Gènes impliqués dans le «shedding» des
protéines GPI chez Saccharomyces cerevisiae
Mémoire
Marc Bélanger
Maîtrise en biochimie
Maître ès sciences (M.Sc.)
Québec, Canada
© Marc Bélanger, 2016
iii
Résumé
Les organismes eucaryotes possèdent plusieurs mécanismes de contrôle qualité en vue d'assurer leur survie en condition de stress. La réponse aux protéines mal repliées (UPR) est un mécanisme à la base de différents volets ayant pour but de ramener l'organisme vers l'homéostasie. L’étude qui suit vise à caractériser plus en profondeur un volet de cette réponse qui est lié non pas à la présence de protéines mal repliées, mais plutôt à des défauts dans l’homéostasie des lipides de la cellule. Notre étude a mis en évidence qu’une forme précise de la protéine à ancrage GPI modèle Yps1p est relâchée de manière importante dans des souches mutantes connues pour déclencher ce volet de la réponse UPR. Nous avons aussi montré que cette relâche ne semble pas être causée par le déclenchement de l’UPR et que des phospholipases sont responsables du clivage de l’ancrage GPI. Une défaillance ou une insuffisance de l’UPR est associée à plusieurs pathologies humaines, dont le diabète de type 2, d'où l'intérêt d'en étudier les différentes facettes.
v
Table des matières
Résumé ... iii
Table des matières ... v
Liste des tableaux ... vii
Liste des figures ... ix
Liste des abréviations ... xi
Remerciements ... xiii
1. Introduction ... 1
1.1. Maturation et transport des protéines ... 1
1.2. Le contrôle qualité des protéines ... 3
1.2.1. La réponse aux protéines mal repliées ... 3
1.2.2. Maladies associées à l’UPR ... 5
1.2.3. L’endoplasmic-reticulum associated degradation ... 6
1.2.4. Les yapsines ... 8
1.3. La membrane plasmique ... 8
1.4. Les protéines à ancrage glycosylphosphatidylinositol ... 10
1.4.1. Le remodelage de l'ancrage GPI ... 10
1.5. Les aspartyl peptidases ... 13
1.6. Les protéines Yps1 et Yps1-DL2 ... 13
1.7. Les mutants de classe 3 ... 13
1.7.1. Lien entre la relâche des protéines GPI et l’UPR ... 14
1.8. Enzymes pouvant mener à la relâche des protéines GPI ... 14
1.8.1. Les phospholipases ... 14
1.8.2. Les peptidases ... 15
Hypothèses et objectifs ... 15
2. Matériel et méthodes ... 17
2.1. Micro-organismes et milieux de culture ... 17
2.1.1. Bactéries utilisées et milieu de culture ... 17
2.1.2. Levures utilisées et milieux de culture ... 17
2.1.3. Construction de souches contenant plusieurs délétions géniques par dissection de tétrades ... 26
2.1.4. Construction de souches contenant plusieurs délétions géniques par Synthetic Genetic Array ... 28
vi
2.2. Acides nucléiques ... 29
2.2.1. Plasmides utilisés ... 29
2.2.2. Préparation d’ADN plasmidique ... 31
2.2.3. Transformations ... 31
2.2.4. Extraction d’ADN génomique de levure ... 31
2.2.5. Amplification PCR ... 32
2.3. Protéomique ... 34
2.3.1. Concentration des protéines du surnageant de culture et extraction des protéines de levures ... 34 2.3.2. Traitement à l’Endo Hf ... 34 2.3.3. SDS-PAGE ... 34 2.3.4. Transfert ... 35 2.3.5. Immunobuvardage Western... 35 2.3.6 Analyse statistique ... 36 3. Résultats ... 37
3.1. Relâche de Yps1p dans des mutants connus pour déclencher l'UPR ... 37
3.2. Relâche de Yps1p dans le milieu extracellulaire en l’absence de l’UPR ... 40
3.3. Création de souches par SGA ou intégration d'un fragment PCR ... 42
3.4. Relâche de Yps1-DL2 dans les mutants de classes 3 en l'absence de certaines phospholipases ... 42
3.5. Relâche de Yps1-DL2 dans les mutants de classes 3 en l'absence de certaines peptidases ... 43
4. Discussion ... 47
4.1. Des problèmes dans l’homéostasie des lipides et le déclenchement de l'UPR semblent liés à la relâche de Yps1-DL2 indépendamment de son activité ... 47
4.2. Le déclenchement de l’UPR n’est pas majoritairement responsable de la relâche de Yps1p dans le milieu extracellulaire ... 48
4.3. La relâche de Yps1-DL2 dans les mutants de classe 3 semble liée à l'action de phospholipases ... 49
4.4. Relâche des protéines à ancrage GPI dans les souches opi3∆... 50
4.5. La relâche de Yps1-DL2 dans les mutants de classe 3 semble indépendante de l’action de peptidases ... 52
5. Conclusion et perspectives ... 55
Bibliographie ... 57
vii
Liste des tableaux
Tableau I : Souches de Saccharomyces cerevisiae utilisées ... 18 Tableau II : Souches de Saccharomyces cerevisiae construites pendant ce projet ... 20 Tableau III : Plasmides utilisés dans ce projet ... 30 Tableau IV : Amorces employées pour amplifier les fragments utilisés pour la délétion
génique et pour la vérification de délétions ... 33 Tableau V : Noms et fonctions des différents gènes utilisés dans cette étude ... 63
ix
Liste des figures
Figure 1 : Représentation schématique de l’entrée d’une protéine dans le RE ... 2
Figure 2 : Représentation schématique de l’activation de l’UPR chez la levure ... 4
Figure 3 : Représentation schématique du mécanisme de l’ERAD ... 7
Figure 4 : Représentation schématique de la composition des radeaux lipidiques... 9
Figure 5 : Représentation schématique d’un ancrage GPI ... 11
Figure 6 : Représentation schématique du remodelage de l’ancrage GPI chez la levure S. cerevisiae ... 12
Figure 7 : La forme endogène de Yps1p est relâchée de manière plus importante dans les mutants déclenchant l’UPR indépendamment du domaine liant les protéines mal repliées de Ire1p... 38
Figure 8 : La forme mutée au site catalytique Yps1-DL2 est relâchée de manière plus importante dans les mutants déclenchant l’UPR indépendamment du domaine liant les protéines mal repliées de Ire1p. ... 39
Figure 9 : La relâche de Yps1-DL2 dans les mutants de classe 3 ne semble pas être une conséquence du déclenchement de l’UPR. ... 41
Figure 10 : La relâche extracellulaire de Yps1-DL2 dans les mutants de classe 3 semble liée à l’action de diverses phospholipases. ... 45
Figure 11 : La relâche extracellulaire de Yps1-DL2 dans les mutants de classe 3 ne semble pas liée à l’action de peptidases. ... 46
Figure 12 : Représentation schématique de l’hydrolyse d’un ancrage GPI par une phospholipase D menant à la production de PA ... 53
xi
Liste des abréviations
°C : Degré Celsius2YT : 2x extrait de levure et tryptone 5-FOA : 5-Fluoroorotic acid
aa : Acides aminés
ADN : Acide désoxyribonucléique ARNm : Acide ribonucléique messagé cm : Centimètre
D.O. : Densité optique DMSO : Diméthylsulfoxyde
dNTPs : Désoxyribonucléotides triphosphates DTT : Dithiothréitol
EDTA : Acide éthylène diamine tetra acétique Endo Hf : Endoglycosidase Hf
ERAD : Endoplasmic reticulum associated degradation g : Gramme
G418 : Généticine
GPI : Ancrage glycosylphosphatidylinositol h : Heure kDa : Kilodalton L : Litre M : Molaire mA : Milliampère min : Minute mL : Millilitre mM : Millimolaire MSG : Glutamate monosodique NaCl : Chlorure de sodium NAT : Nourséothricine nm : Nanomètre
xii
PANTHER : Protein ANalysis THrough Evolutionary Relationships PBST : Tampon phosphate salin contenant du Tween 20
PC : Phosphatidylcholine
PCR : Réaction en chaîne par polymérase PE : Phosphatidyléthanolamine
PEG : Polyéthylène glycol RE : Réticulum endoplasmique rpm : Rotation par minute
S/T : Région riche en sérines et thréonines SC : Synthétique complet
SD : Synthétique dextrose SDS : Sodium dodécyl sulfate
SDS-PAGE : Électrophorèse en gel de polyacrylamide contenant du sodium dodécyl sulfate SGA : Synthetic genetic array
SPM : Milieu de sporulation TGN : Réseau trans-golgien TP : Température pièce U : Unité
UPR : Réponse aux protéines mal repliées
UPRE : Élément de la réponse aux protéines mal repliées URA : Uracile
V : Volt
WT : Type sauvage
YEPA : Extrait de levure, acétate de potassium YNB : Yeast nitrogen base
YPD : Extrait de levure, peptone, dextrose
Yps1-DL : Yps1p dans laquelle la boucle d’insertion a été remplacée Yps1-DL2 : Yps1-DL possédant la mutation D101E
μg : Microgramme μL : Microlitre μm : Micromètre
xiii
Remerciements
Je tiens tout d’abord à remercier mon directeur de recherche, Dr Yves Bourbonnais, pour m’avoir permis de découvrir le monde de la recherche au sein de son laboratoire. Grâce à lui, j’ai pu développer tout au long de ma maîtrise plusieurs compétences essentielles dans ce domaine, tels un esprit critique, un esprit d’analyse et un esprit de réflexion. Ces compétences me suivront tout au long de ma vie et me seront utiles bien au-delà du domaine de la recherche.
Je tiens également à remercier mon co-directeur, Dr Christian Landry, ainsi que les autres membres de mon comité aviseur, Dre Manon Couture et Dr Michel Frenette, pour leurs précieux conseils et le temps qu’ils m’ont accordé tout au long de ma maîtrise.
Je me dois également de remercier Alexandre Dubé, qui m’a formé et m’a appris comment bien travailler dans un laboratoire dès mon arrivée il y a plusieurs années déjà. Il a toujours été présent pour répondre à mes interrogations et pour me fournir des conseils avisés lorsque le besoin se faisait sentir. Bref, je ne serais pas la personne que je suis aujourd’hui s’il n’avait pas été là.
Finalement, je tiens à remercier ma famille et ma conjointe Andrée-Ève pour leur grand support tout au long de mon parcours, surtout dans les moments les plus difficiles. Un merci tout spécial à Andrée-Ève pour son aide constante, pour sa grande compréhension et pour tout le temps qu’elle a pris pour m’aider, que ce soit en lien avec mon projet ou avec tout autre aspect de ma vie. Elle ne m’a jamais laissé tomber et je lui en suis extrêmement reconnaissant.
1
1. Introduction
1.1. Maturation et transport des protéines
Pour qu’une protéine puisse exercer sa fonction convenablement, il est essentiel qu’elle soit non seulement bien repliée, mais aussi qu’elle soit transportée au bon endroit dans la cellule. Pour éviter qu’un nombre important d’aberrations se produisent, toutes les cellules vivantes doivent, par conséquent, avoir des systèmes robustes et efficaces de repliement et de transport des protéines. Dans les cellules eucaryotes, les protéines destinées aux membranes et à la sécrétion entrent dans le réticulum endoplasmique (RE) dès leur synthèse par le ribosome. Cette entrée peut être faite post-traductionnellement si la pro-protéine est synthétisée par un ribosome cytosolique, ou co-traductionnellement si le ribosome est associé directement au RE. Dans ces conditions, le ribosome dirige directement la protéine à l’intérieur du RE pendant son élongation (Brodsky, 1998). L’entrée dans le RE de ces protéines nécessite aussi la présence d’un peptide signal situé en N-terminal de la protéine. Cette séquence de 20 à 30 acides aminés (aa) est reconnue par différentes protéines qui permettent son association au translocon, un canal assurant l’entrée des protéines dans le RE. Le peptide signal sera par la suite enlevé et ne se retrouvera donc pas dans les protéines matures (figure 1) (Haigh et Johnson, 2002). C’est dans le RE que le repliement des protéines a lieu, de même que plusieurs modifications post-traductionnelles telles que l’ajout d’un ancrage glycosylphosphatidylinositol (GPI), un ancrage de lipides et de sucres permettant l’attachement d’une protéine à la membrane plasmique ou à la paroi cellulaire. C’est aussi à cet endroit que débutera la glycosylation (N- ou O-), qui consiste en l’ajout de chaînes glucidiques sur des acides aminés précis. Par la suite, les chaînes glucidiques pourront être modifiées et allongées dans l’appareil de Golgi (Lehle et al., 2006). Les protéines à ancrage GPI, dont il sera question en détail plus loin, sont transportées du RE à l’appareil de Golgi à l’aide de vésicules de transport distinctes de celles utilisées pour les protéines ne possédant pas cet ancrage (Muñiz et al., 2001). Comme tous mécanismes cellulaires, le repliement et le transport des protéines sont loin d’être parfaits. Il a été estimé que, dans des conditions normales, près de 30% des protéines synthétisées sont mal repliées (Schubert et al., 2000).
2
Tirée de: MOLECULAR CELL BIOLOGY 7E, par Harvey Lodish, et al, Copyright 2012 par W.H. Freeman and Company. Utilisé avec l’autorisation de l’éditeur.
3 Pour pouvoir survivre et rester compétitive, la cellule doit posséder des systèmes de contrôle qualité diminuant le stress causé par la présence de telles protéines. En cas de problèmes majeurs, l’un des mécanismes pouvant entrer en jeu est la réponse aux protéines mal repliées (UPR).
1.2. Le contrôle qualité des protéines
1.2.1. La réponse aux protéines mal repliées
La réponse aux protéines mal repliées est une réponse qui permet à la cellule de survivre dans des conditions où le réticulum endoplasmique est sous un stress constant, généralement associé à une accumulation de protéines mal repliées. Dans ces conditions, l’UPR active notamment la transcription de gènes codant pour des protéines chaperonnes, de différents facteurs chargés de la dégradation des protéines causant le stress et d’enzymes de biosynthèse des lipides (Travers et al., 2000; Kimata et al., 2006). Pour activer la transcription de ces gènes, Ire1p, une kinase et une endoribonucléase qui agit comme senseur transmembranaire, est activée soit par la liaison directe de protéines mal repliées ou par la relâche de la protéine chaperonne BiP du domaine luminal de Ire1p (Shamu et al., 1994; Bertolotti et al., 2000). À la suite de son activation, le domaine cytosolique de Ire1p clive une séquence intronique présente dans le pré-ARNm codant pour le facteur de transcription Hac1p, ce qui permet la traduction de cet ARNm en une protéine fonctionnelle (Cox et al., 1993, Mori et al., 1993, Cox et Walter, 1996). Hac1p, en se liant à l’élément de la réponse aux protéines mal repliées (UPRE), activera la transcription des gènes propres à l’UPR mentionnés ci-haut (figure 2) (Cox et Walter, 1996). La régulation de l’UPR semble pouvoir varier et être modulable, permettant à cette réponse de s’adapter plus efficacement à différents stress (Thibault et al. 2011). Cette voie de l’UPR est conservée de la levure aux mammifères et elle est même présente chez la plante (Sidrauski et Walter, 1997; Yoshida et al., 2001, Koizumi et al., 2001, Deng et al., 2011).
4
Vembar et al., 2008
5 Des évidences suggèrent toutefois que les voies de signalisation propres à l’UPR peuvent aussi être déclenchées à la suite d’un défaut dans la composition membranaire, même en absence de protéines mal repliées. En effet, une version mutée d e la protéine Ire1 ne possédant pas de domaine de liaison pour les protéines mal repliées, peut, dans certaines conditions, activer l’UPR presque aussi efficacement que lorsque ce domaine est présent. Il est alors possible de séparer nettement certains gènes déclenchant l’UPR selon l’intensité de la réponse en présence et en absence de ce domaine. Cette séparation permet de remarquer que les mutants déclenchant l’UPR indépendamment du domaine de Ire1p sont des gènes ayant un rôle pouvant mener à un déséquilibre dans l’homéostasie des lipides (Promlek et al., 2011). Chez la levure, seul Ire1p peut déclencher l’UPR, tandis que chez l’homme, il existe deux autres protéines senseurs connues, ATF6 et PERK (Harding et
al., 1999, Shi et al., 1998, Haze et al., 1999). Dans le cas où la cellule serait incapable de
retourner vers l’homéostasie, plusieurs problèmes peuvent survenir chez l’organisme touché.
1.2.2. Maladies associées à l’UPR
Une défaillance ou une insuffisance de l’UPR sont associées à plusieurs pathologies humaines, dont notamment le diabète de type 2 (Doyle et al., 2011, Back and Kaufman, 2012). En temps normal, le corps relâche des quantités importantes d’insuline à la suite de l’entrée de glucose dans le sang. Cette hormone est formée dans le pancréas et a pour but de réduire la quantité de sucre dans le sang et de la conserver à un taux adéquat. Chez un patient atteint de diabète de type 2, les cellules développent une résistance à l’insuline, ce qui empêche, par conséquent, l’entrée du glucose dans les cellules et la réduction du taux de sucre dans le sang. Cette résistance peut être causée par une quantité importante d’acides gras libres, souvent retrouvée chez les individus obèses (Prentki et al., 2006). Dans ces conditions, les cellules du pancréas doivent être constamment actives et à la longue, si elles sont incapables de ramener les taux de glucose à un niveau adéquat, les quantités d’insuline produites diminueront. Cette baisse pourrait être causée par une suractivation de Ire1p, qui se mettrait alors à couper non spécifiquement plusieurs ARNm du RE en conditions de stress importants, incluant celui codant pour l’insuline (Han et al. 2009). Éventuellement, Ire1p agira comme interrupteur pour déclencher l’apoptose des cellules du pancréas (Urano
6
et al. 2000). Il y aura donc de moins en moins de cellules capables de produire de l’insuline. Cette maladie est associée à la réduction de l’espérance de vie moyenne d’une dizaine d’années et il a été estimé en 2013 que cette maladie touchait environ 382 millions de personnes dans le monde (Forouhi et Wareham, 2014). La compréhension des mécanismes sous-jacents à cette maladie est donc très importante. En plus du diabète de type 2, le stress du RE et l’UPR sont aussi associés à plusieurs maladies neurodégénératives, dont la maladie d’Alzheimer, la maladie de Parkinson, la sclérose latérale amyotrophique et les maladies à prions. Tout comme le diabète de type 2, l’incapacité à régler les problèmes de stress au RE finit par entraîner l’apoptose des cellules et résulte, par conséquent, en différentes maladies neurodégénératives (Doyle et al., 2011).
1.2.3. L’endoplasmic-reticulum associated degradation
L'UPR est une réponse qui permet d'augmenter l'efficacité de différents mécanismes de contrôle qualité, dont celui de l’endoplasmic-reticulum associated degration (ERAD) (Friedlander et al., 2000). Les mécanismes ERAD sont des systèmes de contrôle qualité ubiquitaires chez les organismes eucaryotes (Xie et Ng, 2010). Lorsqu'une protéine entre dans le RE, différentes protéines chaperonnes et enzymes de modifications viennent aider à son bon repliement. Si la protéine est repliée adéquatement, elle pourra être triée et transportée à l'endroit approprié. Cependant, si la cellule est incapable de la replier correctement, il y aura rétrotranslocation de cette dernière dans le cytosol, ubiquitination de la protéine et dégradation par le protéasome, un complexe protéique capable de protéolyse (figure 3) (Vembar et Brodsky, 2008). Ces systèmes sont constamment actifs dans les cellules et pourront probablement répondre à la majorité de la demande en absence de stress majeurs. Dans le cas où l’ERAD ne suffit pas à répondre à la demande, d’autres mécanismes devront être utilisés par la cellule pour essayer de se débarrasser des protéines potentiellement nocives (Wang et Ng, 2010). La dégradation post-RE, agissant notamment à l’aide de la vacuole et de certaines protéines membranaires, les yapsines, pourrait agir comme alternative dans ce cas (Hirayama, 2008; Miller, 2010).
7 Vembar et al. 2008
8
1.2.4. Les yapsines
Les yapsines, dont il sera question en détail à la section 1.5., sont des aspartyls peptidases de la levure Saccharomyces cerevisiae qui ont des homologues chez plusieurs autres fungi (Monod et al., 1998; Dujon et al., 2004; Kunihira et al., 2002). Ces enzymes font aussi partie d'un mécanisme de contrôle qualité. Ce mécanisme permet la relâche d'une protéine à ancrage GPI, Utr2p, dans le milieu extracellulaire (Miller et al., 2010). Dans la même étude, le groupe de recherche a démontré l'importance de l'UPR dans l’action des yapsines de même que l’indépendance de leur activité avec les mécanismes ERAD. Ce mécanisme agirait donc comme une voie de secours utilisée par la cellule pour se débarrasser des protéines mal repliées qui auraient échappé à l'ERAD. Ce résultat pourrait expliquer, au moins en partie, le fait que Yps1p, une yapsine, soit surexprimée en conditions d'UPR, signe d'une implication probable de la protéine à cette réponse (Travers et al., 2000).
1.3. La membrane plasmique
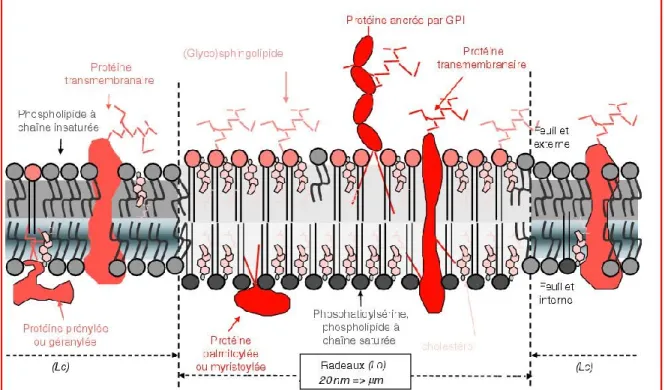
La membrane plasmique est un assemblage de différents lipides, principalement de phospholipides assemblés en bicouche, auquel s’ajoutent des stérols en plus d’une grande variété de protéines et quelques sucres. Elle délimite le cytoplasme du milieu extracellulaire et régule les échanges entre ces deux régions. Elle est présente chez toutes les cellules et est essentielle à leur survie. La membrane n’est pas une organisation fixe, elle est fluide et sa composition peut varier dans le temps. En condition de stress, la cellule cherche à maintenir l’homéostasie de sa membrane dans le but de survivre et d’être aussi compétitive que possible (Alberts et al., 2010). De plus, il existe dans la membrane des microdomaines membranaires enrichis en stérols et en sphingolipides, connus sous le nom de radeaux lipidiques (Simons et Ikonen 1997; Brown et London, 1998). Différentes protéines sont associées spécifiquement à ces assemblages dynamiques, telles les protéines GPI et les yapsines (figure 4) (Bagnat et Simons, 2002).
9 Gerlier et al., 2004
10
1.4. Les protéines à ancrage glycosylphosphatidylinositol
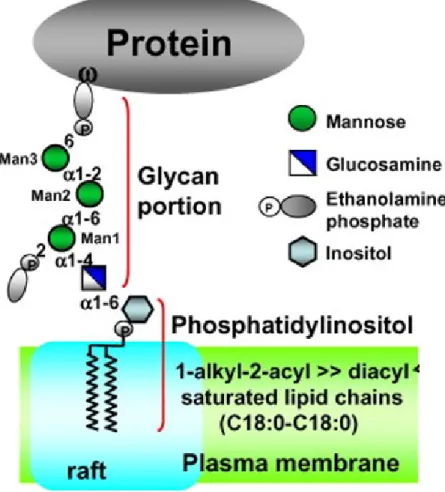
Les protéines à ancrage GPI sont une classe de protéines essentielles à la survie de la levure Saccharomyces cerevisiae et présentes chez tous les organismes eucaryotes. Ces protéines, associées aux radeaux lipidiques, possèdent une grande variété de fonctions, tel un rôle de molécules de signalisation entre l’intérieur et l’extérieur de la cellule. (Mayor et Riezman, 2004). Chez les fungi, plusieurs protéines GPI ont un rôle dans l’assemblage et le remodelage de la paroi cellulaire. Une grande portion de ces protéines n’est cependant pas liée à la paroi cellulaire et se retrouve plutôt ancrée directement à la membrane plasmique (Caro et al., 1997). Les protéines GPI ont en commun quelques caractéristiques. Par exemple, chez la levure, la grande majorité d’entre elles possèdent une région riche en sérine et thréonine (S/T) fortement O-glycosylée (Caro et al., 1997). De plus, on retrouve chez l’ensemble d’entre elles la séquence dirigeant l’ajout du groupement GPI. Malgré ces quelques ressemblances, les chaînes latérales et les portions lipidiques de cet ancrage peuvent varier d’une protéine à l’autre (Mayor et al., 2004). Les principales composantes de cet ancrage sont présentées à la figure 5.
1.4.1. Le remodelage de l'ancrage GPI
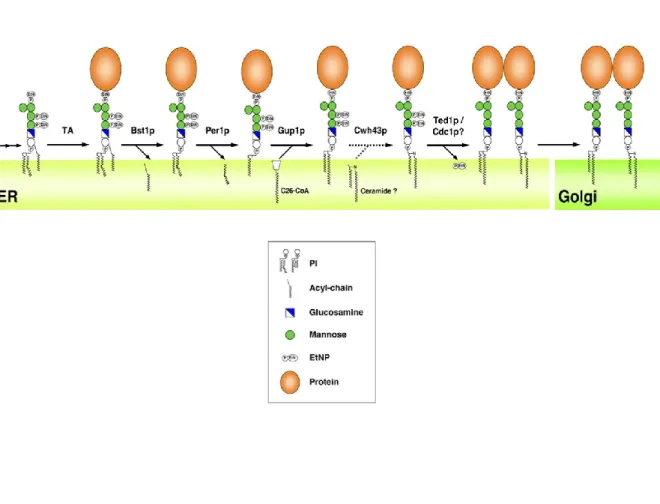
À la suite de l'ajout de l'ancrage GPI à une protéine, différentes étapes de remodelages ont lieu. Tout d'abord, Bst1p est responsable de l'enlèvement d'un groupement acyle présent sur la portion inositol de l'ancrage. Cette étape permet à la cellule de différencier la protéine GPI d'une protéine résidant dans le RE et est essentielle à sa sortie de cet organite (Pittet et Conzelmann, 2007). Après cette étape, l'ancrage peut être modifié de diverses façons. Chez la levure, la majorité des groupements sont modifiés conjointement par Per1p, qui est responsable de l'enlèvement d'un acide gras insaturé, et Gup1p, qui ajoute un acide gras saturé de 26 carbones pour remplacer le groupement enlevé (Pizett et Conzelmann, 2007; Fujita et al., 2006). D’autres étapes peuvent être nécessaires dans le cas des ancrages GPI qui contiennent des céramides (figure 6).
11 Maeda et al. 2011
12
Fujita et al. 2011
Figure 6 : Représentation schématique du remodelage de l’ancrage GPI chez la levure S. cerevisiae
13
1.5. Les aspartyl peptidases
Les aspartyl peptidases sont un groupe d’enzymes protéolytiques qui partagent un même groupe catalytique, soit deux acides aspartiques (Szecsi, 1992). Ces enzymes sont synthétisées sous forme de zymogènes, précurseurs inactifs de la protéine. Le clivage du pro-peptide de ces précurseurs permet de libérer le site actif de l’enzyme, ce qui la rend active. Le substrat peut alors se lier et être clivé par l’aspartyl peptidase (Lazure, 2002). Cette façon de procéder évite l’activation prématurée de ces enzymes dans des compartiments cellulaires inappropriés. Le pro-peptide contribue aussi au repliement, à la stabilité et à l’adressage de la protéine. (Baker, D. et al., 1993)
1.6. Les protéines Yps1 et Yps1-DL2
L’aspartyl peptidase Yps1 est modifiée post-traductionnellement par l’ajout d’un ancrage GPI, ce qui permet son association à la membrane plasmique (Ash et al., 1995). Contrairement à la plupart des aspartyl peptidases, ce groupe de protéines, appelé yapsine, n’est pas soluble. Généralement, Yps1p demeure attachée à la membrane et ne se retrouve qu’en très faible quantité dans le milieu de culture. Elle peut toutefois être relâchée de manière plus importante dans le milieu de culture dans certaines conditions ou à la suite de certaines mutations. Cette protéine a la particularité de posséder une activité de «shedding» intrinsèque, c'est-à-dire qu'elle est capable de se relâcher elle-même dans le milieu de culture (Gagnon-Arsenault et al., 2008). La construction de laboratoire de Yps1-DL2 possède une mutation au site catalytique (D101E) qui empêche cette activité de
«shedding» intrinsèque. Malgré l’absence de cette activité, cette forme mutante de la protéine est elle aussi retrouvée dans le milieu de culture dans certaines conditions, notamment dans les mutants de classe 3 décrits ci-dessous.
1.7. Les mutants de classe 3
Les mutants de classe 3 sont des mutants ayant été identifiés à la suite du criblage de la collection de mutants non-essentiels de Saccharomyces cerevisiae (Dubé, 2011). Le but du criblage était d'identifier les mutations menant à une plus grande relâche des protéines à
14
ancrage GPI dans le milieu de culture. Pour s'assurer d'avoir des résultats reproductibles pour plusieurs protéines, les protéines GPI modèles Gas1p et Yps1p furent détectées et dosées dans le milieu extracellulaire. Des 71 mutants identifiés comme relâchant des quantités plus élevées de ces protéines, cinq mutants (bst1Δ, gup1Δ, opi3Δ, per1Δ et fyv6Δ) entraînaient la relâche d'une forme de Yps1p de poids moléculaire plus élevé que ce qui était retrouvée pour la souche sauvage. De plus, cette forme avait la particularité d'être relâchée indépendamment de l'activité de Yps1p, tel que démontré avec Yps1-DL2. Il est intéressant de noter que de ces cinq mutants, les gènes BST1, GUP1 et PER1 sont tous les trois impliqués dans le remodelage de l'ancrage GPI et qu'OPI3 est responsable de la synthèse de la phosphatidylcholine (PC), une composante importante des membranes. Quant au gène FYV6, il est difficile de le relier aux autres gènes puisque sa fonction est présentement inconnue, bien qu'il ait été proposé qu'il serve à la réparation des cassures d’ADN doubles brins par jonctions d'extrémités non-homologues. Quatre de ces gènes ont donc un lien plus ou moins direct avec la composition des membranes de la cellule (Dubé, 2011).
1.7.1. Lien entre la relâche des protéines GPI et l’UPR
Le lien potentiel entre l'UPR et la relâche de Yps1p par elle-même et indépendamment de son activité est d'autant plus apparent à la suite de ce criblage. En effet, des 71 mutants hypersécréteurs de Gas1p et de Yps1p, 29 sont connus pour déclencher l'UPR, ce qui représente près de 41% (Jonikas et al., 2009). De plus, chacun des mutants de classe 3 déclenche cette réponse.
1.8. Enzymes pouvant mener à la relâche des protéines GPI
1.8.1. Les phospholipases
Les phospholipases sont des enzymes qui ont pour rôle l’hydrolyse des phospholipides et par conséquent, certaines d’entre elles sont susceptibles de cliver la portion phosphatidylinositol des ancrages GPI. Il existe différents types de phospholipases,
15 distincts les uns des autres par leur site actif et, par conséquent, par leur site de coupure. Les phospholipases jouent ainsi plusieurs rôles dans la cellule, notamment dans la méiose et la sporulation. En général, elles permettent le bon fonctionnement du métabolisme des phospholipides (Honigberg et al., 1992; Park et al., 2012; Merkel et al., 1999; Renne et al. 2015; Gibellini et Smith, 2010).
1.8.2. Les peptidases
Les peptidases sont des enzymes qui ont pour rôle l'hydrolyse des liens peptidiques. Les peptidases ont des fonctions très variées et ont, la plupart du temps, des sites de coupures préférentiels dépendamment de la nature des acides aminés les entourant. Les principales peptidases d'intérêt pour ce projet sont les membres de la famille des yapsines, excluant Yps1p, et l'endopeptidase Kex2p (Krysan, et al. 2005; Lesage et al. 2001). Kex2p est une enzyme résidant en permanence dans le réseau trans-golgien (TGN), une section très importante de la voie de sécrétion amenant potentiellement les protéines GPI à la membrane plasmique ou à la paroi cellulaire (Gu et al., 2001; Muñiz et Zurzolo, 2014 ).
Hypothèses et objectifs
Tel que mentionné précédemment, la levure Saccharomyces cerevisiae et les cellules eucaryotes en général possèdent plusieurs mécanismes de contrôle qualité leur permettant de survivre en présence de différents stress. Présentement, la plupart des mécanismes décrits touchant les protéines nécessitent la présence de protéines mal repliées. Cependant, le déclenchement de l'UPR indépendamment de l'accumulation de protéines mal repliées dans les souches mutantes utilisées par le groupe de recherche de Promlek (Promlek et al., 2011) nous amène à penser qu'il existe un autre mécanisme de contrôle qualité qui est activé en présence de défauts membranaires. La présence de certains mutants de classe 3 dans cette liste nous amène à formuler l'hypothèse qu'il existe un autre mécanisme de contrôle qualité, dépendant de l'UPR, qui permettrait à la cellule de se débarrasser de protéines bien repliées, mais retrouvées dans un environnement lipidique inadéquat.
16
L'objectif de ce projet était donc, dans un premier temps, d'essayer d'en apprendre plus sur le lien qui semble exister entre l'UPR et la relâche de la forme de Yps1 de plus haut poids moléculaire. Dans un deuxième temps, nous avons cherché à savoir quelle catégorie d'enzymes, entre les phospholipases et les peptidases, était responsable de cette relâche. Ces deux catégories d'enzymes sont susceptibles de remplir ce rôle puisque leurs zones de clivages potentielles sont très près l'une de l'autre. Par conséquent, il est impossible de discriminer l’enzyme responsable de la relâche de Yps1p sur gel de polyacrylamide. Bien que Kex2p ne réside pas à la membrane plasmique, cette enzyme est un candidat intéressant puisqu’elle est capable de cliver l'ancrage de Yps1p lorsque celui est remplacé par un domaine transmembranaire (construction de laboratoire). Ce clivage, impossible lorsque l’ancrage GPI est intact, devient probablement possible puisque les domaines transmembranaires ne sont pas associés aux radeaux lipidiques. Il est donc possible qu’une mutation créant un défaut membranaire empêche l’association adéquate de Yps1p aux radeaux lipidiques et que Yps1p devienne par le fait même un substrat de Kex2p.
17
2.
Matériel et méthodes
2.1. Micro-organismes et milieux de culture
2.1.1. Bactéries utilisées et milieu de culture
2.1.1.1. Souche de bactérie
La souche d’Escherichia coli utilisée dans ce projet de recherche était MC1061 (hsdR mcrB
araD 139∆ (ara ABC-eu) 7669∆ lacX galK rspL thi) (Sambrook et al., 1989).
2.1.1.2. Milieu de culture
La composition des milieux est donnée en pourcentage masse/volume. Les bactéries étaient ensemencées dans le milieu 2YT (1% extrait de levure, 1,6% tryptone, 0,2% glucose et 0,5% NaCl). Si le milieu avait besoin d’être solide, 2% d’agar était ajouté. Une concentration de 100 μg/mL d’ampicilline était utilisée pour permettre la sélection des bactéries transformées.
2.1.2. Levures utilisées et milieux de culture
2.1.2.1. Souches de levures
Les souches de levures utilisées de sources diverses sont décrites dans le tableau I. Les souches BY4741, BY4742 et les souches de levures ayant une délétion pour un gène non-essentiel et sélectionnées à l’aide du marqueur de résistance KAN ont toutes été obtenues de Open Biosystem. Ces souches ont été omises du tableau I pour en alléger la lecture. Les souches construites pendant ce projet sont présentées dans le tableau II. Ces souches ont été construites à l’aide des souches présentées dans le tableau I.
18
Tableau I : Souches de Saccharomyces cerevisiae utilisées
Souche Génotype Référence
bst1∆ MATa bst1::NAT leu2 his3 ura3 lys2 Ce projet
gup1∆ MATa gup1::NAT leu2 his3 ura3 lys2 Ce projet
opi3∆ MATa opi3::NAT leu2 his3 ura3 lys2 Ce projet
per1∆ MATa per1::NAT leu2 his3 ura3 lys2 Ce projet
fyv6∆ MATa fyv6::NAT leu2 his3 ura3 lys2 Ce projet
las21∆ MATa las21::NAT leu2 his3 ura3 lys2 Ce projet
get1∆ MATa get1::NAT leu2 his3 ura3 lys2 Ce projet
scs3∆ MATa scs3::NAT leu2 his3 ura3 lys2 Ce projet
isc1∆ MATa isc1::NAT leu2 his3 ura3 lys2 Ce projet
arv1∆ MATa arv1::NAT leu2 his3 ura3 lys2 Ce projet
eos1∆ MATa eos1::NAT leu2 his3 ura3 lys2 Ce projet
kex2∆ MATa kex2::NAT leu2 his3 ura3 lys2 Ce projet
yps1∆kex2∆ MATa yps1::KAN kex2::KAN leu2 his3 ura3 lys2 Gagnon-Arsenault et al., 2008
L3262 MATα ura3-52 leu2-3,112 his4-34 Kang et al., 1998
SLH11 MATα ura3-52 leu2-3,112 his4-34
yps1::LEU2 Kang et al., 1998
19 SLH28
MATα ura3-52 leu2-3,112 his4-34 yps1::LEU2 yps2::HIS4 yps3::tc5 yps6::tc5
yps7::tc5 Cho et al., 2010
plb1∆ MATα plb1::KAN can1::STE2pr-his5, lyp1::STE3prLEU2 ura3 his3 leu2 Don du laboratoire de Dr Landry
plb2∆ MATα plb2::KANcan1::STE2pr-his5,
lyp1::STE3prLEU2 ura3 his3 leu2
Don du laboratoire de Dr Landry
plb3∆ MATα plb3::KAN can1::STE2pr-his5, lyp1::STE3prLEU2 ura3 his3 leu2 Don du laboratoire de Dr Landry
spo1∆ MATα spo1::KAN can1::STE2pr-his5, lyp1::STE3prLEU2 ura3 his3 leu2 Don du laboratoire de Dr Landry
spo14∆ MATα spo14::KAN can1::STE2pr-his5,
lyp1::STE3prLEU2 ura3 his3 leu2
Don du laboratoire de Dr Landry
20
Tableau II : Souches de Saccharomyces cerevisiae construites pendant ce projet
Toutes les souches créées par SGA ont été sélectionnées pour chacun des types sexuels. Ces souches sont dans le background BY4741 ou BY4742.
Souches Génotype
bst1∆plb1∆ bst1::NAT plb1::KAN can1::STE2pr-his5,
lyp1::STE3prLEU2 ura3 his3 leu2
bst1∆plb2∆ bst1::NAT plb2::KAN can1::STE2pr-his5,
lyp1::STE3prLEU2 ura3 his3 leu2
bst1∆plb3∆ bst1::NAT plb3::KAN can1::STE2pr-his5,
lyp1::STE3prLEU2 ura3 his3 leu2 bst1∆spo1∆ bst1::NAT spo1::KAN can1::STE2pr-his5, lyp1::STE3prLEU2 ura3 his3 leu2
bst1∆spo14∆ bst1::NAT spo14::KAN can1::STE2pr-his5,
lyp1::STE3prLEU2 ura3 his3 leu2 bst1∆yjl132w∆ bst1::NAT yjl132w::KAN can1::STE2pr-his5, lyp1::STE3prLEU2 ura3 his3 leu2
gup1∆plb1∆ gup1::NAT plb1::KAN can1::STE2pr-his5,
lyp1::STE3prLEU2 ura3 his3 leu2 gup1∆plb2∆ gup1::NAT plb2::KAN can1::STE2pr-his5, lyp1::STE3prLEU2 ura3 his3 leu2
gup1∆plb3∆ gup1::NAT plb3::KAN can1::STE2pr-his5,
lyp1::STE3prLEU2 ura3 his3 leu2 gup1∆spo1∆ gup1::NAT spo1::KAN can1::STE2pr-his5, lyp1::STE3prLEU2 ura3 his3 leu2 gup1∆spo14∆ gup1::NAT spo14::KAN can1::STE2pr-his5, lyp1::STE3prLEU2 ura3 his3 leu2 gup1∆yjl132w∆ gup1::NAT yjl132w::KAN can1::STE2pr-his5, lyp1::STE3prLEU2 ura3 his3 leu2
opi3∆plb1∆ opi3::NAT plb1::KAN can1::STE2pr-his5, lyp1::STE3prLEU2 ura3 his3 leu2
opi3∆plb2∆ opi3::NAT plb2::KAN can1::STE2pr-his5,
21
opi3∆plb3∆ opi3::NAT plb3::KAN can1::STE2pr-his5, lyp1::STE3prLEU2 ura3 his3 leu2 opi3∆spo1∆ opi3::NAT spo1::KAN can1::STE2pr-his5, lyp1::STE3prLEU2 ura3 his3 leu2 opi3∆spo14∆ opi3::NAT spo14::KAN can1::STE2pr-his5, lyp1::STE3prLEU2 ura3 his3 leu2 opi3∆yjl132w∆ opi3::NAT yjl132w::KAN can1::STE2pr-his5, lyp1::STE3prLEU2 ura3 his3 leu2
per1∆plb1∆ per1::NAT plb1::KAN can1::STE2pr-his5,
lyp1::STE3prLEU2 ura3 his3 leu2 per1∆plb2∆ per1::NAT plb2::KAN can1::STE2pr-his5, lyp1::STE3prLEU2 ura3 his3 leu2
per1∆plb3∆ per1::NAT plb3::KAN can1::STE2pr-his5,
lyp1::STE3prLEU2 ura3 his3 leu2 per1∆spo1∆ per1::NAT spo1::KAN can1::STE2pr-his5, lyp1::STE3prLEU2 ura3 his3 leu2
per1∆spo14∆ per1::NAT spo14::KAN can1::STE2pr-his5,
lyp1::STE3prLEU2 ura3 his3 leu2 per1∆yjl132w∆ per1::NAT yjl132w::KAN can1::STE2pr-his5, lyp1::STE3prLEU2 ura3 his3 leu2
fyv6∆plb1∆ fyv6::NAT plb1::KAN can1::STE2pr-his5, lyp1::STE3prLEU2 ura3 his3 leu2 fyv6∆plb2∆ fyv6::NAT plb2::KAN can1::STE2pr-his5, lyp1::STE3prLEU2 ura3 his3 leu2 fyv6∆plb3∆ fyv6::NAT plb3::KAN can1::STE2pr-his5, lyp1::STE3prLEU2 ura3 his3 leu2
fyv6∆spo1∆ fyv6::NAT spo1::KAN can1::STE2pr-his5,
lyp1::STE3prLEU2 ura3 his3 leu2 fyv6∆spo14∆ fyv6::NAT spo14::KAN can1::STE2pr-his5, lyp1::STE3prLEU2 ura3 his3 leu2
fyv6∆yjl132w∆ fyv6::NAT yjl132w::KAN can1::STE2pr-his5,
22
las21∆plb1∆ las21::NAT plb1::KAN can1::STE2pr-his5, lyp1::STE3prLEU2 ura3 his3 leu2 las21∆plb2∆ las21::NAT plb2::KAN can1::STE2pr-his5, lyp1::STE3prLEU2 ura3 his3 leu2 las21∆plb3∆ las21::NAT plb3::KAN can1::STE2pr-his5, lyp1::STE3prLEU2 ura3 his3 leu2 las21∆spo1∆ las21::NAT spo1::KAN can1::STE2pr-his5, lyp1::STE3prLEU2 ura3 his3 leu2
las21∆spo14∆ las21::NAT spo14::KAN can1::STE2pr-his5,
lyp1::STE3prLEU2 ura3 his3 leu2 las21∆yjl132w∆ las21::NAT yjl132w::KAN can1::STE2pr-his5, lyp1::STE3prLEU2 ura3 his3 leu2
isc1∆plb1∆ isc1::NAT plb1::KAN can1::STE2pr-his5,
lyp1::STE3prLEU2 ura3 his3 leu2 isc1∆plb2∆ isc1::NAT plb2::KAN can1::STE2pr-his5, lyp1::STE3prLEU2 ura3 his3 leu2
isc1∆plb3∆ isc1::NAT plb3::KAN can1::STE2pr-his5,
lyp1::STE3prLEU2 ura3 his3 leu2 isc1∆spo1∆ isc1::NAT spo1::KAN can1::STE2pr-his5, lyp1::STE3prLEU2 ura3 his3 leu2 isc1∆spo14∆ isc1::NAT spo14::KAN can1::STE2pr-his5, lyp1::STE3prLEU2 ura3 his3 leu2 isc1∆yjl132w∆ isc1::NAT yjl132w::KAN can1::STE2pr-his5, lyp1::STE3prLEU2 ura3 his3 leu2
scs3∆plb1∆ scs3::NAT plb1::KAN can1::STE2pr-his5, lyp1::STE3prLEU2 ura3 his3 leu2
scs3∆plb2∆ scs3::NAT plb2::KAN can1::STE2pr-his5,
lyp1::STE3prLEU2 ura3 his3 leu2 scs3∆plb3∆ scs3::NAT plb3::KAN can1::STE2pr-his5, lyp1::STE3prLEU2 ura3 his3 leu2
scs3∆spo1∆ scs3::NAT spo1::KAN can1::STE2pr-his5,
23
scs3∆spo14∆ scs3::NAT spo14::KAN can1::STE2pr-his5, lyp1::STE3prLEU2 ura3 his3 leu2 scs3∆yjl132w∆ scs3::NAT yjl132w::KAN can1::STE2pr-his5, lyp1::STE3prLEU2 ura3 his3 leu2
arv1∆plb1∆ arv1::NAT plb1::KAN can1::STE2pr-his5, lyp1::STE3prLEU2 ura3 his3 leu2 arv1∆plb2∆ arv1::NAT plb2::KAN can1::STE2pr-his5, lyp1::STE3prLEU2 ura3 his3 leu2
arv1∆plb3∆ arv1::NAT plb3::KAN can1::STE2pr-his5,
lyp1::STE3prLEU2 ura3 his3 leu2 arv1∆spo1∆ arv1::NAT spo1::KAN can1::STE2pr-his5, lyp1::STE3prLEU2 ura3 his3 leu2
arv1∆spo14∆ arv1::NAT spo14::KAN can1::STE2pr-his5,
lyp1::STE3prLEU2 ura3 his3 leu2 arv1∆yjl132w∆ arv1::NAT yjl132w::KAN can1::STE2pr-his5, lyp1::STE3prLEU2 ura3 his3 leu2
bst1∆kex2∆ MATa bst1::KAN kex2::NAT leu2 his3 ura3 lys2
gup1∆kex2∆ MATa gup1::KAN kex2::NAT leu2 his3 ura3 lys2
opi3∆kex2∆ MATα opi3::KAN kex2::NAT leu2 his3 ura3 lys2
per1∆kex2∆ MATα per1::KAN kex2::NAT leu2 his3 ura3 lys2
24
Les souches suivantes sont dans le background L3262
Souches Génotype
bst1∆ MATα bst1::NAT ura3-52 leu2-3,112 his4-34
bst1∆yps1∆ MATα bst1::NAT ura3-52 leu2-3,112 his4-34
yps1::LEU2
bst1∆yps1∆yps2∆yps3∆ MATα bst1::NAT ura3-52 leu2-3,112 his4-34 yps1::LEU2 yps2::HIS4 yps3::tc5 bst1∆ yps1∆yps2∆yps3∆
yps6∆yps7
MATα bst1::NAT ura3-52 leu2-3,112 his4-34 yps1::LEU2 yps2::HIS4 yps3::tc5 yps6::tc5 yps7::tc5
gup1∆ MATα gup1::NAT ura3-52 leu2-3,112 his4-34
gup1∆yps1∆ MATα gup1::NAT ura3-52 leu2-3,112 his4-34 yps1::LEU2
gup1∆yps1∆yps2∆yps3∆ MATα gup1::NAT ura3-52 leu2-3,112 his4-34
yps1::LEU2 yps2::HIS4 yps3::tc5 gup1∆yps1∆yps2∆yps3∆
yps6∆yps7
MATα gup1::NAT ura3-52 leu2-3,112 his4-34 yps1::LEU2 yps2::HIS4 yps3::tc5 yps6::tc5 yps7::tc5
opi3∆ MATα opi3::NAT ura3-52 leu2-3,112 his4-34
opi3∆yps1∆ MATα opi3::NAT ura3-52 leu2-3,112 his4-34 yps1::LEU2
opi3∆yps1∆yps2∆yps3∆ MATα opi3::NAT ura3-52 leu2-3,112 his4-34
yps1::LEU2 yps2::HIS4 yps3::tc5 opi3∆yps1∆yps2∆yps3∆
yps6∆yps7 yps1::LEU2 yps2::HIS4 yps3::tc5 yps6::tc5 yps7::tc5 MATα opi3::NAT ura3-52 leu2-3,112 his4-34
per1∆ MATα per1::NAT ura3-52 leu2-3,112 his4-34
25
2.1.2.1. Milieux de culture
La composition des milieux est donnée en pourcentage masse/volume. Lorsqu’aucune sélection n’était nécessaire, le milieu riche utilisé pour faire croître les différentes souches était le YPD (1% extrait de levure, 2% tryptone, 1% glucose) liquide ou solide (2% agar). Lorsqu’un gène de résistance à un antibiotique était utilisé pour sélectionner des clones possédant une mutation d’intérêt, le milieu était supplémenté de généticine (G418, 250 µg/mL) et/ou de nourséothricine (NAT, 200 µg/mL).
Les transformants étaient sélectionnés en utilisant le milieu minimal SC (0,67% "yeast nitrogen base without amino acid" (YNB sans aa), 2% glucose) auquel s’ajoutait un «drop-out» contenant presque tous les acides aminés ainsi que deux bases azotées (adénine et uracile) auquel il est possible d’enlever une ou plusieurs composantes lorsqu’une sélection est nécessaire (SC - URA dans le cas d’une sélection plasmidique, SC avec un drop-out complet lorsque la souche est non-transformée). Les quantités de chaque constituant du «drop-out» sont décrites dans Methods in Yeast Genetics (Kaiser, 1994). De l’agar (2%) pouvait être ajouté pour obtenir un milieu solide. Ce milieu pouvait être non-tamponné (pH 3.0) ou tamponné avec 50 mM succinate pour pouvoir ajuster le pH à 6.0.
Le milieu SD (0,67% YNB sans aa, 2% glucose, 2% agar) permettait la détermination du type sexuel de spores par croisement avec des souches complétant toutes les auxotrophies des levures à la suite de la conjugaison.
Le milieu 5-Fluoroorotic acid (5-FOA) servait à sélectionner les souches ne possédant pas le gène de complémentation d’auxotrophie URA3, et donc, dans le cadre de ce projet, à sélectionner les levures n’ayant pas de plasmide. Il était composé de 0,67% (YNB sans aa), 2% glucose, 0,1% de 5-fluoroorotic acid ainsi qu’un drop-out - URA auquel s’ajoutait 50 µg/mL d’uracile. Cette portion du milieu devait être filtrée avec un filtre 0,2 µm tandis qu’un deuxième mélange contenant 2% agar devait être autoclavé. Lorsque le mélange autoclavé avait refroidi, il était incorporé avec le reste du milieu qui pouvait maintenant être coulé dans les boîtes de pétris.
26
Le milieu YEPA (2% acétate de potassium, 2% tryptone, 1% extrait de levure) servait d’intermédiaire entre le milieu de sélection des diploïdes et le milieu SPM dans le but de construire des souches de levures contenant plusieurs mutations d’intérêt.
Le milieu SPM (acétate de potassium 0,3%, raffinose 0,02%, «drop-out» ne contenant que les acides aminés et les bases azotées essentielles) (Kaiser, 1994) est un milieu pauvre entraînant la sporulation des levures diploïdes. Pour ce projet, le «drop-out» contenait de l’histidine, de l’uracile et de la leucine.
Les milieux utilisés pour les sélections du synthetic genetic array (SGA) étaient composés de 0,1734% YNB sans aa, de 0,428% de glutamate monosodique (MSG), de 2% glucose et de 2% agar. L’utilisation du MSG au lieu de l’ammonium sulfate est nécessaire puisque ce dernier nuit à la fonction du G418 et de NAT en milieu synthétique (Cheng et al., 2000). Les différents drop-out et antibiotiques utilisés à chaque étape de sélection sont décrits dans Diss et al. (2013).
2.1.3. Construction de souches contenant plusieurs délétions géniques par dissection de tétrades
2.1.3.1. Croisements
Une souche MATa d’intérêt était placée dans 5 mL de milieu YPD avec une souche MATα contenant en son génome une deuxième mutation d’intérêt. Le tube était incubé à 30°C pour une nuit avec agitation. Le lendemain, 5 µL du milieu étaient déposés sur un milieu YPD + G418 + NAT permettant la sélection des levures diploïdes.
2.1.3.2. Sporulation
La première étape du protocole de sporulation était de répartir les cellules diploïdes dans 5 mL de milieu sélectif. Après une incubation d’une nuit à 30°C, les cultures étaient diluées pour obtenir une D.O.600 de 0,6 dans 10 mL de milieu YPD + G418 + NAT frais. Les
27 étaient centrifugées à température pièce (TP) et lavées avec 20 mL d’eau stérile. Elles étaient par la suite remises en suspension dans 10 mL de milieu YEPA et incubées pour la nuit à 30°C avec agitation. Le lendemain, les cellules étaient lavées à 2 reprises dans 20 mL d’eau stérile puis elles étaient resuspendues dans 25 mL de milieu SPM. L’incubation à 30°C dans ce milieu durait entre 3 et 5 jours.
2.1.3.3. Dissection
Une aliquote de 150 µL de cellules sporulées était centrifugée et resuspendue dans 100 µL de zymolyase 20T (200 µg/mL). Les cellules étaient laissées à TP pour environ 20 min. Elles étaient centrifugées un maximum de 20 s à 13000 rpm et remises en suspension dans 50 µL de sorbitol 1 M (le volume peut être ajusté selon la concentration des spores). Une ligne de cellules était ensuite étalée sur un pétri YPD au niveau à l’aide d’un fil à boucle. Un microscope à dissection était utilisé pour séparer les spores de plusieurs tétrades à des endroits distincts sur la gélose. Les pétris de dissections étaient incubés à 30°C pour 3 jours.
2.1.3.4. Sélection des souches contenant les mutations d’intérêt
La dissection des tétrades mène à trois types de patrons. Les spores pouvaient être de type parental, de type non parental ou finalement être sous forme de tétratype. Ces différents patrons permettent de déterminer le génotype d’une spore avant de le confirmer par réaction en chaîne par polymérase (PCR). En faisant des répliques velours sur milieu YPD + G418 et YPD + NAT, il est simple d’identifier quel gène est inactivé dans quelle souche. Si les spores sont de type parental, les 4 spores pourront croître, deux sur un des milieux sélectifs et les deux autres, sur l’autre. Si les levures sont de type non-parental, seulement deux des spores pourront croître, mais elles pousseront sur chacun des milieux de sélection. Pour ce qui est des tétratypes, 3 des 4 spores vont croître, une d’entre elles contenant les deux mutations d’intérêt.
28
2.1.3.5. Identification du type sexuel
Pour déterminer le type sexuel des spores d’intérêt, une gélose YPD était inoculée avec celle-ci et les pétris étaient incubés à 30°C jusqu’au lendemain. Ces souches étaient croisées avec un tapis de cellules his1∆ MATa d’une part et avec un tapis de cellules his1∆
MATα d’autre part. Ces croisements étaient incubés toute la nuit à 30°C et des répliques
étaient ensuite faites sur milieu SD. La formation de levures diploïdes permettait la complémentation de l’ensemble des auxotrophies des spores haploïdes et de la souche
his1∆. Il était donc possible de déduire le type sexuel d’une souche à partir de celui de la
souche his1∆ de départ.
2.1.4. Construction de souches contenant plusieurs délétions géniques par Synthetic Genetic Array
La construction de souche par SGA a été faite selon la méthode décrite dans Tong et Boone en 2006 (Tong et Boone, 2006). Les différentes souches utilisées sont décrites dans le tableau I et le marqueur de résistance de la deuxième souche était le marqueur NAT.
2.1.5. Délétion de gènes par intégration d’un fragment PCR
2.1.5.1. Amplification du marqueur de sélection
Les marqueurs de sélections utilisés pouvaient être amplifiés de deux façons distinctes. Dans un premier temps, les plasmides de la série pUG, notamment pAG25 contenant le marqueur NAT, pouvaient être utilisés. La région d’intérêt du plasmide était amplifiée par PCR à l’aide d’amorce possédant 40 nucléotides d’homologies avec les régions 5’ et 3’ du gène dont on veut supprimer la fonction et 20 nucléotides d’homologie avec des régions communes à tous les plasmides pUG situées de part et d’autres du marqueur de sélection. Un cycle PCR standard était effectué selon les conditions suivantes : 5 min de dénaturation initiale à 94°C, suivi de 35 cycles identiques de 1 min à 94°C, 30 s à 55°C et 3 min à 72°C, finalement, une élongation de 10 min à 72°C avait lieu.
29
2.1.5.2. Préparation et transformation de cellules de levures compétentes
Les cellules de levures compétentes ont été préparées et transformées selon un protocole basé sur celui de Gietz et Woods (2002). La préparation de cellules compétentes est essentielle à l’intégration du fragment PCR dans le génome d’une souche d’intérêt. La transformation de cellules compétentes était effectuée en ajoutant 8 µL d’ADN de l’amplicon PCR contenant les régions d’homologie avec le gène d’intérêt à 20 µL de cellules compétentes. Par la suite, 100 µL de «plate mixture» (acétate de lithium 100 mM, Tris 10 mM pH 7,5, EDTA 1 mM, PEG 3350 45%) était ajouté aux cellules et l’incubation était poursuivie à température pièce pendant 30 minutes. À la suite de cette incubation, 15 µL de DMSO était ajouté et les cellules étaient placées à 42°C pour 15 à 20 min. Une fois le choc thermique terminé, le mélange était centrifugé à 2000 rpm pour 3 min et le surnageant était séparé du culot par aspiration. Le culot était remis en suspension dans 100 µL de YPD et incubé à 30°C pour 4 h. Finalement, les cellules étaient étalées sur un milieu sélectif et incubées de 2 à 3 jours à 30°C.
2.2. Acides nucléiques
2.2.1. Plasmides utilisés
30
Tableau III : Plasmides utilisés dans ce projet
Nom Description Source
pRS426 Origine de réplication de 2µ, marqueur
URA3
Christianson et al., 1992, ATCC 77107
pRSYPS1 pRS426 contenant YPS1 exprimé avec
son promoteur naturel Tremblay et al., 2001
pRSYPS1-DL2
pRSYPS1 dans lequel la séquence codante pour la boucle de Yps1p a été remplacée par la séquence codante de
la boucle de YPS3 en plus de la mutation D101E
Mémoire d’Alexandre Dubé,
2011
pAG25 Utilisé pour amplifier le marqueur
31
2.2.2. Préparation d’ADN plasmidique
L’isolement d’ADN plasmidique de bactéries était effectué avec la trousse «GenElute Plasmid Miniprep Kit» de Sigma-Aldrich. Le protocole du manufacturier était suivi.
2.2.3. Transformations
2.2.3.1. Bactéries
Les transformations de bactéries ont toutes été faites selon le protocole du CaCl2 (Kaiser,
1994). La sélection des transformants se faisait sur milieu 2YT avec ampicilline (100 μg/mL)
2.2.3.2. Levures
Les souches de levures ont été transformées selon le protocole de la méthode à l’acétate de lithium tirée de Methods in Yeast Genetics (Kaiser, 1994).
2.2.4. Extraction d’ADN génomique de levure
L’extraction d’ADN génomique a été faite selon 2 protocoles distincts dépendamment de la pureté d’ADN souhaitée.
Dans un premier temps, une extraction classique phénol : chloroforme pouvait être effectuée selon la méthode décrite dans Methods in Yeast Genetics (Kaiser, 1994).
Un deuxième protocole d’extraction plus rapide pouvait aussi être utilisé lorsqu’une pureté élevée n’était pas nécessaire. Le protocole s’inspire du protocole créé par Looke et al. en 2011. Un volume de 100 µL d’une culture liquide était suspendu dans 100 µL d’une solution de LiOAc 200 mM et SDS 1%. Les cellules étaient placées à 70°C pour 10 min
32
puis 300 µL d’éthanol 95% froid étaient ajoutés. Une centrifugation de 3 min à 13000 rpm avait lieu ce qui faisait précipiter l’ADN ainsi que les débris cellulaires présents. Le surnageant était retiré et le culot était nettoyé avec 300 µL d’éthanol 70% froid. Une deuxième centrifugation avait lieu et le culot était ensuite séché. Ce culot était repris dans 100 µL d’eau et une centrifugation de 15 s à 13000 rpm avait lieu. Celle-ci faisait précipiter les débris cellulaires tout en gardant l’ADN en suspension. Pour un PCR, 1 µL de cette suspension était utilisé.
2.2.5. Amplification PCR
2.2.5.1. Pour confirmation de délétions géniques
La PCR a été utilisée pour confirmer les délétions des souches créées pour ce projet. Pour vérifier ces délétions, deux amorces devaient être utilisées (Tableau IV). L’une spécifique au gène d’intérêt et une deuxième s’appariant au brin opposé et spécifique à la cassette de résistance ou de complémentation d’auxotrophie utilisée pour la sélection.
Le mélange réactionnel standard était composé de : 1 μL d’ADN génomique, 10 pmoles de chaque amorce, 2,5 μL de tampon ThermoPol 10x (NEB), 2,5 μL de dNTPs à une concentration de 2,5 μM et de 1U de VENT DNA polymérase (NEB) pour un volume final de 25 μL. Le mélange réactionnel était ensuite placé dans le thermocycleur et un programme similaire à celui retrouvé dans le mémoire de maîtrise d’Alexandre Dubé (2011) était utilisé.
2.2.5.2. Amplification de séquences
L’amplification de séquences servait principalement à la délétion d’un gène par intégration d’un fragment PCR telle que décrite à la section 2.1.5.
33
Tableau IV : Amorces employées pour amplifier les fragments utilisés pour la délétion génique et pour la vérification de délétions
Nom Séquence (5’ → 3’) Bst1A GCATAACTCACTAGGTACCCTCAAA Bst1D CAAAATTTACGGCTTTGAAAAAGTA Gup1A ATCAGCTCAATCGGACATAAAAATA Gup1D TTAGAAATGCTCAAGGATGGATTAG Opi3A CGTATAATTCGACTTCCTTGTTGTT Opi3D TAGCTGGTAATTCTCCATCACTTTC Per1A ATTGAACATAGAGGCAATTGCTAAG Per1D AATGCATGTTCTCTCAATAAGGAAG Fyv6A GATATACCTGGGAAGTTACAATGGA Las21A GCTGTTCTTCTCTCCTCTTTTGTAA Scs3A CAGATCTGCTGTTGAGTGAGTAAGT Isc1A GTTTGGTTTGATTTATTTTGGTTTG Arv1A GAGACATTTTCCAATGCTGATTACT Plb1D ATCTTCGATGAAAGTACTGCATGAAC Plb2A AGCTTGTTTTTCCTTAGTGTGCCTTC Plb3D AGTCCGTGAAAAATTACATTACCAA Spo1A TTTAACAAATATGGGCCAAAACTAA Spo14A TAAGCAACACTTGCTGATTACAGTC Yjl132WA ATGAATGTGCAAAACATTTCCTAAT Kex2A ATAATCAATGAGGGTCATTTTCTGA Hac1A ATACATTTATGAGGGTTGTAAGGCA NatB CGGTAAGCCGTGTCGTCAAG KanB CTGCAGCGAGGAGCCGTAAT KanC TGATTTTGATGACGAGCGTAAT
34
2.3. Protéomique
2.3.1. Concentration des protéines du surnageant de culture et extraction des protéines de levures
Les protocoles utilisés pour les concentrations des protéines du surnageant de culture et l’extraction des protéines intracellulaires de levures ont été fait selon les protocoles retrouvés dans le mémoire d’Alexandre Dubé (2011). Cependant, des cultures de 30 mL des différentes souches de levures étaient généralement utilisées et elles pouvaient être incubées avec agitation pour 24 ou 48 h à 30°C selon la vitesse de croissance. Du volume de 30 mL initial, 5 mL étaient retirés et utilisés pour l’extraction des protéines intracellulaires. Dans tous les cas, un équivalent de 13,5 x 106 cellules était déposé sur gel.
Le 25 mL de culture restant était concentré à l’aide de membrane Amicon (Millipore) d’une limite d’exclusion de 10 kDa.
2.3.2. Traitement à l’Endo Hf
L’Endo Hf permet de simplifier l’analyse sur gel de protéines hyperglycosylées puisqu’elle
enlève la N-glycosylation qui est composée de chaînes glucidiques de longueurs très variables. Ceci permet de rendre la masse de ces protéines homogène. Le traitement était effectué selon la méthode décrite par la compagnie (NEB).
2.3.3. SDS-PAGE
L’analyse des protéines par SDS-PAGE a été effectuée selon la méthode décrite par Sambrook et al. (1989). Des volumes de 2,5 µL de DTT 1,0 M et de 5 µL de tampon d’échantillon (Tris-HCl pH 6,8 50 mM, DTT 100 mM, 2% SDS, 0,1% Bromophénol Bleu, 10% glycérol) étaient ajoutés aux échantillons traités à l’Endo Hf. Les échantillons étaient
bouillis pendant 5 min avant d’être déposés sur gel de polyacrylamide. Des gels de séparation de 10% ont été utilisés pour séparer les protéines. La migration s’effectuait à 175 V pour une période d’environ 1 h dans un tampon de cuve (Tris base 25 mM, glycine 250 mM, 0,1% SDS)
35
2.3.4. Transfert
Les protéines étaient transférées sur une membrane de nitrocellulose selon le protocole fourni avec l’appareil de transfert semi-humide LKB Nova Blot (Amersham Pharmacia Biotech). La membrane était immergée dans le tampon de transfert (glycine 2,93 g/L, Tris base 5,81 g/L, SDS 0,375 g/L, 20% méthanol). La membrane était ensuite déposée entre 8 morceaux de papier Whatman, 4 au-dessus et 4 au-dessous de celle-ci. Le gel était déposé directement au-dessus de la membrane et une tension de 0,8 mA par cm2 était appliquée
pendant 1 h.
2.3.5. Immunobuvardage Western
Les immunobuvardages ont été effectués selon la méthode décrite dans la trousse Western Blotting Kits (Odyssey). La membrane était bloquée dans le tampon de blocage d’Odyssey pendant un minimum d’1 h. À la suite de cette incubation, 0,1% Tween 20 était ajouté à la solution et l’anticorps primaire était incubé 2 h à une dilution de 1 : 5000. Les anticorps utilisés étaient dirigés vers Yps1p, étaient polyclonaux et provenaient de lapins. L’anticorps anti-Yps1p (lot 294, 3è ou 4è injection) était dirigé contre la boucle de la protéine (aa 35 à 246) et n’était utilisé que pour détecter des formes de la protéine possédant cette boucle. L’anticorps anti-Ysp1p (lot 268, 6è injection), était quant à lui utilisé pour détecter la forme ne possédant pas la boucle. Cet anticorps était dirigé contre les aa 245 à 532 de Yps1p. Dans les deux cas, les aa d’intérêt était fusionnés avec la glutathion S-transférase (Ash et
al., 1995). Après l’incubation, la membrane était lavée 3 fois 5 min dans du tampon
phosphate salin contenant du Tween 20 (PBST) (NaH2PO4 20 mM, Na2HPO4 80 mM, NaCl
100 mM pH 7,5, 0,1% Tween 20) et une nouvelle solution du tampon de blocage était utilisé par la suite. À celle-ci venait s’ajouter 0,1% de Tween 20 et 0,01% de SDS. L’anticorps secondaire était un anti-lapin couplé à un fluorochrome émettant un signal à 800 nm (Licor-Mendel). Il était utilisé à une concentration de 1 : 10000 pendant 1 h. Le signal des immunobuvardages était quantifié à l’aide du logiciel image studio (LI-COR).
36
2.3.6 Analyse statistique
La comparaison de la relâche de Yps1-DL2 dans les mutants de classe 3 a été effectuée à l’aide d’un test de Student (unilatéral, variances différentes). Dans tous les cas, l’hypothèse nulle stipulant que les moyennes sont égales a été vérifiée. Les moyennes ont été considérées comme significativement différentes si la valeur-p était inférieure à 0,05.
37
3. Résultats
3.1. Relâche de Yps1p dans des mutants connus pour déclencher
l'UPR
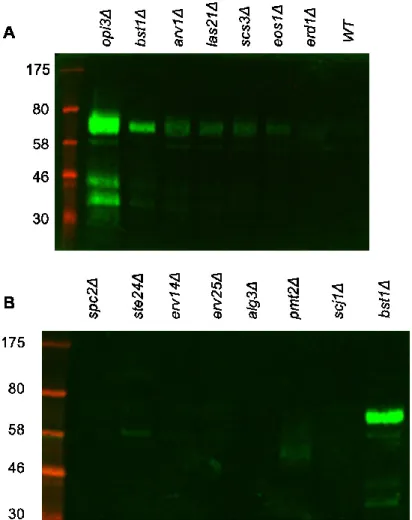
Tel que mentionné précédemment, il semble exister un lien entre l'UPR et la relâche de Yps1p dans le milieu de culture. Dans le but de mieux comprendre ce lien potentiel, nous avons utilisé les différents mutants déclenchant l'UPR sélectionnés et séparés en deux catégories par Promlek et al. en 2011. Les résultats présentés à la figure 7 montre que la bande de plus haut poids moléculaire, située entre 58 et 80 kDa (environ 64 kDa), est présente pour plusieurs des mutants déclenchant l'UPR à la suite d'un défaut membranaire (opi3Δ, bst1Δ, las21Δ, arv1Δ et scs3Δ), alors que ce n'est pas le cas pour les mutants déclenchant cette réponse à la suite d'une accumulation de protéines mal repliées (spc2Δ,
ste24Δ, erv14Δ, erv25Δ, alg3Δ, pmt2Δ, scj1Δ, eos1Δ et erd1Δ). Certains mutants
déclenchant la réponse UPR à la suite d’une accumulation de protéines mal repliées relâchent tout de même la bande de plus haut poids moléculaire de Yps1p. C’est le cas pour la souche eos1Δ (figure 7A). Les résultats obtenus pour la détection de la forme originale de Yps1p (figure 7) et de la version plasmidique ayant le site catalytique muté Yps1-DL2 (figure 8) sont similaires, bien que quelque peu différents parfois. Par exemple, la souche pmt2Δ donne une relâche assez importante de Yps1-DL2, alors que cette relâche n’est que très peu détectable pour la forme endogène de Yps1p. Les formes de plus hauts poids moléculaires des souches pmt2Δ et get1Δ migrent plus loin sur gel de polyacrylamide, potentiellement à cause de la glycosylation plus faible de Yps1p dans ces souches. Encore une fois, la souche eos1Δ relâche la forme de plus haut poids moléculaire dans le milieu de culture. Contrairement à ce qui était observé à la figure 7, la souche
erv14Δ, qui nécessite le domaine liant les protéines mal repliées de Ire1p pour déclencher
l’UPR, semble également relâcher la forme de plus haut poids moléculaire. La présence de bandes supplémentaires dans les souches exprimant Yps1-DL2 peut potentiellement être expliquée par la moins grande spécificité de l’anticorps utilisé pour révéler la présence de cette forme mutée de Yps1p. Il est également possible que la surexpression de la protéine dans ces souches permette la détection de formes supplémentaires de cette dernière.