 2013, a comparative analysis of P. flocculosa to the genomes
18
of U. maydis, Sporisorium reilianum and Ustilago hordei highlighted similarities and differences with respect to effector proteins (Lefebvre et al., 2013). The comparative genomic analysis of phylogenetically‐related species revealed a higher conservation of virulent secreted proteins in the three pathogens and a near complete loss in P. flocculosa. These results highlighted that the main difference between phytopathogenic Ustilaginales and P. flocculosa could be attributed to a few specific effector proteins, thus confirming the important role of such proteins in defining plant‐pathogen interactions. The focusing on potential effectors revealed that, in comparison to U. maydis, the genome of P. flocculosa has nearly the same number of predicted secreted proteins. For its part, P. flocculosa possesses 200 specific CSEPs where no orthologs are found in closely related species.
The centrality of effector proteins in the biology of plant pathogenic microbes is demonstrated by the presence of vast arrays of effector‐like genes that are found in practically all pathogen genomes. This is particularly striking in the genomes of the obligate biotrophic fungi that cause powdery mildews (Spanu et al., 2010; Wicker et al., 2013). On the other hand, Lefebvre et al. (2013) identified some unusual genes unique to P. flocculosa that could account for the elusive properties linked to its biocontrol activity. P. flocculosa also possesses two NPP1 containing proteins, absent in all pathogenic Ustilaginales. These observations led us to the hypothesis that features unique of P. flocculosa would be responsible for its specificity towards barley powdery mildew.
In order to define and understand the spectrum of genes involved in the biocontrol properties of P. flocculosa, transcriptomic analysis could represent a powerful tool to identify the specific factors defining the interaction between P. flocculosa and powdery mildew. The technique is essential as a first step to implicate novel genes and pathways that may be involved or associated with a particular condition. Transcriptomic data from next generation sequencing technology gives us information about the activity of genes that change their expression pattern in response to a signal originating from the host plant or in the host tissue and may reveal mechanisms of pathogenesis and biocontrol activity as initiated by fungal pathogens and fungal BCAs. This study aimed to identify the differential gene expression pattern in P. flocculosa in response to barley powdery mildew (Bgh) by transcriptome analysis. The main
19 objective of the work was to uncover genes responsible for the biocontrol activity of P. flocculosa and specificity towards barley powdery mildew with a specific emphasis on those coding for effector proteins. MATERIALS AND METHODS Plant material Barley (Hordeum vulgare) plants cv. Foster displaying high susceptibility to Bgh were used for this study. Barley seeds were sown in the greenhouse. Three weeks later, the seedlings were exposed to natural infection with Bgh. The fungus was allowed to grow on the surface of leaves under moist conditions for 4‐5 days until it covered 30‐40% of the leaf surface. Fungal material Pseudozyma flocculosa was grown in yeast malt peptone dextrose broth (YMPDB) and harvested at four different time points: 2h, 8h, 18h and 30h spanning the different growth phases of the fungus (Hammami et al., 2011). This fungal material was used as control for P. flocculosa grown in biological control condition. Inoculation with Pseudozyma flocculosa
From a 3‐day‐old culture, sporidia titer was adjusted to 1 x 107 cfu/ml and inoculated on the plants highly infected with Bgh. Water from the sporidia solution was left to evaporate for 20 minutes and the plants were covered with plastic bags to maintain a high humidity level. Barley mildew leaf samples were collected at 12h, 24h and 36h post inoculation. Water sprays were used as control. RNA isolation Total RNA was isolated from control, powdery mildew infected, and P. flocculosa growing on powdery mildew of barley infected leaves, and flask cultures of P. flocculosa at different time points using trizol followed by RNeasy mini kit from Qiagen. Concentration and purity of the extracted RNAs were subsequently measured by Nanodrop and also evaluated by gel electrophoresis on 0.8% agarose gels at 130V and stained with ethidium
20
bromide. The RNA was checked for integrity on a Bioanalyzer 2100 algorithm (Agilent Technologies) before making the cDNA libraries.
cDNA library construction
Library construction was done for all the samples with the Illumina® TruSeq® RNA Sample Preparation Kit v2. This kit was used to convert the mRNA in total RNA into a library of template molecules suitable for subsequent cluster generation and DNA sequencing. The first step in library preparation involves purifying the poly‐A containing mRNA molecules using poly‐T oligo‐attached magnetic beads. Following purification, the mRNA is fragmented into small pieces using divalent cations under elevated temperature. The cleaved RNA fragments are copied into first strand cDNA using reverse transcriptase and random primers. This is followed by second strand cDNA synthesis using DNA polymerase I. These cDNA fragments then go through an end repair process, the addition of a single ‘A’ base, and then ligation of the adapters. The products are then purified and enriched by PCR to create the final cDNA library. The cDNA was checked for integrity before performing the sequencing process on a Bioanalyzer 2100 (Agilent Technololgies). RNA sequencing Replications and randomization are essential components of a well‐planned and properly analyzed RNA‐seq design. In our study we used five replications for each sample at different time points and before library preparations all the samples were randomized to different groups and each group contained six individual libraries. RNA sequencing was performed on 48 cDNA library samples. We performed multiplexing and each cDNA library was labeled or barcoded with sample specific sequences that allow multiple samples to be included in the same sequencing reaction while maintaining high fidelity sample identities downstream. Six libraries were pooled together while maintaining a proper ratio of RNA quantity for each library and then sequenced in one lane of a flow cell using Illumina HiSeq 2000 sequencing technology. Two sequencing runs were performed at the McGill University and Génome Québec Innovation Centre (McGill University, Montréal, Canada).
21 Mapping reads to the reference genome
Raw sequences in FASTQ format obtained from the sequencing platform were analyzed using CLC Genomics Workbench v8.0.1 (CLC bio, Aarhus, Denmark). Low‐quality bases (Q < 15) were trimmed from both ends of the sequences and the adapters were trimmed and the processed reads were used for further analysis. The sequences were mapped to the respective reference genomes of P. flocculosa, Bgh and barley using a series of programs, including Bowtie2 for short‐read mapping and TopHat v1.3.3 for defining exon–intron junctions. The updated genome sequences and annotations of Bgh can be found in http://www.blugen.org/. The complete genome sequence of P. flocculosa, assembled into 1,281 scaffolds, was used for protein identification based on homology with U. maydis sequences (Lefebvre et al., 2013). The whole‐genome shot gun sequencing information was obtained from GenBank under the accession number AOUS00000000.
The recent genome sequence of barley was obtained from
http://plants.ensembl.org/Hordeum_vulgare. The principal component analysis (PCA) was performed by CLC Genomics Workbench v8.0.1 after mapping as a measure of quality control to check whether the overall variability of the samples reflect their grouping.
Genome annotation
Gene functions were predicted using InterproScan v4.8 (database v38.0) (Quevillon et al., 2005). Annotation of CAZymes was performed using the dbCAN Web server (Yin et al., 2012). A search for genes involved in the biosynthesis of secondary metabolites was performed using JCVI Secondary Metabolite Unique Regions Finder Web server (SMURF) (Khaldi et al., 2010). Annotation of secreted proteins and CSEPs was accomplished according to the method described by Mueller et al. (2008). Secreted proteins were selected based upon SignalP v3.0 (Bendtsen et al., 2004) D‐value and Dmax cutoffs, TargetP v1.1 (Emanuelsson et al., 2000) predicted location of proteins, TMHMM v2.0 (Krogh et al., 2001) predicted the number of transmembrane domains and position according to cleavage site, and finally, correlation to LocDB or PotLocDB ProtComp v9.0 (http://www.softberry.com) databases. Based on InterproScan‐assigned domains, proteins lacking enzymatic functions were classified as candidate effectors (CSEP).
22
Identification of differentially expressed genes
The Illumina HiSeq reads were normalized and the expression level of each transcript was expressed as the number of reads per transcript kilobase per million fragments mapped (RPKM) value, which was calculated based on the number of mapped reads. A fold change >4 and FDR‐corrected p‐value <0.05 was used as a parameter to detect differentially expressed genes at different conditions in each library based on RPKM values. A gene ontology (GO) term was assigned to each transcript based on the GO annotations for biological process in each of the reference genome. The GO annotations of P. flocculosa can be found in from GenBank under the accession number AOUS00000000.
23 RESULTS
Characteristic growth of Pseudozyma flocculosa in culture conditions.
Pseudozyma flocculosa growth was analyzed in YMPD culture conditions and characterized morphologically. The different growth stages of P. flocculosa showing germination, mycelial growth and sporidia formation were monitored at various time points (Figure 1). Time 0 corresponds to the biological status of P. flocculosa in a subculture from an actively growing, 72h‐old liquid culture. At this stage, the fungus is present only in the form of ovoid spores called sporidia. After 4 h, in a fresh medium containing the necessary nutrients for their development, sporidia germinate and develop hyphae. At 8 h, the culture expands in mycelial growth, with no residual sporidia. After 12 h, mycelial filaments started producing new sporidia, a process that continued over the next 24 h. After 18h, the presence of needle‐like structures was observed, a clear indication of flocculosin production by the fungus. After 36 h, only sporidia could be observed. Based on these observations, the spore phase (2h), exponential growth phase (8h) and flocculosin production phase (18h) were considered as distinctive growth phases of P. flocculosa and selected for transcriptomic studies. Figure 1: Characterization of Pseudozyma flocculosa in culture condition. Growth and developmental phases of P. flocculosa cultured in YMPD medium over 72 h. Calibration of P. flocculosa in response to powdery mildew fungus Barley plants cv. Foster grown in a greenhouse showed clear signs of infection after two weeks (Figure 2). For their part, powdery mildew colonies inoculated with P.
24 flocculosa were covered by the BCA as early as 12 h after inoculation (Figure 3). Over time, the powdery mildew fungal structures appeared to be embedded in a mycelial network that looked like a "spider web" (24h), which led to their progressive and complete collapse at 36h.
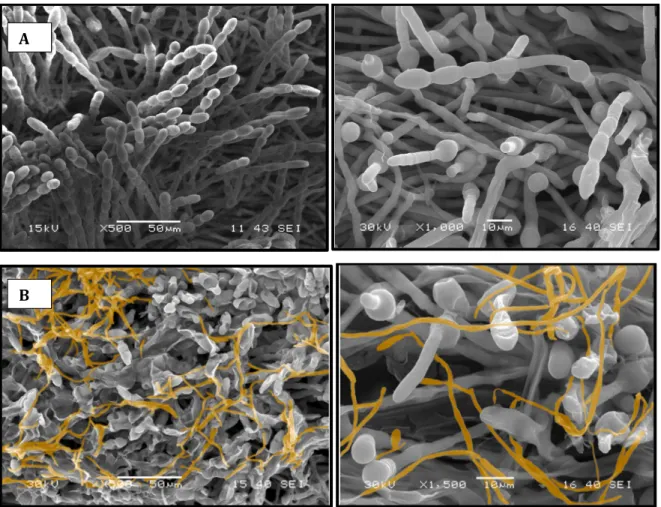
Electron microscopy results clearly highlighted the destruction of powdery mildew conidia by P. flocculosa over time (Figure 4). These results clearly showed that P. flocculosa progressively invaded the colonies of powdery mildew leading to their collapse over time. Figure 2: Development of powdery mildew disease on barley plants. (a) Two week‐ old Hordeum vulgare plants cv. Foster grown in greenhouse (b) plants exposed to natural infection with Blumeria graminis f.sp. hordei.
25 A B Figure 3: Antagonism of Pseudozyma flocculosa on barley powdery mildew colonies over time. (A) Barley leaf samples sprayed with water as a control; (B) barley leaf samples inoculated with biocontrol agent P. flocculosa. T0h T12h T24h T36h T0h T12h T24h T36h
26
Figure 4: Scanning electron microscopy observations of barley powdery mildew fungus (A) before and (B) after treatment with Pseudozyma flocculosa. Transcriptional dynamics of powdery mildew fungus in response to P. flocculosa RNA integrity, cDNA library validation and sequencing The leaf samples collected at various time points pre and post‐inoculation with P. flocculosa are presented in Table 1. Quality of the samples was assessed with a bioanalyser and found to be very high (Figure 5). In total, 3200 million reads, each 100 nucleotides long, were generated, with approximately 200 million reads from each lane. In total, 2% of the reads aligned to rRNA and were removed prior to mapping to reference genomes.
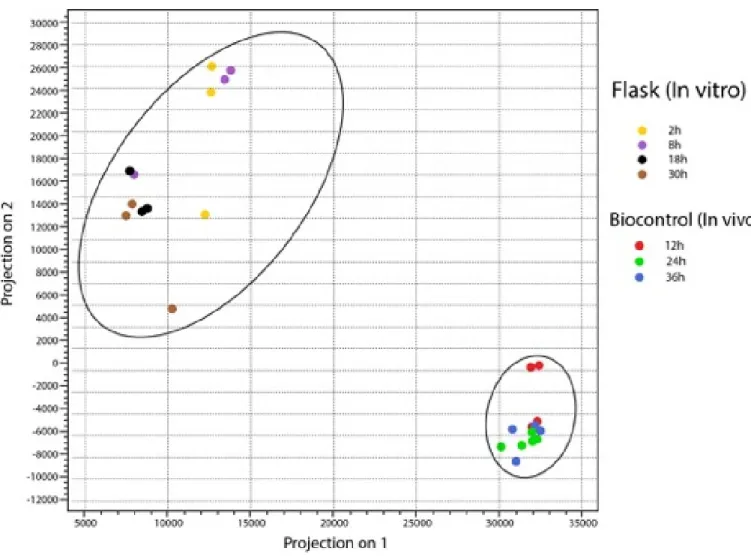
The principal component analysis (PCA) performed on P. flocculosa samples revealed a distinct difference between the in vitro and biocontrol conditions (Figure 6 a). In the same manner, there was a clear clustering of the different treatments along with the replications associated with each interaction (Figure 6 b).
A
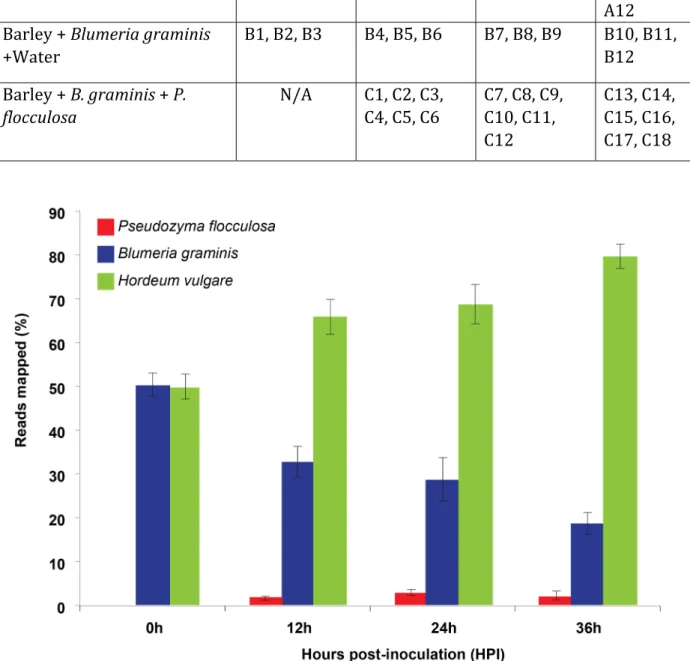
27 The processed reads were then aligned to the P. flocculosa reference genome. The remaining reads were mapped against the genome of B. graminis and the process repeated against the barley reference genome (Figure 6). At time 0 h, before inoculation with P. flocculosa, the reads mapped in equal proportions between B. graminis and barley. At 12 h, the reads that mapped to the P. flocculosa genome represented around 1% and this was accompanied by a decrease and increase in the respective reads of B. graminis and barley. The same trend was observed at 24 h for the pathogen and the plant, while P. flocculosa reads more than doubled. Interestingly, the differences between B. graminis and barley amplified at 36 h but reads associated with P. flocculosa receded (Figure 7).
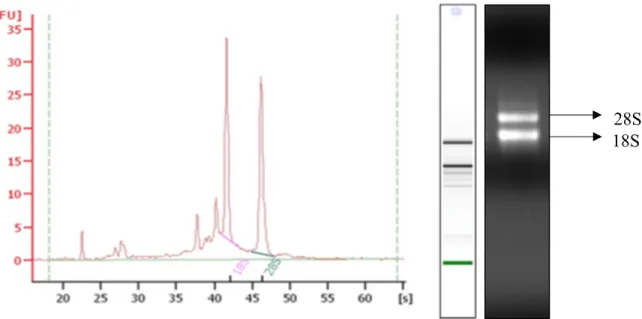
Figure 5. Total RNA isolation from barley leaf samples and integrity check using bioanalyser. Total RNA isolation was performed on barley leaf samples collected at various time points using trizol followed by RNeasy Mini Kit from Qiagen. The integrity of the RNA was measured with an Agilent bioanalyzer.
28S 18S
28 Figure 6 a. Principal Component Analysis (PCA) of P. flocculosa samples grown in vitro and in vivo. Principal component analysis generated from the gene expression data of P. flocculosa in artificial culture media in comparison with biological control conditions. Each color dot in the plot represents the samples and their replicates collected at different time points.
29 Figure 6 b. Principal Component Analysis (PCA) of barley leaf samples collected at various conditions. Principal component analysis generated from the gene expression data of the samples of barley, the interaction Blumeria graminis f.sp. hordei ‐barley and P. flocculosa‐B. graminis‐barley collected under different conditions. Each color dot in the plot represents the samples and their replicates collected at different time points.
30
Table 1. Sample collection pre and post‐inoculation of barley leaves with Pseudozyma flocculosa and Bgh. The leaf samples were collected at four different time points under three different conditions. At each time point 3‐6 biological replicates were collected for statistical significance. Figure 7. Tripartite interaction mapping. Distribution of the total reads (rRNA reads removed) on to the reference genome of Hordeum vulgare, Blumeria graminis f.sp. hordei and Pseudozyma flocculosa before the inoculation with the biocontrol agent (sterile water
Samples Time 0 Time 12
hours
Time 24 hours
Time 36 hours
Barley (Green leaf) A1, A2, A3 A4, A5, A6 A7, A8, A9 A10, A11,
A12 Barley + Blumeria graminis +Water B1, B2, B3 B4, B5, B6 B7, B8, B9 B10, B11, B12 Barley + B. graminis + P. flocculosa N/A C1, C2, C3, C4, C5, C6 C7, C8, C9, C10, C11, C12 C13, C14, C15, C16, C17, C18
31 used as control) and hours post‐inoculation (hpi) with the biocontrol agent P. flocculosa at 12, 24 and 36 hours.
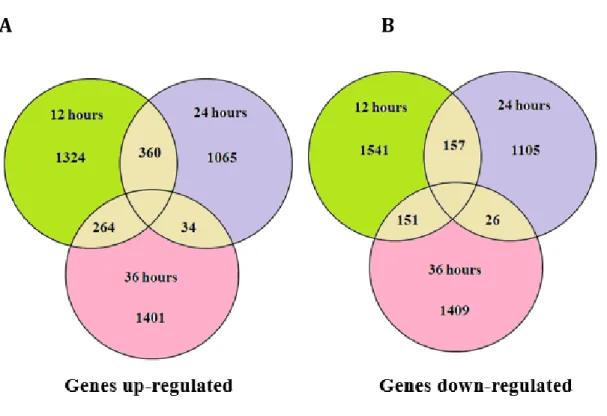
Differential gene expression pattern in P. flocculosa
Differentially expressed genes (DEGs) were detected between in vitro grown P. flocculosa and P. flocculosa growing on Bgh. Genes with a false discovery rate (FDR‐BH) less than 0.05 and fold change greater than 4 were considered to be differentially expressed. Our analysis revealed that 1948 genes, 1459 genes, and 1699 genes were differentially expressed and upregulated in P. flocculosa in biocontrol conditions at 12, 24 and 36 h, respectively (Figure 8a). At the same time, 1541, 1105 and 1409 genes were differentially expressed and down regulated in P. flocculosa in biocontrol conditions at 12, 24 and 36 h, respectively (Figure 8b).
A
B
Figure 8. Differential gene expression pattern of Pseudozyma flocculosa genes. Differential gene expression pattern of P. flocculosa genes in biological control conditions against Blumeria graminis f.sp. hordei in comparison with in vitro cultures. The Venn diagram indicates the number of genes that are differentially expressed at each specific time point. Intersection region in the Venn diagram indicates the number of genes
32
expressed in common between the time points. A. Differentially upregulated genes B. Differentially down regulated genes.
Differential gene expression pattern of genes involved in flocculosin production Flocculosin is a cellobiose lipid with antifungal properties, and initially thought to be involved in the biocontrol activity of P. flocculosa. The expression level of 11 genes involved in flocculosin production by P. flocculosa is shown in Figure 9. Compared to in vitro conditions, no genes of the flocculosin cluster appear to be highly expressed on Bgh, although a lower but consistent level of expression is seen for all genes.
Figure 9. Differential gene expression pattern of flocculosin producing genes. Differential gene expression pattern of genes found in the flocculosin cluster between biological control conditions against Blumeria graminis f.sp. hordei (red) and in vitro
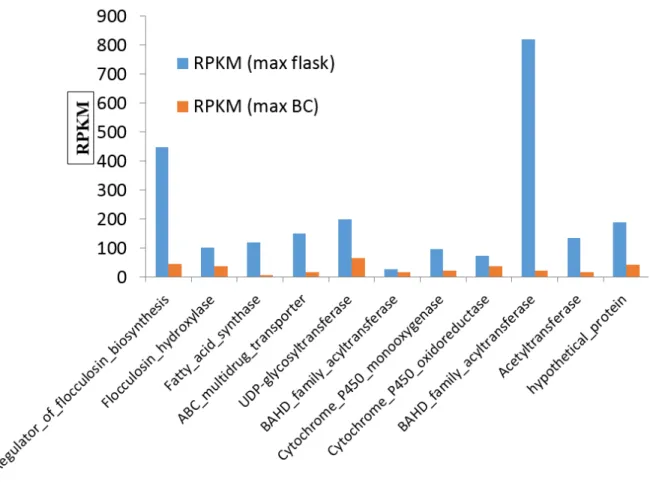
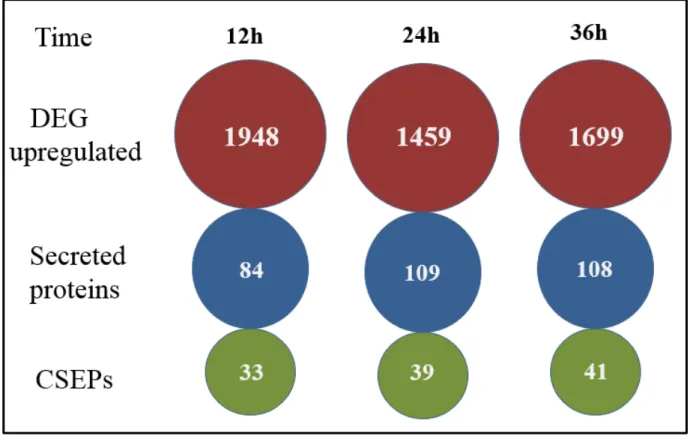
33 cultures (blue). The expression level of the 11 genes is expressed in RPKM (Reads Per Kilobase per Million mapped reads). P. flocculosa effector candidates are differentially expressed during infection with B. graminis f.sp. hordei Effector proteins are key factors in establishing the interaction between a plant and a pathogen but little is known of their role between two interacting fungi. Here, we present results derived from the analysis of the 200 unique CSEPs in P. flocculosa reported by Lefebvre et al. (2013). Among the effector genes, 33, 39 and 41 CSEPs were found to be specifically upregulated at 12, 24 and 36 h, respectively (Figure 10). Of this group, pf02826 had the highest fold change reaching nearly 1000. In total, 27 CSEPs had more than a six‐ fold change, suggesting that CSEPs played an important role in the interaction of P. flocculosa with Bgh (Figure 11).
Figure 10. Upregulation of effector candidates at different time points. Upregulation of candidate secreted effector proteins (CSEPs) in Pseudozyma flocculosa in biological
34 control condition against Blumeria graminis f.sp. hordei at different time points of 12, 24 and 36 hours post inoculation with P. flocculosa.
Figure 11. Differential gene expression pattern of effector candidates. Differential gene expression pattern of candidate secreted effector proteins (CSEPs) in Pseudozyma flocculosa between biological control condition against Blumeria graminis f.sp. hordei (BCA) and in vitro cultures (flask) condition.
35 Discussion
Transcriptomic analysis of the molecular interactions between a plant (Hordeum vulgare) – a pathogen (Blumeria graminis) – a biological control agent (Pseudozyma flocculosa) showed the differentially expressed genes involved in the biological control activity of P. flocculosa. This atlas of differentially expressed genes revealed the role and importance of candidate secreted effector proteins (CSEPs) in the antagonism of P. flocculosa toward a powdery mildew fungus. These results are part of a comprehensive effort aimed at understanding the molecular crosstalk in the tripartite interaction between a plant, pathogen and biocontrol agent.
Pseudozyma flocculosa is an effective antagonist of powdery mildews but its specific activity toward this particular group of plant pathogens is intricate and complex. It cannot parasitize plants but is a powerful antagonist of powdery mildews (Jarvis et al., 1989; Hajlaoui and Bélanger, 1993; Clement‐Mathieu et al., 2008). In earlier classification, all smut fungi were ecologically characterized by their ability to infect plants and shared a similar life cycle with a yeast‐like haploid phase and a parasitic dikaryophase, culminating in the production of numerous powdery black teliospores, hence, their common name (Begerow et al., 2006). However, a number of anamorphic fungi lacking sexual development, initially placed in deuteromycetous taxa, were found to be morphologically and phylogenetically related to the Ustilaginales. In order to integrate these anamorphs into the general phylogenetic system of Ustilaginomycetes, Begerow et al. (2000) analyzed and compared diagnostic ribosomal DNA sequences of teleomorphic and anamorphic species of Ustilaginomycetes. Their analyses confirmed that species of Pseudozyma and Ustilaginales parasitizing grasses form a monophyletic group. Pseudozyma species thus represent the sole known members of the Ustilaginales that cannot parasitize plants.
Pseudozyma flocculosa was grown in artificial media for morphological characterization at different time points. Obligate biotrophs like B. graminis present a number of challenges since they cannot be cultured outside their host; their life cycle is closely tied with the infection process and the host’s response. P. flocculosa only grows as an epiphyte on the leaf surface, but will rather develop extensively when in presence of powdery mildew colonies. With these challenges in hand, it was difficult to optimize the
36
time points for the growth of P. flocculosa in biological control conditions. Preliminary experiments and small‐scale sequencing (MiSeq) were performed to optimize a bioassay so it could be used for transcriptomic analysis. Based on the results of these experiments and the growth of P. flocculosa in in vitro conditions, the fungus was inoculated on plants and the samples were collected 12, 24 and 36 hours post‐inoculation (hpi) with P. flocculosa. The microscopy results showed distinct differences between the leaf samples inoculated with water and P. flocculosa. Destruction of conidia of Bgh by P. flocculosa was clearly visible as early as 12 hpi and complete at 36 hpi in electron microscopy. These results justified our time points chosen for sample collection for transcriptomic analysis. Earlier studies about P. flocculosa showed that flocculosin is an active molecule involved in the mode of action of P. flocculosa (Mimee et al., 2005). Following the discovery of flocculosin and its structural similarity with ustilagic acid, a cluster of 11 genes responsible for flocculosin production was identified (Teichmann et al., 2010). These results highlight that the production of unusual glycolipids by two related yet disparate organisms is the result of an intricate and well‐conserved enzymatic process exclusive to the two studied fungi. From a biological or evolutionary point of view, one has to assume that conservation of this gene cluster serves a distinct purpose, even though evidence to that effect is still lacking. In our transcriptomic analysis the expression level of the 11 genes within the gene cluster responsible for flocculosin production was high when P. flocculosa was cultured in artificial media in comparison with the biocontrol condition. These results proved that P. flocculosa relies on mechanisms other than flocculosin production to antagonize barley powdery mildew as previously suggested by Marchand et al., (2007).
Following the release of the first fungal genomes and the development of reliable bioinformatics tools to predict secretion signals in protein sequences, emphasis has been placed on the study of effector proteins as determinants of pathogenicity (Torto et al., 2003). In a variety of plant pathogens, including smut fungi, powdery mildews, rusts, and oomycetes, effectors have been found to affect virulence, suppress plant defense responses, dictate host specificity, and/or to maintain a biotrophic interaction. Based on the method described by Mueller et al. (2008), 547 secreted proteins were identified in P.
37 flocculosa. Among them, 345 could not be assigned an enzymatic function and were therefore considered as CSEPs. Of those, 200 were found to be unique to P. flocculosa. In our differential gene expression analysis, we closely followed the 200 CSEPs that are unique to P. flocculosa. Our results showed that many of them are upregulated when P. flocculosa antagonizes powdery mildew structures. One of them had nearly a 1000‐fold change, which would classify it as a good candidate to investigate further for functional studies. Taken together, these results strongly suggest that effector proteins play a very important role in the interaction of P. flocculosa with barley powdery mildew.
In conclusion this is the first study of transcriptomic analysis of a tripartite interaction plant‐pathogen‐biocontrol agent. RNA‐seq results highlighted the global changes in the gene expression pattern in P. flocculosa in response to the pathogen and the host. In particular, DGE pattern analysis gave us the atlas of effector proteins possibly involved in the interaction between P. flocculosa and Bgh. This investigation of the role of effector proteins in such interactions opens new opportunities toward understanding the potential factors involved in biological control. To our knowledge, this is the first report linking effector proteins in fungal‐fungal interactions. ACKNOWLEDGEMENTS We thank Institut de Biologie Integrative et des Systems (IBIS) at Laval University for their technical assistance. This work was supported by grants from the Natural Sciences and Engineering Research Council of Canada, Centre SÈVE and the Canada Research Chairs Program to R.R.B
REFERENCES
Avis, T.J. and Bélanger, R.R. (2001). Specificity and mode of action of the antifungal fatty acid cis‐9‐heptadecenoic acid produced by Pseudozyma flocculosa. Applied and Environmental Microbiology, 67: 956‐60.
Begerow, D., Bauer, R. and Boekhout, T. (2000). Phylogenetic placements ustilaginomycetous anamorphs as deduced from nuclear LSU rDNA sequences. Mycological Research, 104: 53‐60.
Begerow, D., Stoll, M. and Bauer, R. (2006). A phylogenetic hypothesis of Ustilaginomycotina based on multiple gene analyses and morphological data. Mycologia, 98: 906‐91.