L'impact de la mutation UBQLN2 sur la protéinopathie
de TDP-43 dans la sc
lérose latérale amyotrophique et
la démence fronto-temporale
Mémoire
Laurence Renaud
Maîtrise en neurobiologie - avec mémoire
Maître ès sciences (M. Sc.)
Québec, Canada
i
L’impact de la mutation UBQLN2 sur la protéinopathie de TDP-43
dans la sclérose latérale amyotrophique et la démence
fronto-temporale
Mémoire
Laurence Renaud
Sous la direction de :
ii
Résumé
La sclérose latérale amyotrophique (SLA) est une maladie neurodégénérative affectant les neurones moteurs supérieurs et inférieurs menant éventuellement à une paralysie générale du patient. Le décès du patient survient généralement entre 2 à 5 ans subséquemment à l’apparition des premiers symptômes. La SLA consiste en la maladie neurologique causant le plus décès chez l’adulte et il est estimé qu’environ 10% des cas sont familiaux (fSLA) et 90% sont sporadiques (sSLA). De plus, 15% des patients souffrant de la SLA développent également une démence fronto-temporale (DFT) s’illustrant par des troubles de comportements ainsi qu’un changement de personnalité majeur. Il a été démontré que la SLA et la DFT partagent un spectre génétique commun et les patients atteints de DFT démontrent une protéinopathie caractérisée par une accumulation anormale de certaines protéines dans le cytoplasme des neurones et des cellules gliales, tout comme pour les patients souffrant de SLA. Plusieurs gènes mutés ont été identifiés au cours des dernières années pour la forme fSLA, notamment superoxide dismutase 1 (SOD1), TAR DNA-binding protein 43 (TDP-43), ubiquilin-2 (UBQLN2), Fused in sarcoma (FUS), optineurin (OPTN), etc. Un des gènes le plus étudié et le mieux décrit est celui codant la protéine TDP-43. Cette protéine nucléaire, lorsque mutée, est délocalisée dans le cytoplasme, où elle forme des inclusions anormales et persistantes. Ces agrégats, lorsque formés, renferment également plusieurs autres composés, notamment d’autres protéines telles qu’UBQLN2, différentes nucléoporines, ubiquitine, etc. UBQLN2 est une protéine ayant un rôle primordial dans le système de dégradation du protéasome (UPS) ainsi que pour l’autophagie. Cette protéine est responsable de la liaison entre les protéines destinées à être dégradées avec l’UPS. Il a été démontré dernièrement in vitro et in vivo que la mutation d’UBQLN2 est liée à l’agrégation de TDP-43. Cependant, le mécanisme exact de ce phénomène reste grandement incompris et nécessite encore beaucoup d’attention et de travail.
Dans ce mémoire, nous avons utilisé pour notre étude des cellules en culture afin de surexprimer les formes natives et mutantes d’UBQLN2 humain (hUBQLN2) pour étudier l’effet d’UBQLN2 sur la protéine TDP-43. Notre équipe a réussi à démontrer dernièrement dans les cellules de neuroblastome de souris (Neuro2a) que la surexpression de l’UBQLN2 entraînait une délocalisation de TDP-43 du noyau vers le cytoplasme en plus de son accumulation anormale dans
iii
des agrégats. De plus, l’effet synergique entre les formes mutées de TDP-43 et UBQLN2 a également été démontré dans un modèle murin dans notre article paru en 2018 comme quoi la mutation d’UBQLN2 influence grandement la protéinopathie de TDP-43.
À la suite de ces constats, nous avons orienté nos études sur le phénomène synergique entre UBQLN2 et TDP-43 qui est encore grandement méconnu. Pour ce faire, une analyse complète et exhaustive de la littérature a été effectuée afin de bien comprendre la mutation UBQLN2 et consiste en la première revue littéraire couvrant la totalité des ouvrages publiés sur la mutation d’UBQLN2 dans la SLA. Par la suite, nous avons convenu d’étudier l’effet de la mutation
d’UBQLN2P497H sur le transport nucléo-cytoplasmique dans les cellules Neuro2a ainsi que dans
les tissus de souris transgéniques. Les souris utilisées sont les mêmes que pour l’article Picher-Martel et al., 2018, soit des souris simple transgénique UBQLN2P497H et TDP-43G348C ainsi que la première souris double transgénique arborant UBQLN2P497H/TDP-43G348C. Les souris doubles transgéniques se sont avérées très intéressantes. En effet, elles ont développé les caractères typiques retrouvés chez les patients SLA/DFT avec une perte motoneuronale accompagnée de dégénérescence axonale, atrophie musculaire, gliose, trouble moteur ainsi que cognitif en plus d’agrégations cytoplasmique de TDP-43 importantes. À l’aide de ce modèle unique nous avons approfondi notre compréhension des déficits observés au niveau du transport nucléo-cytoplasmique, déficit grandement observé chez les patients SLA.
Nous avons observé que le transport nucléo-cytoplasmique était davantage et significativement altéré dans les cellules co-transfectées avec les gènes encodant pour les deux protéines mutées plutôt que transfectées avec une seule. Nous avons également observé pour les souris double transgéniques comparées aux souris simples pour l’une ou l’autre de ces protéines. Nos résultats suggèrent donc que la mutation d’UBQLN2 exacerbe significativement la protéinopathie de TDP-43 et engendre une perturbation importante du transport nucléo-cytoplasmique ainsi que des complexes du pore nucléaire.
iv
En conclusion, ce mémoire démontre un rôle important de la mutation d’UBQLN2 sur TDP-43 et leur grande affinité à augmenter les déficits du transport nucléo-cytoplasmique. Ceci suggère donc qu’UBQLN2 et TDP-43 sont intimement liés et peuvent jouer un rôle synergique dans la physiopathologie de la SLA. Le modèle murin double transgénique pourra indubitablement être utilisé en laboratoire afin de tester de nouvelles approches thérapeutiques.
v
Table des matières
RÉSUMÉ ... II LISTES DES FIGURES ... VIII LISTE DES ABRÉVIATIONS ... IX REMERCIEMENTS ... XII AVANT-PROPOS ... XIV
INTRODUCTION ... 1
1.1 LA SCLÉROSE LATÉRALE AMYOTROPHIQUE ... 1
1.1.1 Généralités ... 1
1.1.2 Épidémiologie ... 1
1.1.3 La sclérose latérale amyotrophique au niveau clinique ... 3
1.1.4 La démence fronto-temporale ... 4
1.2 MÉCANISMES MOLÉCULAIRES DE LA SLA ... 5
1.2.1 TAR DNA binding protein 43 (TDP-43) ... 5
1.2.1.1 Métabolisme des ARNs ... 6
1.2.1.2 Modifications post-traductionnelles ... 8 1.2.1.3 Granules de stress ... 9 1.2.1.4 Délocalisation et agrégation de TDP-43 ... 10 1.2.2 Ubiquiline-2 (UBQLN2) ... 11 1.2.2.1 Structure ... 11 1.2.2.2 UBQLN2 et l’UPS ... 12 1.2.2.3 UBQLN2 et l’autophagie ... 14
1.2.2.4 UBQLN2 dans la sclérose latérale amyotrophique ... 16
1.2.3 Transport nucléo-cytoplasmique ... 19
1.2.3.1 Généralités ... 19
1.2.3.2 Dysfonction du transport nucléo-cytoplasmique dans la SLA ... 22
1.3 HYPOTHÈSES ET OBJECTIFS DU MÉMOIRE ... 25
1.4 RÉSUMÉ ... 29
1.5 ABSTRACT ... 29
1.6 BACKGROUND ... 31
1.7 UBQLN2 ... 32
1.8 BIOLOGICAL FUNCTIONS ... 32
1.8.1 UBQLN2 and the ubiquitin-proteasome system ... 32
vi
1.9 UBQLN2 MUTATIONS IN FAMILIAL ALS ... 34
1.9.1 Proteasome and autophagy impairment ... 35
1.9.2 Direct interaction with TDP-43 promoting aggregation ... 37
1.9.3 UBQLN2 in neuroinflammation ... 38
1.9.4 Stress granules ... 39
1.10 ALS ANIMAL MODELS OF UBQLN2 ... 41
1.11 CONCLUSION AND FUTURE DIRECTIONS ... 42
1.12 REFERENCES ... 44
1.13 FIGURES LEGEND ... 48
CHAPITRE 3. UBQLN2 MUTATION EXACERBATES TDP-43 PROTEINOPATHY AND DISRUPT NUCLEAR PORE COMPLEXES ... 54
1.14 RÉSUMÉ ... 56
1.15 ABSTRACT ... 57
1.16 INTRODUCTION ... 58
1.17 METHODS ... 60
1.17.1 Cell Cultures and transfection ... 60
1.17.2 Generation of transgenic mice ... 60
1.17.3 Protein extraction and Western blot for cells and mice ... 60
1.17.4 Immunofluorescence on cells and tissues ... 62
1.17.5 Immunoprecipitation ... 62
1.17.6 Statistical analysis ... 63
1.18 RESULTS ... 63
1.18.1 Co-transfection of mutant UBQLN2P497H and TDP-43G348C causes a significant perturbation of the Ran cycle 63 1.18.2 The co-transfection of mutant UBQLN2P497H and TDP-43G348C causes mislocalization of RanGAP1 and NUPs as well as changes in their cellular levels in Neuro2a cells. ... 64
1.18.3 UBQLN2 mutation modifies NPCs proteins expression in vivo in the spinal cord. ... 65
1.19 DISCUSSION ... 66 1.20 CONCLUSION ... 71 1.21 DECLARATIONS... 71 1.22 ABBREVIATIONS ... 72 1.23 REFERENCES ... 73 1.24 FIGURE ... 76 DISCUSSION GÉNÉRALE ... 82
vii
CONCLUSION ... 91 BIBLIOGRAPHIE ... 93
viii
Listes des figures
FIGURE 1SCHÉMA DE LA RELATION ENTRE L’ORGANISATION DES VOIES DE SIGNALISATION MOTRICE ATTEINTES, DES DIFFÉRENTES
MALADIES ET CERTAINS GÈNES IMPLIQUÉS DANS LES MALADIES AFFECTANT LES NEURONES MOTEURS.(JAMES,2006) ... 3
FIGURE 2REPRÉSENTATION SCHÉMATIQUE DES DOMAINES DE TDP-43 ET DES MUTATIONS RATTACHÉES (PRASAD ET AL.,2019) ... 7
FIGURE 3FONCTIONS NORMALES DE TDP-43(LAGIER-TOURENNE,POLYMENIDOU AND CLEVELAND,2010) ... 7
FIGURE 4REPRÉSENTATION SCHÉMATIQUE DES DOMAINES D’UBQLN2(RENAUD ET AL.2019) ... 12
FIGURE 5RÔLES D’UBQLN2 DANS LE SYSTÈME UBIQUITIN-PROTÉASOME ET L’AUTOPHAGIE (RENAUD ET AL.,2019) ... 14
FIGURE 6UBQLN2 INDUIT L’AUTOPHAGIE D’AGRÉGATS DE PROTÉINES GRÂCE À HSP70 JUSQU’AU PROTÉASOME (HJERPE ET AL.,2016) ... 16
FIGURE 7LES DOMAINES D’UBQLN2 AVEC LES DIFFÉRENTES MUTATIONS OBSERVÉES SUR CHACUN DES SITES (DILLEN,2013). ... 17
FIGURE 8NUCLÉOPORINES CONSTITUANT LE COMPLEXE DU PORE NUCLÉAIRE ET LEURS DIFFÉRENTES CONSÉQUENCES RÉSULTANT DE LA MUTATION DE TDP-43 DANS LA SLA(CHOU ET AL.,2018) ... 20
FIGURE 9SCHÉMATISATION DES DIVERS MÉCANISMES NÉCESSAIRES À L’IMPORTATION ET EXPORTATION DE PROTÉINES ENTRE LE NOYAU ET LE CYTOPLASME ... 22
FIGURE 10DÉFICITS DU TRANSPORT NUCLÉO-CYTOPLASMIQUE DANS LA PROTÉINOPATHIE DE TDP-43 POUR LA SLA(GASSET-ROSA ET AL.,2019B) ... 25
FIGURE 11SCHEMATIC REPRESENTATION OF THE DOMAIN ARCHITECTURE OF THE HUMAN UBIQUILIN-2 GENE. ... 48
FIGURE 12ROLES OF UBQLN2 IN PROTEIN DEGRADATION IN BOTH THE UPS AND THE AUTOPHAGY SYSTEMS. ... 49
FIGURE 13THE INTEGRATIVE MODEL FOR WT AND MUTANT UBQLN2 PATHOLOGY IN ALS. ... 50
FIGURE 14THE DOUBLE MUTATION EXACERBATES NUCLEOCYTOPLASMIC TRANSPORT DEFICITS IN NEURO2A CELLS. ... 76
FIGURE 15CO-TRANSFECTION OF MUTANT PCMV-UBQLN2P497H/PCMV-TDP-43G348C SIGNIFICANTLY DISRUPTS RANGAP1 AND NPCS ... 78
FIGURE 16DOUBLE TRANSGENIC MICE MUTATION UBQLN2P497H/TDP-43G348C DISRUPTS NCT ... 78
FIGURE 17ABNORMAL MORPHOLOGY OF THE NUCLEUS OF MOTOR NEURONS IS MORE IMPORTANT FOR THE DOUBLE TRANSGENIC (UBQLN2P497H/TDP-43G348C) MICE THAN THE OTHER GROUPS. ... 81
ix
Liste des abréviations
SLA sclérose latérale amyotrophique TDP-43 TAR-DNA binding protein 43 SOD1 superoxyde dismutase 1
FUS Fused in Sarcoma UBQLN2 Ubiquiline-2 OPTN optineurin
MNS motoneurone supérieur MNI motoneurone inféfieur DFT démence fronto-temporale EMG électromyographie
AMPA alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid NF neurofilaments
RE réticulum endoplasmique PERK PKR-like ER kinase
IRE1 inositol-requiring transmembrane Kinase ATF6 activating transcription factor 6
XBP1 X-Box-binding protein 1
eIF2α eukaryotic initiation factor 2 alpha GFAP glial fibrillary acidic protein NF-κB Nuclear factor kappa-B
hnRNPs heterogeneous nuclear ribonucleoproteins NTD domaine N-terminal
CTD domaine C-terminal GS granules de stress
TIA-1 T-cell restricted intracellular antigen-1 PABP poly (A)-binding protein
G3BP1 Ras Gap SH3 domain binding protein 1 UPS ubiquitin-proteasome system
x UBL N-terminal ubiquitin-like
UBA C-terminal ubiquitin-associated STI1 stress-induced protein1-like
Ubxd8 ubiquitin regulatory X domain-containing protein 8 AMC autophagie médiée par des chaperonnes
LC3 microtubule-associated protein 1A/1B-light chain 3 APG9A autophagy-related protein 9A
ATG16 autophagy-related 16
TNC transport nucléo-cytoplasmique
MN membrane nucléaire
xi
Je dédicace ce mémoire à mon cher ami Louis-Charles Béland, qui a sauvé ce manuscrit en me
recommandant fortement de changer le mot concordant pour conformément à la page 31. Merci. Que nenni.
xii
Remerciements
J’aimerais tout d’abord remercier mon directeur de recherche sans qui ma maîtrise et tous mes projets rattachés n’auraient pas été possible. Merci au Dr Jean-Pierre Julien de m’avoir accueillie et surtout de m’avoir donné ma chance au sein de votre laboratoire. J’ai apprécié votre confiance, votre support et l’encouragement dont vous m’avez témoigné au cours de ces deux dernières années. Vous avez été un mentor qui a su me transformer professionnellement en scientifique et je suis grandement reconnaissante.
Je voudrais par la suite remercier mon ami Louis-Charles Béland de m’avoir supporté pendant 2 ans. C’est une longue période où tu as subi mes excès de leadership négatif à rouler ma chaise à côté de toi et à te déranger dans ton travail pour te parler de choses insignifiantes. Tu as rendu ma maîtrise beaucoup plus amusante et je n’ai pas (encore) sombré dans la dépression grâce à toi et à tout le plaisir que nous avons eu ensemble. C’est difficile à avouer mais je vais beaucoup m’ennuyer.
Je ne voulais pas remercier Vincent Picher-Martel, mais je n’ai pas trop le choix. Merci là.
Je remercie également une personne qui, malgré son cours passage de 4 mois ou moins au laboratoire, a su être un ami et un professeur marquant pour moi. Merci Patrick Cordeau pour ta patience avec mes absurdités/vulgarités, nos séances de ping-pong/balle au mur pour me vider de mon excès de leadership négatif et pour tout ce que tu m’as appris dans le grand domaine qu’est la science. Tu es un français pô pire. Je suis très excitée par tous les projets qui nous empêchent de profiter de la vie et j’ai toujours eu confiance que tu m’amènerais où je le désire dans ma carrière. Pardon de me coller à toi comme une sangsue.
Je remercie également une jeune femme exceptionnelle, Tereza Ljutić (la grosse Thérèse), en qui j’ai découvert une grande et profonde amitié. Merci d’être venue au Québec dans notre laboratoire
xiii
et de m’avoir porté si souvent compagnie à jouer au volleyball, manger des sushis et venir relaxer chez moi. Je m’ennuie terriblement de toi. Je suis enchantée de penser que nous allons nous voir plus souvent dans le futur. Je t’aime.
Je remercie également mes parents de tout le support dont ils m’apportent depuis que j’existe. Pardon de parfois être un parasite avec vous, votre soutient a toujours été extrêmement important pour moi. Je sais que vous êtes toujours derrière moi peu importe mes folles décisions et que malgré la distance vous allez toujours m’épauler. Merci. Je vous aime de tout mon cœur. Vous êtes mes humains préférés.
J’aimerais également remercier mes matantes préférées Christine Bareil et Geneviève Soucy, non seulement pour leur travail dans le laboratoire mais aussi pour leur accueil chaleureux et leur soutien au cours de ces deux dernières années. Vous êtes non seulement mes matantes mais aussi des amies avec qui j’ai eu tellement de plaisir à travailler avec. Merci pour cette maîtrise en si belle compagnie.
J’aimerais remercier tous les autres membres des laboratoires de Jean-Pierre et Jasna pour leur amitié/aide dans les dernières années : Silvia, Pierre, Romina, Sai, Sunny, Amélie, Banshi, Poojah, Daniel, Reza, Hejer et Revathy.
xiv
Avant-Propos
Dans le chapitre 2, l’article « Key role of UBQLN2 in pathogenesis of amyotrophic lateral sclerosis and frontotemporal dementia » a été publié dans le journal Acta Neuropathologica Communications le 18 juillet 2019 (doi: 10.1186/s40478-019-0758-7). Il n’y a eu aucune modification entre la version publiée et la version intégrée dans ce mémoire. Je suis l’auteure principale de cet article.
Auteurs : Laurence Renaud, Vincent Picher-Martel, Philippe Codron and Jean-Pierre Julien
Contribution des auteurs : Laurence Renaud a réalisé la revue de la littérature, l’écriture de l’article et la réalisation des figures. Philippe Codron a aidé à la structure du papier et à la correction du document. Vincent Piché-Martel et Jean-Pierre Julien ont conçu, supervisé le projet et corrigé l’article.
Dans le chapitre 3, l’article « Impact of ALS-related UBQLN2P497H mutation on
nucleocytoplasmic transport » n’a pas encore été publié. Il manque quelques expériences avant de pouvoir être soumettre à un journal. Je suis l’auteure principale de cet article.
Auteurs : Laurence Renaud, Vincent Picher-Martel, Philippe Codron and Jean-Pierre Julien.
Contribution des auteurs : Laurence Renaud a effectué 95% des expériences de l’article. Vincent Picher-Martel a fait la caractérisation des souris dans un papier antérieur. Philippe Codron a effectué quelques immunofluorescences sur tissus et cellules, corrigé l’article et conseillé pour la réalisation du projet. Jean-Pierre Julien a conçu et supervisé le projet.
1
Introduction
1.1 La sclérose latérale amyotrophique
1.1.1 GénéralitésLa sclérose latérale amyotrophique (SLA), ou la maladie de Lou Gehrig en l’honneur du joueur de baseball des Yankees de New-York, se caractérise par une perte progressive des motoneurones supérieurs et inférieurs menant à une faiblesse progressive ainsi qu’une paralysie. L’espérance de vie est très courte, soit de 2 à 5 ans suivant l’apparition des premiers symptômes. Malgré le développement de deux médicaments pour la maladie, aucun traitement efficace pouvant ralentir de manière significative ou même arrêter la maladie n’existe à ce jour. Nous connaissons maintenant des mutations dans plusieurs gènes impliqués dans la forme familiale de la maladie, notamment les gènes codant pour la superoxyde dismutase 1 (SOD1), TAR-DNA binding protein 43 (TDP-43), C9ORF72, Fused in Sarcoma (FUS) et ubiquiline-2 (UBQLN2). Plusieurs mécanismes ont pu être mis en évidence suite à l’identification de ces gènes mais encore beaucoup d’études devront être conduites requises afin de comprendre avec précision le lien entre ces mécanismes et la SLA.
1.1.2 Épidémiologie
Une méta-analyse systématique a été effectuée en 2017 sur plus de 13 000 patients dans 45 études différentes afin de recenser l’incidence de patients atteints de la SLA par continent. En Asie, environ 0,83 nouveaux cas/100 000 habitants ont été dénombrés tandis qu’en Europe et en Amérique du Nord l’incidence est plus élevée, soit de 1,81 nouveaux cas/100 000 habitants (Marin et al., 2017). Au Canada, il est estimé par l’organisme SLA Canada qu’environ 3000 personnes vivent avec la maladie actuellement et qu’à chaque année environ 1 000 Canadiens meurent de la SLA. Un nombre similaire de Canadiens reçoivent un diagnostic de SLA à chaque année (Canada, 2018). L’âge moyen de l’apparition des premiers symptômes varie de 61,8 ± 3,8 ans pour les hommes et de 64.4 ± 2.9 ans pour les femmes au diagnostic (Chio et al., 2013).
2
Plusieurs facteurs de risques pouvant possiblement mener au développement de la SLA ont été proposés mais très peu peuvent être confirmés. En effet, outre les facteurs génétiques décrivant la forme familiale de la SLA (fSLA), l’analyse des possibles facteurs pouvant mener à la forme sporadique n’es pas conclusive. Cependant, bien que le facteur génétique soit mieux décrit, cela n’affecte que 5 à 10% des patients atteints de la SLA. Nous comptons 126 gènes impliqués dans la fSLA jusqu’à ce jour, dont ceux mentionnés précédemment. Parmi les facteurs non-génétiques, il a été proposé que les traumatismes crâniens subis par le sport puissent avoir un rôle important dans le développement de la maladie. En effet, suite au décès du célèbre joueur de baseball Lou Gehrig, les chercheurs se sont concentrés sur l’incidence possible du sport et le lien entre les traumatismes crâniens répétés et le développement de la SLA (Schmidt et al., 2010; Sundman, Hall and Chen, 2014). Une méta-analyse a également confirmé une augmentation significative de l’incidence de la SLA chez des joueurs professionnels de soccer et football (Lacorte et al., 2016). Le tabagisme a également été proposé comme une source potentielle d’incidence de la SLA chez les patients mais les études sont plutôt partagées sur le sujet. Certaines proposent en effet un risque augmenté à développer la SLA (Nelson et al., 2000; Armon, 2003; Armon, 2009; Weisskopf et al., 2010; Sutedja et al., 2007) tandis que d’autres ne montre pas de corrélation significative entre le tabagisme et la SLA (Pamphlett and Ward, 2012; Alonso, Logroscino and Hernan, 2010). D’autres études sont donc nécessaires afin d’élucider le lien exact entre le tabagisme dans le développement de la SLA.
Un facteur de risque environnemental davantage étudié mais dont les résultats sont encore une fois contradictoires, est celui sur l’exposition à des métaux lourds ou des toxines environnementales. En effet, plusieurs études supportent une corrélation positive entre l’exposition au plomb et le développement de la SLA (Felmus, Patten and Swanke, 1976; Roelofs-Iverson et al., 1984; Chancellor et al., 1993; Armon, 2003) tandis que d’autres n’ont démontré aucune relation entre les deux (Gresham et al., 1986; McGuire et al., 1997; Gait et al., 2003).
3
1.1.3 La sclérose latérale amyotrophique au niveau clinique
La SLA est caractérisée par une perte progressive et rapide des neurones moteurs autant supérieurs (MNS) qu’inférieurs (MNI). Initialement, la majorité des cas cliniques débutent par des faiblesses au niveau des mains ou jambes unilatéralement pour ensuite progresser vers le membre collatéral et éventuellement aux autres parties du corps. Les patients montrent initialement de la lenteur, de la rigidité et de l’incoordination du mouvement avant de progresser rapidement vers l’atrophie et la paralysie. L’évolution de la maladie varie énormément d’un patient à l’autre mais en moyenne la durée de vie demeure très courte, soit de 2 à 5 ans suivant le début des symptômes. Il s’agit cependant d’une moyenne et les extrêmes sont également possibles, soit une évolution rapide sur moins de 6 mois ou au contraire une progression très lente pouvant s’étaler sur plusieurs années, comme par exemple le cas du Dr Stephen Hawking, célèbre physicien ayant vécu plus de 50 ans avec la
maladie. Plusieurs facteurs peuvent expliquer cette variation importante de l’espérance de vie des patients, soit l’âge, la présence de symptômes bulbaires, la progression de l’atrophie musculaire, le degré d’atteinte respiratoire, le type de mutation de la maladie, l’état nutritionnel du patient, etc. (Cudkowicz et al., 1997); (Desport et al., 1999).
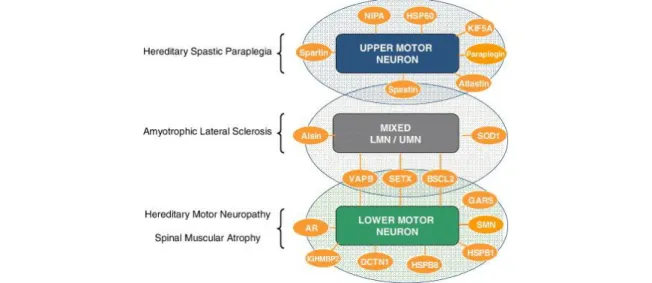
Figure 1 Spectre des gènes impliqués entre les maladies affectant les neurones moteurs.
Schéma de la relation entre l’organisation des voies de signalisation motrice pour la paraplégie héréditaire spastique, la SLA, la neuropathie motrice héréditaire et l’atrophie musculaire spinale (James, 2006)
4
La SLA, la sclérose latérale primaire ainsi que l’atrophie musculaire progressive sont trois maladies du même spectre touchant les neurones moteurs. Il n’est cependant pas encore certain si ces maladies consistent en une variante d’une seule maladie ou si ce sont trois maladies indépendantes (Al-Chalabi et al., 2016). Au moment du diagnostic, il peut être complexe pour les médecins lors de l’apparition des premiers symptômes de déterminer avec exactitude laquelle de ces trois maladies est en cause, les symptômes étant similaires et que les trois maladies partagent un spectre important de symptômes et gènes mutés (Fig. 1) (James, 2006). Il est donc primordial de pouvoir différencier ces différentes formes de maladies motrices pour les essais cliniques, le début des traitements et pour évaluer le pronostic des patients puisque, par exemple, les patients atteints de la sclérose latérale primaire ont une espérance de vie plus importante vis-à-vis les patients SLA (Jackson et al., 2009).
1.1.4 La démence fronto-temporale
Au début des années 1990, 70 cas de patients avec la SLA dans le cadre d’une étude démontraient des troubles cognitifs de démence fronto-temporale (DFT). Depuis, il est bien connu que la SLA et la DFT partagent un grand spectre de gènes communs mutés (Mitsuyama, 1993). En effet, des patients SLA/DFT partageaient des aspects neuropathologiques communs au niveau des tissus, dont des inclusions renfermant ubiquitine, et cette caractéristique devint par la suite le principal critère pathologique du continuum de la SLA/DFT (Leigh et al., 1988). Une seconde découverte renforça le spectrum entre la SLA ainsi que la DFT en 2006 par la présence de la protéine TDP-43 dans les inclusions cytoplasmiques comme principal composant pour les deux pathologies (Neumann et al., 2006). De plus, comme mentionné précédemment, les deux maladies partagent un large éventail de gènes mutés, soit C9ORF72, TDP-43, UBQLN2 et SQSTM1 (Ringholz et al., 2005). Il existe trois formes cliniques de la DFT observée chez les patients ; l’aphasie primaire progressive sémantique, l’aphasie primaire progressive non-fluente et finalement la DFT comportementale (DFTc). À l’histopathologie, des inclusions de TDP-43, de tau ou d’autres protéines sont fréquemment observées dans chacune de ces formes et la DFTc représente la catégorie la plus fréquente chez les patients SLA. Cette dernière est caractérisée
5
par des changements de comportement ainsi que de personnalité, soit une déshinibition de comportements sociaux menant à un comportement inapproprié. Les patients démontrent également une perte d’empathie, de l’inertie, de l’hyperoralité ainsi que des changements alimentaires, des comportements stéréotypés ou compulsifs/ritualistes sont également possibles. Des troubles de mémoire sont également notés chez certains patients mais ne constituent pas un trouble majeur de la pathologie. Finalement, la prévalence de la DFT parmi les patients SLA fluctue beaucoup, il est estimé qu’environ 50% des patients SLA démontrent des troubles cognitifs et qu’uniquement 10 à 15% peuvent être considéré comme répondant aux critères de démence et que seulement 15% des patients avec la DFT développent la SLA (Neary, Snowden and Mann, 2000).
1.2 Mécanismes moléculaires de la SLA
Nous devons une grande partie de nos connaissances des mécanismes pathologiques grâce à la forme familiale de la SLA, qui représente malheureusement seulement 5 à 10% des cas répertoriés. La SLA est encore bien incomprise et malgré la découverte de certains mécanismes moléculaires menant à la pathologie, la SLA est extrêmement complexe et fortement soupçonnée d’être multifactorielle. Parmi la multitude de mécanismes pathologiques de la SLA, ceux pertinents pour cette étude consiste principalement en une dérégulation des ARNs, un déficit important du transport nucléo-cytoplasmique, délocalisation et formation d’agrégats intracytoplasmiques et une diminution de l’efficacité au niveau de la dégradation des protéines par autophagie et le système ubiquitine du protéasome (UPS).
1.2.1 TAR DNA binding protein 43 (TDP-43)
TAR DNA-binding protein 43 (TDP-43) est encodée par le gène TARDBP ou ALS10 et est impliquée dans l’épissage, la traduction, la dégradation et le transport des ARNm (Buratti and Baralle, 2001; Buratti et al., 2001; Hefferon et al., 2004). Au cours des années 2000, la présence de TDP-43 dans des agrégats ubiquitinés de patients SLA/DFT a été découverte et plusieurs mutations au niveau du gène ont été découvertes peu de temps après (Neumann et
6
al., 2006; Arai et al., 2006; Kabashi et al., 2008; Kwiatkowski et al., 2009). Cette découverte fut marquante pour la recherche, non seulement pour la SLA et la DFT, mais également pour l’Alzheimer, la maladie d’Huntington et la maladie de Parkinson dont les patients ont démontré les mêmes caractéristiques de protéinopathie de TDP-43 (Amador-Ortiz et al., 2007; Schwab et al., 2008; Chanson et al., 2010). Les dites caractéristiques de la protéinopathie de TDP-43 consistent principalement en la délocalisation cytoplasmique et l’agrégation de TDP-43.
1.2.1.1 Métabolisme des ARNs
TDP-43 fait partie de la grande famille des ribonucléoprotéines hétérogènes liant l’ARN (hnRNPs) et possède un domaine N-terminal (NTD), deux motifs de reconnaissance de l’ARN (RRM1-2) en plus d’un domaine C-terminal riche en glycine (CTD) (Fig. 2) (Gao et al., 2018; Berning and Walker, 2019; Prasad et al., 2019b). TDP-43 a multiples rôles, notamment dans la stabilité des ARNm, leur transcription, l’épissage alternatif et TDP-43 possède également les signaux nécessaires afin d’effectuer son transport vers le cytoplasme (Fig. 3) (Fiesel et al., 2012; Shiga et al., 2012; Mohagheghi et al., 2016; Ayala et al., 2011; Polymenidou et al., 2011). De récentes études suggèrent que TDP-43 joue un rôle important au niveau de la répression de la transcription des exons cryptiques non-conservés, exons impliqués dans la création de codons STOP prématurés ou de codons faux-sens lorsqu’introduit dans des ARNm (Ling et al., 2015). TDP-43 régularise également les microARNs en se localisant dans les fibres périchromatiques où les miRNA sont synthétisés (Lee, Lee and Trojanowski, 2011). Finalement, TDP-43 joue également un rôle majeur au niveau de la formation des granules de stress cytoplasmiques que nous aborderons au paragraphe 1.2.1.3 (Fig 3).
7
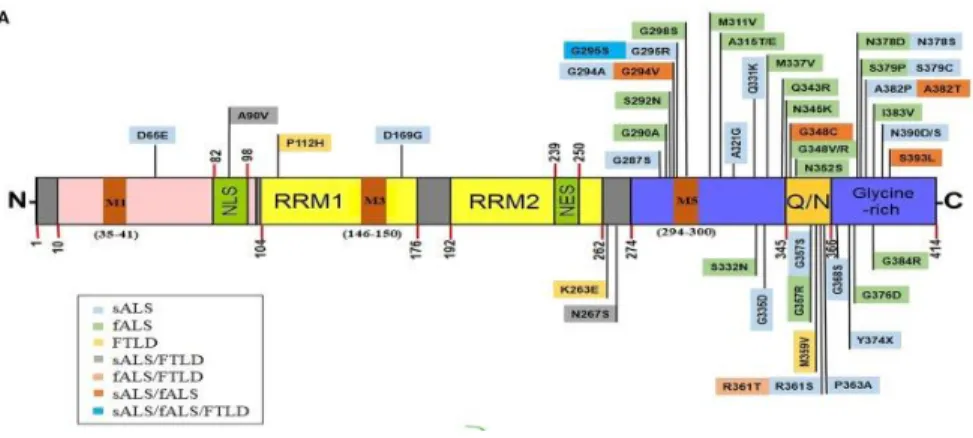
Figure 2 Représentation schématique des domaines de TDP-43 et des mutations rattachées.
TDP-43 est constitué d’un domaine NTD, de deux domaines RRM, un signal d’exportation nucléaire (NES), un signal de localisation nucléaire (NLS), un domaine de type prion sur le segment C-terminal et des motifs de localisation mitochondriaux (Prasad et al., 2019)
Figure 3 Fonctions normales de TDP-43.TDP-43 est impliqué dans: (A,B,C,D) la transcription; (E,F,G) l’épissage; (H) le traitement des micro-ARN; (I) le transport nucléo-cytoplasmique (J) les granules de stress et (K) le transport des ARN et leur translation locale (Lagier-Tourenne, Polymenidou and Cleveland, 2010)
Une caractéristique importante des mutations de TDP-43 trouvées dans la SLA est que celles-ci sont assocelles-ciées à sa délocalisation cytoplasmique de la protéine et son assemblage en
8
agrégats anormaux. Cette délocalisation nucléaire de la protéine suggère fortement une perte des fonctions nucléaires de TDP-43. TDP-43 possède un domaine de localisation nucléaire N-terminal (NLS) ainsi qu’une séquence d’exportation nucléaire (NES) localisé dans le RRM2 (Fig. 2) (Lee, Lee and Trojanowski, 2011). En raison de sa séquence NES, TDP-43 ne se retrouve localisé qu’à 30% au niveau cytoplasmique plutôt que nucléaire dans des conditions normales (Barmada et al., 2010). Les résultats restent mitigés et sujet de débats scientifiques, certains suggérant que la perte de TDP-43 au niveau nucléaire n’est pas nécessaire en soi afin de créer l’environnement cellulaire toxique causant la SLA, mais qu’il s’agit plutôt de la localisation cytoplasmique anormale des mutants TDP-43 qui seraient responsables de cette toxicité (Barmada et al., 2010; Arnold et al., 2013; Austin et al., 2014). Ceci suggère qu’en plus d’une perte des fonctions normales, TDP-43 pourrait également agir par gain de fonctions toxiques dans la pathologie. De plus, dans la pathogénèse de la SLA, TDP-43 a été observée comme quoi sa mutation induisait une perte de capacité à épisser certains ARNm (Prasad et al., 2019b). Les mutations de TDP-43 ont également été démontrées comme diminuant l’expression de FUS et augmentant celle de progranuline (Polymenidou et al., 2011). Même si ces résultats suggèrent une perte des fonctions de TDP-43 à jouer son rôle au niveau de l’épissage des ARN pré-messager, d’autres études sont nécessaires afin de bien comprendre la problématique et déterminer si ces conditions peuvent affecter le développement et l’évolution de la SLA. De plus il ne faut pas négliger la possibilité que TDP-43 subit une perte de fonctions normales en plus d’un gain de fonctions toxiques.
1.2.1.2 Modifications post-traductionnelles
Les modifications post-traductionnelles, notamment la phosphorylation, les coupures aberrantes protéiques et l’ubiquitination, participent à la formation des inclusions cytoplasmiques et jouent un rôle important au niveau de l’agrégation (Sreedharan et al., 2008). La phosphorylation des sérines 379, 403, 404 et 409 de TDP-43 induit son oligomérisation et agrégation anormale mais nos connaissances par rapport à la phosphorylation de TDP-43 sont malheureusement limitées (Hasegawa et al., 2008). La mutation des résidus 409 et 410 empêcherait leur phosphorylation et diminuerait donc par le
9
fait même la toxicité de TDP-43 lorsque surexprimé (Liachko, Guthrie and Kraemer, 2010). Par ailleurs, la phosphorylation de TDP-43 accroitrait sa durée de vie, contribuant donc par le fait même à son accumulation anormale (Zhang et al., 2010). Chez les patients atteints de SLA ainsi que dans les modèles animaux de la forme familiale de la maladie, la coupure aberrante du fragment C-terminal de TDP-43 mène à la formation de structures beta amyloïdes et participe activement à la formation d’inclusions cytoplasmiques (Igaz et al., 2009; Zhang et al., 2009; Conicella et al., 2016). Dans des conditions normales, ces inclusions sont éliminées grâce au système ubiquitin-protéasome (UPS) et par autophagie. Cependant, un dysfonctionnement de l’UPS ou l’autophagie par UBQLN2 est soupçonné d’être un facteur important de l’accumulation et la persistance des agrégats cytoplasmique de TDP-43 dans les tissus de patients (Neumann et al., 2006).
1.2.1.3 Granules de stress
Les granules de stress (GS) consistent en des formations cytoplasmiques se formant en réponse au stress, par exemple en présence de stress oxydatif, d’infection, stress du réticulum endoplasmique (RE), stress osmotique, d’inhibition du protéasome, etc. (Aulas and Vande Velde, 2015). Ces inclusions contiennent des protéines ainsi que des ARNm non-traduits, des marqueurs classiques comme T-cell restricted intracellular antigen-1 (TIA-1), Ras Gap SH3 domain binding protein 1 (G3BP1), poly (A)-binding protein (PABP) et des facteurs d’initiation de la traduction (eIF2α, eIF3 ou eIF4A/B/G) (Decker and Parker, 2012). Il a été démontré que TDP-43 est recruté dans les GS suivant divers stress, notamment le stress du RE, le stress oxydatif et particulièrement l’inhibition du protéasome. Son recrutement dans les GS dépend de sa liaison aux ARNs par son domaine RRM1 en plus de sa liaison aux protéines par son domaine C-terminal (Colombrita et al., 2009). Toutefois, le rôle exact et l’impact de la participation de TDP-43 dans la formation des GS est encore grandement incompris et mérite plus d’étude. Il a toutefois été démontré que la délétion de TDP-43 ne prévenait pas la formation de GS mais pouvait toutefois retarder le processus (Colombrita et al., 2009; Aulas, Stabile and Vande Velde, 2012).
10
Au niveau clinique, les tissus de certains patients atteints de la SLA/DFT montrent une colocalisation de TDP-43 avec TIA-1 et PABP dans les inclusions cytoplasmiques, suggérant donc que les GS peuvent être les précurseurs des agrégats retrouvés dans la pathologie (McGurk et al., 2014). Toutefois, ce sujet est grandement discuté et débattu par les chercheurs. En effet, certains ont démontré que l’inhibition des GS et la réduction de la formation d’agrégats était possible grâce à des inhibiteurs de traduction et que l’ablation d’ataxin-2, protéine essentielle à la formation des GS, réduisait la toxicité induite par TDP-43 (Liu-Yesucevitz et al., 2010; Becker et al., 2017). Cependant, d’autres études supportent plutôt l’hypothèse que des mutations dans TDP-43 n’apportent aucun changement ni de diminution au niveau du nombre de GS, niant ainsi le rôle de TDP-43 dans la formation des GS (McDonald et al., 2011; Orru et al., 2016). De plus, chez certains patients SLA/DFT possédant une mutation au niveau de TIA-1 aucune colocalisation entre TDP-43 et TIA-1 n’a été observé malgré un nombre important d’agrégats positifs pour TDP-43 (Hirsch-Reinshagen et al., 2017). Ainsi, la question à savoir si les GS sont recrutées lors de la formation des agrégats ou plutôt si les GS représentent un précurseur d’agrégats n’est toujours pas démystifié et nécessite davantage de recherche.
1.2.1.4 Délocalisation et agrégation de TDP-43
Comme mentionné plus haut, la plupart des fonctions connues de TDP-43 sont effectuées dans le noyau de la cellule. Toutefois, sa séquence contient les signaux nécessaires à son transport entre le noyau et le cytoplasme, phénomène fréquent au sein d’une cellule normale. Dans la pathologie de la SLA, TDP-43 mutante est amenée à être délocalisée du noyau et à se retrouver en quasi-totalité dans le cytoplasme, formant par la suite des agrégats potentiellement toxiques. Ces agrégats ne sont toutefois pas uniquement limités au soma, on en retrouve également dans les neurites ainsi que dans le noyau des cellules, même si ces agrégats sont moins fréquents et moins bien décrits (Mackenzie et al., 2011; Neumann et al., 2006). Les études se sont davantage concentrées au niveau des agrégats cytoplasmiques à savoir si ceux-ci causaient une toxicité pouvant mener à la mort neuronale. Il a été déterminé que des mutations au niveau du NLS déclenchaient la délocalisation cytoplasmique en plus de l’agrégation de TDP-43, autant pour sa forme native que pour celles mutées (Igaz et al.,
11
2011; Winton et al., 2008). De plus, grâce à la microscopie en temps réel, il a été possible d’observer que la neurotoxicité est directement liée avec la quantité de TDP-43 délocalisé dans le cytoplasme plutôt qu’à la spécificité de la mutation ou même qu’à la présence d’agrégats (Barmada et al., 2010).
1.2.2 Ubiquiline-2 (UBQLN2)
UBQLN2 est une protéine impliquée dans le transport des protéines ubiquitinées jusqu’au protéasome où elles sont dégradées. Il s’agit d’ailleurs du sujet d’étude le plus travaillé en ce qui à trait à l’UBQLN2, soit le mécanisme par lequel la mutation d’UBQLN2 affecte les fonctions normales de l’UPS et prévient la dégradation normale des protéines. Également, l’implication d’UBQLN2 dans la délocalisation et la formation d’agrégats insolubles cytoplasmiques de TDP-43 est de mieux en mieux décrit dans la littérature. En effet, de récentes études démontrent un effet synergique entre les deux protéines mutées et une augmentation de la protéinopathie de TDP-43 chez les patients et modèles animaux par la présence d’UBQLN2 (Deng et al., 2011; Picher-Martel et al., 2018). Toutefois, UBQLN2 mutée n’affecte pas seulement TDP-43 ou l’UPS, mais a également été démontrée comme étant impliquée dans une dysfonction de l’autophagie (Hjerpe et al., 2016), la neuroinflammation (Picher-Martel et al., 2015; Picher-Martel et al., 2018) et finalement la formation de GS (Dao et al., 2018). Ces données illustrent donc le rôle important joué par UBQLN2 au sein de l’accumulation protéique anormale ainsi que les déficits des voies de dégradation au sein des pathogénèses de la SLA et DFT.
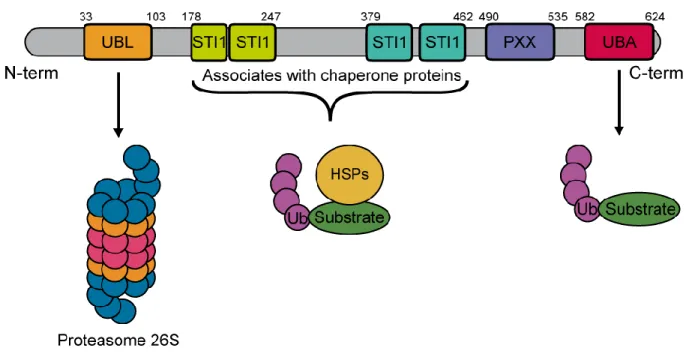
1.2.2.1 Structure
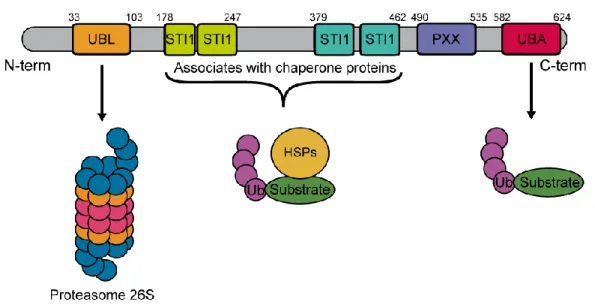
Le gène UBQLN2, également nommé Chap1/Dsk2 ou PLIC, est localisé sur le chromosome Xp11.21. UBQLN2 est une protéine faisant partie de la famille des ubiquiline de 66 kDa (Deng et al., 2011; Kaye and Shows, 2000). La protéine UBQLN2 contient un domaine ubiquitin-like (UBL) localisé à l’extrémité N-terminal qui a la capacité d’interagir avec le protéasome grâce à la sous-unité S5a du complexe 19S du protéasome 26S. UBQLN2 contient également un domaine ubiquitin-associated (UBA) sur son segment C-terminal qui
12
peut, quant à lui, s’attacher aux protéines marquées par des chaînes polyubiquitine (Walters et al., 2002). De plus, UBQLN2 contient 4 domaines stress-induced protein 1-like (STI-1 like) qui sont impliqués dans l’interaction avec des médiateurs d’autophagie ainsi que des protéines heat-shock. Finalement, le dernier domaine de la protéine consiste en un domaine contenant 12 répétitions PXX et est impliqué dans l’interaction protéine-protéine (Fig. 4) (Aitio et al., 2010). C’est ce domaine unique qui différencie UBQLN2 des autres membres de sa famille, notamment UBQLN1, 3 et 4. De plus, de manière intéressante, la plupart des mutations identifiées au sein du gène UBQLN2 sont localisées dans le domaine PXX. Il est toutefois nébuleux à savoir comment les mutations influencent la structure secondaire et les fonctions du domaine PXX de manière précise.
Figure 4 Représentation schématique des domaines d’UBQLN2. Le gène UBQLN2 possède un domaine ubiquitin-like (UBL) sur le segment N-terminal qui interagit avec le protéasome et un domaine ubiquitin-associated (UBA) sur le segment C-terminal pour l’activité de l’UPS. UBQLN2 a également quatre domaine stress-induced protein 1 (STI-1) et un domaine proline-rich repeat contenant 12 répétitions de PXX (Renaud et al., 2019)
1.2.2.2 UBQLN2 et l’UPS
UBQLN2 est normalement localisée dans le cytosol et UBQLN2 est principalement exprimée dans le cerveau, le cœur, le rein, le pancréas, la rate et d’autres tissus (Zhang and Saunders, 2009). UBQLN2 est essentiellement impliquée au niveau de l’homéostasie protéique, en guidant les protéines repliées ou redondantes vers le protéasome afin de se faire dégrader. L’UPS est un système indispensable et hautement régulé pour toutes les cellules. Le
13
protéasome 26S est composé d’un centre 20S, en forme de tonneau, et d’une ou deux particules régulatrices 19S. Le centre 20S est composé de deux anneaux heptagonaux α et deux anneaux β, les anneaux α étant responsables de dicter l’entrée protéique dans le tonneau de dégradation tandis que les anneaux β sont responsables de la coupure des peptides (Groll et al., 2000). Les protéines marquées par ubiquitine, un polypeptide très conservé de 76-résidues, sont les cibles afin de se faire dégrader par une cascade enzymatique comprenant trois étapes (Walters et al., 2002). Tout d’abord, l’enzyme activatrice d’ubiquitine E1 lie l’ubiquitine à la protéine d’intérêt grâce à un lien intermédiaire thiol-ester de haute énergie. Par la suite, l’ubiquitine est transférée à E2, soit l’enzyme de conjugaison d’ubiquitine. Celle-ci permet la liaison entre elle-même et E3, une enzyme ligase, et cette liaison permet le transfert final de l’ubiquitine sur un résidu lysine de la protéine destinée à se faire dégrader. Les protéines ainsi dites polyubiquitinées sont par la suite reconnues par le domaine UBL d’UBQLN2 et amenées à la structure S5a du protéasome 26S où elles sont dégradées (Fig.
5A) (McKinnon and Tabrizi, 2014).
De plus, UBQLN2 peut être activée par les heat shock protein 70 (HSP70). En effet, UBQLN2 est inactive sous des conditions normales de repos mais devient activée lorsque HSP70 se lie à des protéines, libérant un site de liaison sur la protéine UBQLN2. L’activation d’UBQLN2 induit sa liaison au protéasome 26S formant un complexe de dégradation (Hjerpe et al., 2016). Toutefois, en plus de sa participation active dans le transport des protéines ubiquitinées vers le protéasome, UBQLN2 joue également un rôle au niveau de la dégradation des protéines associées au réticulum endoplasmique (RE). UBQLN2 a la capacité d’interagir avec l’ubiquitin regulatory X domain-containing protein 8 protein (Ubxd8) qui se trouve à être localisé dans la membrane du RE et qui facilite le transport de substrats du endoplasmic reticulum-associated protein degradation (ERAD) jusqu’au cytosol (Xia et al., 2014). UBQLN2 peut également interagir avec Herp (homocysteine-induced ER protein), une autre protéine membranaire du RE qui est activée en condition de stress et qui induit une protection cellulaire en stabilisant l’homéostasie calcique et mitochondriales (Fig. 5A) (Kim et al., 2008).
14
Figure 5 Rôles d’UBQLN2 dans le système ubiquitin-protéasome et l’autophagie.A) UBQLN2 cible des substrats ubiquitinés et interagit avec des protéines du RE impliquées avec le ERAD, comme Herp et Ubxd8 afin d’amener les substrats à se faire dégrader par le 26S protéasome. B) UBQLN2 est impliquée dans la macro-autophagie et interagit avec LC3 indirectement par un mécanisme inconnu et peut délivrer les substrats ubiquitinés au domaine UBA. (Renaud et al., 2019)
1.2.2.3 UBQLN2 et l’autophagie
L’autophagie, dans les cellules des mammifères, se retrouve sous trois différentes formes : la micro-autophagie, la macro-autophagie et l’autophagie médiée par des chaperons (AMC). Chacune de ces voies livre sa cargaison aux lysosomes (Parzych and Klionsky, 2014). La micro-autophagie est la forme la moins connue des trois, nos connaissances actuelles permettent de décréter qu’elle implique l’invagination de la membrane du lysosome afin de capturer le substrat à dégrader (Mijaljica, Prescott and Devenish, 2011). La macro-autophagie est la mieux connue des trois formes. Celle-ci implique la formation de novo de vésicules cytoplasmiques ne provenant pas d’évagination d’autres organelles. Ces vésicules portent le nom d’autophagosomes et une caractéristique unique de celles-ci est qu’elles peuvent être formées à différents endroits dans le cytoplasme selon leurs cibles. L’autophagosome va fusionner avec le lysosome afin de former l’autolysosome et ainsi causer la dégradation de son contenu (Hayashi-Nishino et al., 2009; Yla-Anttila et al., 2009). Finalement, l’AMC nécessite l’intermédiaire de chaperonnes spécifiques à un pentapeptide
15
(KFERQ) et les protéines contenant KFERQ sont dépliées par l’action des chaperonnes et amenées au niveau du lysosome pour dégradation (Dice, 1990); (Orenstein and Cuervo, 2010). La chaperonne la plus couramment retrouvée pour accomplir ces fonctions est la protéine de choc thermique 70 kDa 8 (HSPA8/HSC70) (Chiang et al., 1989).
L’implication d’UBQLN2 dans l’autophagie est bien établie et décrite. Tout d’abord, une première étude a démontré l’interaction entre UBQLN2 et un marqueur d’autophagosome, soit le microtubule-associated protein 1A/1B-light chain 3 (LC3) (Fig. 5B) (N'Diaye et al., 2009a). Le mécanisme exact de cette interaction entre UBQLN2 et LC3 n’est pas encore connu. De plus, UBQLN2 a pu être observée dans des vésicules en compagnie d’optineurine (OPTN) (Osaka et al., 2015). OPTN est une protéine adaptatrice multifonctionnelle de liaison de l’ubiquitine et est principalement impliquée dans l’autophagie. Il a cependant été proposé que ce soit un facteur important impliqué dans la régulation de l’inflammation et a récemment été associée avec la SLA (Markovinovic et al., 2018). Ces vésicules positives pour UBQLN2 et OPTN l’étaient également pour Rab11 (ras-related protein), un marqueur d’endosome ainsi que p62, microtubule-associated protein 1A/1B-light chain 3 (LC3), autophagy-related protein 9A (APG91) et l’autophagy-related genes 16 (ATG16), qui sont toutes des molécules régulatrices de l’autophagie. Une étude a démontré que des mutations au sein d’UBQLN2 ou même d’OPTN pouvaient bloquer la formation de ces vésicules et ainsi affecter l’autophagie (Osaka et al., 2015). De plus, UBQLN2 peut également dégrader des agrégats ubiquitinées grâce à son domaine STI-1 qui peut interagir avec HSP70. Lorsque dans des conditions de repos, UBQLN2 est sous sa forme inactive mais peut être activée grâce à la liaison de HSP70 avec une cible destinée à se faire dégrader. UBQLN2, HSP70 et la cible sont par la suite dirigés au protéasome (Fig. 6) (Hjerpe et al., 2016).
16
Figure 6 UBQLN2 induit l’autophagie d’agrégats de protéines grâce à HSP70 jusqu’au protéasome. UBQLN2 peut interagir avec HSP70 par son domaine STI-1. Dans des conditions de repos, UBQLN2 est sous sa forme inactive mais peut être activée grâce à la liaison de HSP70 avec une cible destinée à se faire dégrader. UBQLN2, HSP70 et la cible sont par la suite dirigés au protéasome pour dégradation (Hjerpe et al., 2016)
1.2.2.4 UBQLN2 dans la sclérose latérale amyotrophique
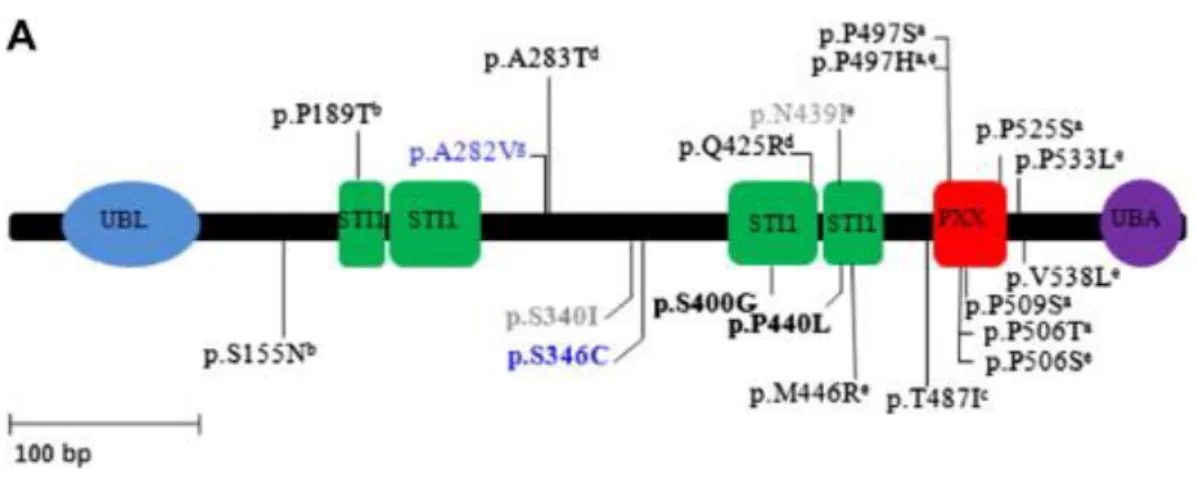
La découverte du premier gène muté d’UBQLN2 dans un cas de SLA remonte en 2011 lors d’une étude sur une famille de cinq générations porteuses de la SLA-DFT, découverte suivie de celles de quatre autres mutations suite à l’analyse de 188 autres familles (Deng et al., 2011). Étonnement, ces cinq mutations étaient toutes situées au niveau du domaine PXX d’UBQLN2 et affectaient des résidus proline (P497H, P497S, P506T, P509S et P525S). Depuis cette publication, plus de 10 autres mutations différentes affectant UBQLN2 dans les cas de SLA ont été découvertes et publiées (Daoud et al., 2012; Williams et al., 2012; Synofzik et al., 2012; Gellera et al., 2013; Fahed et al., 2014). La plupart des mutations ont été observées dans le domaine PXX d’UBQLN2, mais certaines ont également été découvertes dans le domaine STI-1 et même en dehors des domaines connus (Fig. 7) (Dillen, 2013). Les mutations retrouvées dans le domaine STI-1 sont toutes à caractère dominant avec
17
une pénétrance importante d’environ 90% et affectent autant les hommes que les femmes (Vengoechea et al., 2013). Les patients SLA ayant une mutation dans le gène UBQLN2 développent une pathologie TDP-43 fortement similaire aux cas sporadiques de la SLA. D’ailleurs, l’étude marquante réalisée en 2011 qui fit découvrir au monde l’importance d’UBQLN2 démontra également la présence d’agrégats cytoplasmiques contenant UBQLN2 co-localisant avec TDP-43 et ubiquitine au sein de tous les patients sporadiques de la maladie (Deng et al., 2011). Ceci suggère un rôle important joué par UBQLN2 au niveau de la pathogénèse de la SLA, non seulement dans les cas familiaux mais aussi dans les cas sporadiques.
Figure 7 Les domaines d’UBQLN2 avec les différentes mutations observées sur chacun des sites.
La plupart des mutations connues se retrouvent au niveau du domaine PXX, dont P497H. Toutefois ce n’est pas le seul domaine limité aux mutations et des mutations sont mêmes présentes dans des domaines inconnus (Dillen, 2013).
Malgré ces découvertes excitantes sur UBQLN2, son implication exacte dans le développement ou la progression de la SLA est encore très incompris. La dégradation de protéines redondantes ou repliées est critique pour la maintenance de la santé cellulaire, particulièrement au sein des neurones qui ne sont pas des cellules proliférantes. C’est pourquoi l’étude du dysfonctionnement du protéasome occasionné par une mutation d’UBQLN2 est un sujet de recherche fortement prisé et étudié. Tout d’abord, une accumulation d’un rapporteur d’efficacité du protéasome, soit UbiquitinG76V_GFP, a été
18
Toutefois, ceci ne démontre pas directement l’efficacité du protéasome, mais peut seulement suggérer qu’ubiquitine est accumulée dans les agrégats. Un autre groupe a utilisé un autre rapporteur d’efficacité du protéasome, soit le rapport endogène Myc, comme celui-ci est rapidement dégradé par le protéasome lorsque la synthèse de nouvelles protéines est bloquée par de la cycloheximide (Chang and Monteiro, 2015). Un délai a été observé dans la dégradation de Myc dans les cellules HeLA lorsque celles-ci avaient été transfectées avec UBQLN2 muté (P497H, P497S, P506T, P509S ou P525S). Cependant, lorsqu’un test chymotrypsine-like a été effectué afin de mesurer l’efficacité du protéasome, aucune dysfonction de celui-ci n’a été détecté, réduisant l’impact de l’étude et de ses conclusions (Chang and Monteiro, 2015). D’autres études ont plutôt suggéré que la forme mutante IVm-UBQLN2 pouvait entraver les fonctions de l’UPS en réduisant la livraison de protéines ubiquitinées au protéasome (Chang and Monteiro, 2015; Osaka, Ito and Suzuki, 2016). En effet, les mutations d’UBQLN2 dans la pathologie de la SLA mènent à une accumulation d’ubiquitinated high-molecular-weight complexes (HMWCs) et une délétion du domaine UBL séquestre les substrats ubiquitinés autant de l’UPS que de l’autophagie (mATG9 et ATG16L1). Ceci résulte en une augmentation de l’accumulation des HMWCs et participe donc à la pathogénèse de la SLA (Osaka, Ito and Suzuki, 2016). Récemment, notre groupe a démontré que la surexpression du mutant UBQLN2P497H a la capacité de séquestrer l’ubiquitine lys48_bound, résultant en une diminution de l’efficacité du protéasome, supportant les hypothèses que les mutations d’UBQLN2 sont grandement impliquées au sein de la voie de signalisation de l’UPS et que sa mutation entraîne une dysfonction significative de celui-ci (Picher-Martel et al., 2018).
Il a été démontré dernièrement que les inclusions cytoplasmiques contenant TDP-43 muté co-localisaient avec non seulement la forme mutée d’UBQLN2 mais également sa forme native, soit WT, dans des cellules Neuro2a co-transfectées avec les deux gènes mutés (Deng et al., 2011). Il a été démontré ultérieurement qu’UBQLN2 interagit directement avec les fragments de TDP-43 (Cassel and Reitz, 2013). Conformément avec ces résultats, notre
laboratoire a également démontré que la surexpression de hUBQLN2WT ou hUBQLN2P497H
TDP-19
43 (Picher-Martel et al., 2015). Il est important de noter toutefois que la forme mutée d’UBQLN2 induit une délocalisation et une accumulation plus importante de TDP-43 que la forme WT. Ces observations concordent avec d’autres études qui ont également observé une grande affinité entre TDP-43 et UBQLN2 à former des inclusions cytoplasmiques ensemble (Deng et al., 2011; Fahed et al., 2014; Cassel and Reitz, 2013; Jantrapirom et al., 2018; Le et al., 2016). La formation de ces inclusions a également été démontrée comme étant dose-dépendante, soit que les cellules avec une faible expression de WT ou UBQLN2 mutée ne montraient pas d’inclusions cytoplasmiques d’UBQLN2 dans les agrégats de TDP-43 (Deng et al., 2011; Picher-Martel et al., 2015).
Des stratégies afin d’augmenter l’ubiquitination ont été proposées afin de renforcer les fonctions de l’UPS (Cozzolino et al., 2012). En se basant sur cette idée, notre laboratoire a proposé que les mutations d’UBQLN2 pourraient séquestrer les protéines ubiquitines et par le fait même réduire la dégradation de TDP-43 muté par l’UPS, augmentant donc son accumulation anormale dans le cytoplasme des cellules (Picher-Martel et al., 2018). La
co-expression d’un vecteur ubiquitine avec les mutants hUBQLN2P497H, hTDP-43G348C dans des
cellules Neuro2a a mené à une réduction significative des niveaux d’UBQLN2 et TDP-43 mutants, suggérant qu’une régulation à la hausse d’ubiquitine peut devenir une potentielle cible thérapeutique intéressante pour la pathologie de la SLA.
1.2.3 Transport nucléo-cytoplasmique
1.2.3.1 Généralités
Le transport nucléo-cytoplasmique (TNC) est crucial et essentiel pour les fonctions cellulaires et a dernièrement été démontré comme étant défectueux dans les pathologies SLA/DFT. Le transport entre le noyau et le cytoplasme est possible grâce à plus de 30 nucléoporines (NUPs) formant le complexe du pore nucléaire (CPN). Les CPN sont situés sur la membrane nucléaire de la cellule et ont pour fonction d’être une porte filtrant le trafic protéique/ARNs entre le noyau et le cytoplasme (Lim, Aebi and Fahrenkrog, 2008). Certaines nucléoporines sont situées sur la portion cytoplasmique des filaments du pore nucléaire, d’autres sont situées plutôt au niveau du canal central du pore, plusieurs sont
20
transmembranaires et finalement le reste des NUPs sont situées dans le panier nucléaire du pore (Fig. 8). Il a été démontré dernièrement que la pathologie de TDP-43 a un impact important sur les nucléoporines et induit une perturbation importante du TNC, soit en séquestrant les NUPs dans des agrégats TDP-43, soit en les délocalisant ou soit en les faisant former des inclusions (Chou et al., 2018).
Figure 8 Nucléoporines constituant le complexe du pore nucléaire et leurs différentes conséquences résultant de la mutation de TDP-43 dans la SLA. Représentation du pore nucléaire résumant comment les agrégats de TDP-43 interagissent avec différents composants de la machinerie du transport nucléo-cytoplasmique (Chou et al., 2018)
Le TNC est un processus très rapide, directionnel et énergie-dépendante (Lui and Huang, 2009). Ce processus, en présence de RanGTPase ainsi que de karyophérines, peut atteindre un taux maximal de quelques centaines de macromolécules par seconde par CPN (Kubitscheck, 2005). Les karyophérines sont classées en deux groupes selon leurs fonctions, soit les importines pour l’importation nucléaire de cargaisons et exportines pour l’exportation nucléaire. Ces deux groupes de karyophérines possèdent chacun trois domaines fonctionnels, incluant un domaine de liaison de Ran au N-terminal, un domaine de liaison aux nucléoporines au centre et finalement un domaine de liaison au cargo au segment C-terminal (Lui and Huang, 2009). En général, les importines et les exportines reconnaissent la localisation d’un peptide signal sur la cargaison et peuvent ainsi former un complexe avec
21
les protéines cibles possédant le signal de localisation nucléaire (NLS) ou un signal d’exportation nucléaire (NES) (Boulikas, 1993).
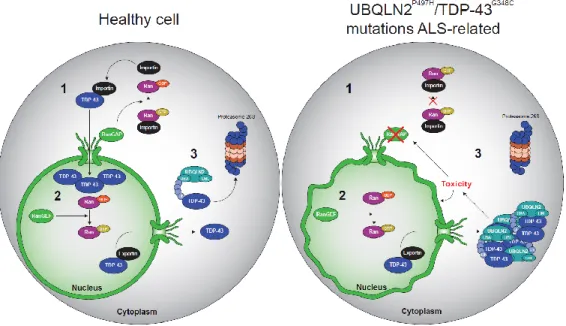
La protéine Ran (Ras-related Nuclear protein) est une petite ras GTPase qui médie les échanges nucléo-cytoplasmiques des macromolécules au travers de l’enveloppe nucléaire et se retrouve principalement au niveau du noyau de la cellule. Plusieurs études ont également démontré que Ran agit à titre de commutateur afin de réguler l’assemblage ou le désassemblage de complexes de transport de cargaison nucléaire (Lui and Huang, 2009; KB et al., 2014). Ran GTPase-activating enzyme 1 (RanGAP1), dans les cellules de mammifères, est enrichie dans le cytoplasme des cellules et au niveau de la membrane nucléaire dans les filaments du CPN (Zhang et al., 2015). Celle-ci agit sur l’importation de protéine du cytoplasme vers le noyau en hydrolysant la protéine RanGTP en RanGDP, permettant la séparation de celle-ci avec une protéine importine. La protéine importine peut, par la suite, se lier à une cargaison et permettre son transport jusque dans le noyau. L’analogue nucléaire de RanGAP1, RanGEF (Ran Guanine nucleotide Exchange Factor), également connu sous le nom de regulator of chromosome condensation 1 (RCC1), agit plutôt sur Ran afin de permettre l’exportation de protéines. RanGEF hydrolyse RanGDP en RanGTP, permettant ainsi la liaison d’une protéine exportine avec son cargo et l’exportation du complexe en milieu cytoplasmique (Fig 9) (Wu et al., 2018).
RanGAP1 et RanGEF travaillent de concert afin de maintenir un cycle fonctionnel de Ran au sein des cellules. Ainsi, RanGAP1, RanGEF, Ran, le CPN, les NUPs et les karyophérines sont responsables du TNC normal TNC et il a été démontré que ce processus est couramment observé dans les maladies neurodégénératives, notamment la SLA (Freibaum et al., 2015; Zhang et al., 2015; Chou et al., 2018; Shang et al., 2017), la maladie d’Huntington (Grima et al., 2017; Gasset-Rosa et al., 2019a), l’Alzheimer (Eftekharzadeh et al., 2018) et même simplement l’âge (D’Angelo et al., 2009; Pujol, Söderqvist and Radu., 2002).
22
Figure 9 Schématisation des divers mécanismes nécessaires à l’importation et exportation de protéines entre le noyau et le cytoplasme. Dans une cellule saine RanGAP1 cytoplasmique hydrolyse
RanGTP en RanGDP, libérant la protéine importine. Celle-ci peut se lier à un cargo et permettre son transport nucléaire. RanGEF nucléaire hydrolyse RanGDP en RanGTP et permet la liaison d’une exportine à son cargo et son exportation au niveau cytoplasmique. UBQLN2 fonctionnelle peut donc mener les protéines défectueuses à se faire dégrader au protéasome. Pour un cas SLA avec les mutants UBQLN2P497H/TDP-43G348C l’hypothèse
est que RanGAP1 ne peut plus hydrolyser RanGTP et donc empêche la libération de la protéine importine. Ceci empêche l’importation nucléaire mais pas l’exportation et c’est pourquoi une accumulation anormale est observée. Comme UBQLN2 est également mutée, elle ne peut donc pas accomplir ses fonctions de dégradation protéiques et se retrouvent liée à TDP-43G348C sans pourvoir le mener au protéasome.
1.2.3.2 Dysfonction du transport nucléo-cytoplasmique dans la SLA
1.2.3.2.1 C9orf72
Des mutations dans le gène C9orf72 (chromosome 9, open reading frame 72) découvertes dans des cas de SLA ont fait l’objet d’études récemment par rapport au déficit du TNC. Une expansion constituée de multiples répétitions de la séquence de nucléotides GGGGCC du HRE (hexanucleotide repeat expansion) dans le premier intron de C9orf72 a récemment été observée dans plus de la moitié des cas fSLA et un quart des cas familiaux de DFT, devenant donc un sujet prisé de recherche (DeJesus-Hernandez et al., 2011). Le mécanisme précis par lequel les expansions de la séquence GGGGCC cause la SLA/DFT est encore grandement incompris malgré d’intenses investigations mais trois hypothèses ont été émises sur la contribution de ces expansions à la toxicité de C9orf72 : 1) une perte de fonctions de C9ORF72 en raison d’une perturbation de la transcription; 2) un gain de fonction toxique
23
causés par les expansions correspondantes dans l’ARNm; 3) gain de fonctions toxiques causés par la présence de dipeptide protein repeats (DPRs) que ces expansions encodent (Donnelly et al., 2013; Ling, Polymenidou and Cleveland, 2013; Suzuki et al., 2013). Jusqu’à présent, la plupart des études supportent plutôt un gain de propriétés toxiques comme étant le principal contributeur à la pathogénèse, bien que la cause cette toxicité soit encore incomprise, à savoir si elle est causée par les ARNs ou plutôt par les DPRs.
C’est pourquoi C9orf72 est un modèle très étudié dans la SLA et particulièrement au niveau du TNC. En effet, une étude sur la drosophile a démontré que RanGAP1 était un potentiel modificateur de la neurodégénération médiée par GGGGCC. La surexpression de RanGAP1 ou simplement une seule copie d’un allèle avec un gain de fonction de RanGAP1 a supprimé de manière significative la neurodégénération liée à GGGGCC tandis qu’un knockdown de RanGAP1 l’a plutôt augmenté. Ceci suggère donc, considérant le rôle important de RanGAP1 au niveau du TNC, que le transport noyau-cytoplasme est un important modulateur de la pathologie C9orf72 dans la SLA. Il a été observé que RanGAP1 formait des agrégats co-localisant avec des foci d’ARN dans des tissus post-mortem (soit des cerveaux) et dans des cellules iPS (induced pluripotent stem) de patients C9-SLA causée par des HRE dans C9orf72 (Zhang et al., 2015). Ces observations suggèrent une accumulation et délocalisation de RanGAP1 qui devient incapable d’activer RanGTPase par hydrolyse et est donc incapable d’activer l’importation nucléaire de cargaisons. De surcroît, dans cette même étude, une délocalisation nucléaire importante de Ran vers le cytosol a été observée. Grâce au live imaging, l’équipe de recherche a également constaté que le TNC était perturbé en utilisant FRAP (fluorescence recovery after photobleaching) avec une protéine fluorescente tagguée NLS causant une diminution de plus de 50% de l’importation protéique dans les neurones iPS, démontrant concrètement un déficit de l’importation protéique.
1.2.3.2.2 TDP-43
Une étude récente sur la pathologie de TDP-43 dans la SLA et le transport nucléo-cytoplasmique a dévoilé 6 nouvelles découvertes intéressantes : 1) les protéines impliquées
24
dans le TNC sont des composés majeurs des agrégats cytoplasmiques TDP-43; 2) les agrégats de TDP-43 mutée causent la délocalisation et/ou l’agrégation des NUPs et des facteurs de transport (TF); 3) TDP-43 mutant et agrégée cause des défauts morphologiques dans la membrane nucléaire (MN) et les CPN en plus de déficits fonctionnels de l’importation protéique et exportation d’ARNm; 4) plusieurs gènes Nup agissent comme modificateurs génétiques de la toxicité de TDP-43 chez la drosophile; 5) l’inhibition pharmacologique et moléculaire de la toxicité de TDP-43 parvient à ramener le TNC à la normale, 6) la pathologie des pores nucléaires est également présente non seulement dans le cortex moteur des cas sporadiques et familiaux de patients SLA avec des inclusions positives pour pTDP-43, mais également dans le cortex frontal, suggérant qu’un déficit du TNC représente une caractéristique neuropathologique commune dans la SLA et potentiellement d’autres protéinopathies liées à TDP-43 (Chou et al., 2018).
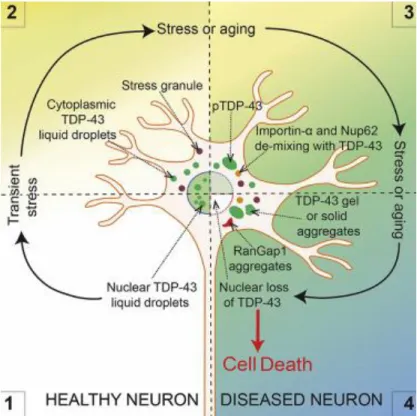
Une seconde étude encore plus récente démontre qu’une perturbation du TNC augmente la déplétion nucléaire de TDP-43 (Gasset-Rosa et al., 2019b). Ces derniers ont démontré, dans des cellules neuronales SH-SY5Y, qu’une exposition transitoire à des particules semblables à l’amyloïde a provoqué une rétention graduelle dans le cytoplasme de composés essentiels au TNC. Une délocalisation de RanGAP1 a également été observée dans les cellules en moins d’une semaine et augmentait de manière dépendante au temps. En moins d’un mois suivant l’exposition aux fibrilles, près de 70% des cellules avaient accumulé RanGAP1 dans de larges inclusions cytoplasmiques, en plus d’une accumulation progressive de TDP-43 cytoplasmique. De plus, des NUPs ainsi que les karyophérines importine α et β ont également été observées comme étant délocalisées et co-localisant avec les agrégats cytoplasmiques de TDP-43 (Fig. 10).