THESE
Pour l'obtention du grade de
DOCTEUR DE L'UNIVERSITÉ DE POITIERS UFR des sciences fondamentales et appliquées
Institut de chimie des milieux et matériaux de Poitiers - IC2MP (Diplôme National - Arrêté du 7 août 2006)
École doctorale : Sciences pour l'environnement - Gay Lussac Secteur de recherche : Chimie organique, minérale, industrielle
Cotutelle : Universidade Técnica (Lisboa, Portugal)
Présentée par :
Nuno Miguel Rocha Batalha
Optimization of the balance between activity and selectivity on a hydroisomerization catalyst
Directeur(s) de Thèse : Yannick Pouilloux, Francisco Lemos Soutenue le 08 octobre 2012 devant le jury Jury :
Président João Bordado Professor, Universidade Técnica de Lisboa Rapporteur Avelino Corma Profesor, Universidad Politécnica de Valencia Rapporteur Johan Martens Professeur, Katholieke Universiteit Leuven Membre Yannick Pouilloux Professeur des Universités, Université de Poitiers Membre Francisco Lemos Professor, Universidade Técnica de Lisboa Membre Ludovic Pinard Docteur, Université de Poitiers
Membre Christophe Bouchy Ingénieur, IFP énergies nouvelles, Lyon Membre Vicenzo Calemma Ingegnere, ENI, Milano
Pour citer cette thèse :
Nuno Miguel Rocha Batalha. Optimization of the balance between activity and selectivity on a hydroisomerization
catalyst [En ligne]. Thèse Chimie organique, minérale, industrielle. Poitiers : Université de Poitiers, 2012.
THESE
pour l’obtention du Grade de
DOCTEUR DE L’UNIVERSITE DE POITIERS
(Faculté des Sciences Fondamentales et Appliquées) (Diplôme National - Arrêté du 7 août 2006)
Ecole Doctorale : Sciences pour l’Environnement Gay-Lussac Secteur de Recherche : Chimie Organique, Minérale, Industrielle
Présentée par :
Nuno Miguel Rocha BATALHA
************************
Optimization of the balance between activity and selectivity on a
hydroisomerization catalyst
************************
Directeurs de Thèse : Professeur Francisco LEMOS, Professeur Yannick POUILLOUX, ************************
Soutenance le 8 octobre 2012
devant la Commission d’Examen
************************
JURY
J. BORDADO, Professeur, Universidade Técnica de Lisboa Président
J. MARTENS, Professeur, Katholieke Universiteit Leuven Rapporteurs
A. CORMA, Professeur, Universidad Politécnica de Valencia
V. CALEMMA, Docteur – Ingénieur, eni, Milan Examinateurs
C. BOUCHY, Docteur – Ingénieur, IFP energies nouvelles, Lyon
L. PINARD, Docteur, Université de Poitiers
F. LEMOS, Professeur, Universidade Técnica de Lisboa
Optimization of the balance between activity and selectivity on a hydroisomerization catalyst
i
Abstract
One of the main challenges when developing adequate catalysts for the catalytic dewaxing process (hydroisomerization) is to maximize the isomerization products yield and the catalyst activity, while maintaining a low selectivity towards light cracking products. Indeed, shape selective catalysts based on medium pore zeolites, e.g. Pt/ZSM-22, were proven to produce high yields of isomerization products, whereas larger pore zeolites were more active but less selective. The main objective of this thesis was, then, to study and develop a catalyst with both high activity and selectivity towards the production of isomerization products. For that two parallel studies were made: the first based on the impact of the proximity between the active sites on the reaction (Part I); and the second, focused on the development of a high performance hydroisomerization catalyst using BEA zeolite nanocrystals as an acid support (Part II).
The participation of the spilt-over hydrogen (Hsp) species on the hydroisomerization reaction
mechanism played a major role on the study performed on the first part of this manuscript. Indeed, when the active sites were near enough the Hsp species were able to diffuse into the vicinity of the
acid sites promoting the direct hydrogenation of the carbenium ions. Due to this fact, a reaction mechanism was proposed using this reaction as an alternative to the classical mechanism proposed by Weisz, where the hydrogenation reaction takes place on the metallic sites. This phenomenon justified the higher activity and selectivity observed on the catalysts where the active sites were side by side.
On the second part of this manuscript nanocrystals of BEA zeolite were used to develop a high performance hydroisomerization catalyst. However, when synthesized the BEA nanocrystals tend to agglomerate. This phenomenon was proven to create mass transfer limitations responsible for a high reduction on the catalysts activity and selectivity. To avoid the nanocrystals agglomeration, the BEA nanocrystals were germinated on an alumina support. The resulting composite catalysts were tested on the n-hexadecane hydroisomerization, where they showed high activity and selectivity when compared to the zeolite alone, i.e. up to 2.8 times more active and the isomers yield increased from 35 wt.% to 80 wt.%. Moreover, the composite catalyst revealed a quite interesting cracking products distribution with a higher occurrence of heavier products (C9+). Finally, the composite
catalyst showing the best performance was compared to the Pt/ZSM-22 (reference), showing that for an equal selectivity it was 36 times more activity.
Keywords: Hydroisomerization, bifunctional catalyst, reaction mechanism spilt-over hydrogen,
Optimization of the balance between activity and selectivity on a hydroisomerization catalyst
ii
Resumo
Um dos principais desafios no desenvolvimento de catalisadores adequados para o processo de desparafinação catalítica (hidroisomerisação) é o de maximizar o rendimento em isómeros e a atividade do catalisador, mantendo uma baixa seletividade em produtos de craqueamento. Catalisadores com seletividade de forma à base de zeólitos de poros intermédios, como Pt/ZSM-22, mostraram a capacidade de gerar rendimentos elevados em isómeros, enquanto que zeólitos de poros largos são mais ativos mas menos seletivos. O principal objetivo desta tese foi, então, estudar e desenvolver um catalisador com alta atividade e seletividade para a produção de isómeros. Para isso dois estudos foram efectuados em paralelo: o primeiro com base no impacto da proximidade entre os centros ativos na reação (Part I), e a segunda, voltada para o desenvolvimento de um catalisador de alta eficiência para a reação de hidroisomerisação utilizando nano-cristais de zeólito BEA como suporte ácido (Part II).
A participação do “spilt-over” de hidrogénio (Hsp) no mecanismo da reação de hidroisomerisação
foi estudada na primeira parte deste manuscrito. Efetivamente, quando os centros activos estão suficientemente próximos, as espécies de Hsp foram capazes de se difundir até à vizinhança dos
centros ácidos e aí promover a hidrogenação direta dos iões carbénio. Um mecanismo foi, então, proposto utilizando esta reação como uma alternativa para o mecanismo proposto por Weisz, onde a reação de hidrogenação tem lugar nos centros metálicos. Este fenómeno justifica a maior atividade e seletividade observada nos catalisadores em que os centros ativos estão próximos.
Na segunda parte deste manuscrito, nano-cristais de zeólito BEA foram utilizados para desenvolver um catalisador de hidroisomerisação de alta eficiência. Contudo, quando sintetizados, os nano-cristais BEA tendem a aglomerar. Este fenómeno cria limitações de transferência de massa, levando a uma redução elevada da atividade e seletividade dos catalisadores. Para evitar esta aglomeração, os nano-cristais de BEA foram germinados sobre um suporte de alumina. Os catalisadores resultantes foram testados na hidroisomerisação de n-hexadecano, mostrando elevadas atividade e seletividade quando comparado com o zeólito puro, ou seja, até 2,8 vezes mais ativo e o rendimento máximo em isómeros aumentou de 35 m/m% para 80 m/m% . Além disso, este catalisador revelou uma distribuição de produtos de craqueamento surpreendente, com uma maior ocorrência de produtos mais pesados (C9+). Finalmente, o catalisador de maior desempenho foi comparado com o
Pt/ZSM-22 (referência), mostrando igual seletividade mas uma atividade 36 vezes superior.
Palavras-chave: hidroisomerisação, catalisador bifuncional, mecanismo de reação, “spilt-over” de
Optimization of the balance between activity and selectivity on a hydroisomerization catalyst
iii
Résumé
Un des principaux défis lors de l'élaboration des catalyseurs adéquats pour le procédé de déparaffinage catalytique (hydroisomérisation) est de maximiser le rendement en isomères et l'activité du catalyseur, tout en maintenant une faible sélectivité en produits de craquage. En effet, des catalyseurs avec sélectivité de forme à base de zéolithes à taille de pore intermédiaire, par exemple Pt/ZSM-22, sont sélectives en isomères, tandis que les zéolithes à large pore sont plus actifs, mais moins sélectif. L'objectif principal de cette thèse était, alors, d’étudier et de développer un catalyseur à la fois actif et sélectif en isomères. Deux études parallèles ont été réalisées: la première basée sur l'impact de la proximité entre les sites actifs sur la réaction (Part I), et la seconde, portant sur le développement d'un catalyseur d'hydroisomérisation de haute performance en utilisant des nanocristaux de zéolithe BEA comme support acide (Part II).
La participation de l’épandage d’hydrogène (Hsp) sur le mécanisme de la réaction
d'hydroisomérisation a été démontrée. En effet, lorsque les sites actifs sont proches, les espèces Hsp
diffusent au voisinage des sites acides provocant l'hydrogénation directe des ions carbénium. Un mécanisme de réaction a, alors, été proposé utilisant ce phénomène comme une alternative au mécanisme classique proposé par Weisz, où la réaction d'hydrogénation a lieu uniquement sur les sites métalliques. Ce phénomène justifie l'activité et la sélectivité plus élevées observées sur les catalyseurs, où les sites actifs sont proches.
Sur la deuxième partie de ce manuscrit, des nanocristaux de zéolithe BEA ont été utilisés pour développer un catalyseur d’hydroisomérisation à haute performance. Toutefois, lorsque de la synthèse, les nanocristaux BEA ont tendance à s'agglomérer. Ce phénomène crée des limitations de transfert de masse responsables d’une forte réduction de l'activité et de la sélectivité des catalyseurs. Pour éviter cette agglomération, les nanocristaux ont été germinés sur un support d'alumine. Ces catalyseurs ont été testés sur l’hydroisomérisation du n-hexadécane, où ils ont montré une activité et une sélectivité élevée par rapport à la zéolithe seule : ils sont 2,8 fois plus actif et le rendement en isomères a augmenté de 35 pds.% à 80 pds.%. De plus, le catalyseur composite a montré une distribution de produits de craquage très particulière, avec une génération plus élevée de produits lourds (C9+). Le catalyseur composite le plus performant, comparé avec le catalyseur Pt/ZSM-22
(référence), a été aussi sélectif mais 36 fois plus actif.
Mots clés: Hydroisomérisation, catalyseur bifonctionnel, mécanisme de réaction, épandage d’hydrogène, BEA, nanocristaux, limitations diffusionelles, zéolithes hiérarchisées
Optimization of the balance between activity and selectivity on a hydroisomerization catalyst
iv
Acknowledgements
I would like to begin by expressing my gratitude to Professor Fernando Ramôa Ribeiro for the excellent opportunity given to accomplish my Ph. D. thesis. I am also thankful to him for believing in my capabilities, for all the encouragement, for the consideration that he always expressed for me and for my work and for his kindness and attention.
To Professor Francisco Lemos, Dr. Patrick Magnoux, Professor Yannick Pouilloux and Dr. Ludovic Pinard, I present my sincere thanks for their scientific and bureaucratic guidance, and for the availability throughout all these years. Without their interest and patience this Ph. D. would not have been possible. Moreover, I present my thanks to Jean-Louis Lemberton for all his contributions.
In particular, I would like to express my gratitude to Ludovic Pinard for all the time, support and dedication he has given me all these years. “Thank you so much Ludo, I could not have chosen
someone better to do my Ph. D. with.”
A special thanks to Sophie Morisset for being so nice and available all the time, as well as for the major help she gave me on the catalysts synthesis. I am also grateful: to all the personnel from the analysis group of the IC2MP for all the help and availability during all these years; to the glass blowers of the IC2MP for being able to quickly repair everything that I broke; and to Michel Chauveau for being so nice, available and for fixing everything, always.
I would like to thank Dr. Thomas Belin for all the time and patience that he spent interpreting and explaining me how the RF-GC technique works.
I wish to thank my lab co-workers of the IC2MP / LACCO research lab, in particular to those of the zeolites group, for all the help you have given me during this thesis. A special thanks to all the friends that I have made, in particular to those with which I have shared my office, on the IC2MP / LACCO during this thesis for their help, good spirit and for all the good memories from the times spent together.
I wish to thank my lab co-workers and friends from the research lab at IST for being so nice on every one of my short visits.
I express my gratitude to all my Brazilian friends, in particular Marcelo, which I have made at Rio de Janeiro during the internship in the LACES research group (UFRJ). Also, I thank Henrique Cerqueira for giving me the opportunity of making this internship.
Optimization of the balance between activity and selectivity on a hydroisomerization catalyst
v
I would like to express my gratitude to the Fundacão para a Ciência e a Tecnologia (FCT) for my Ph.D. grant (ref. SFRH/BD/43551/2008).
To Leila my deepest thanks for receiving me in your house and for adopting me in your family. To all my roommates, special thanks for sharing a part of your life with me and for all the good memories from the time we have spent together.
To all my family and friends, thank you for being part of my life.
Most important, I am more than grateful to my parents, Rosária and Manuel, and to my brother, Carlos, for the unconditional support and affection.
Poitiers, 20 June 2012 Nuno Batalha
Optimization of the balance between activity and selectivity on a hydroisomerization catalyst
vi
General Index
Context
1
1
Diesel Fuel Emissions ... 4
2
Fischer-Tropsch to diesel... 6
2.1 Fischer-Tropsch process ... 6
2.2 Diesel fuel specifications ... 7
2.3 Upgrading LTFT products ... 9
3
Objective ... 11
P
ART
I:
HYDROISOMERIZATION:IMPACT OF THE PROXIMITY OF THE ACTIVE SITESChapter I
– Literature Review:
Reaction mechanism
15
1
Introduction ... 18
2
Reactions involved in hydroisomerization ... 18
2.1 Acidic function reactions ... 19
2.2 Metallic function reactions... 26
2.3 Bifunctional mechanism ... 27
3
Problematic ... 31
Chapter II
– Experimental Techniques:
Acid sites characterization and
hydroisomerization
33
1
Catalysts ... 37
Optimization of the balance between activity and selectivity on a hydroisomerization catalyst
vii
1.2 Catalyst preparation ... 37
2
Catalyst characterization ... 39
2.1 Induced coupled plasma (ICP-AES) ... 39
2.2 Infrared spectroscopy ... 40
2.3 Toluene Hydrogenation... 44
2.4 Hydrogen chemisorption ... 46
2.5 Pyridine hydrogenation followed by IR spectroscopy ... 47
3
Catalytic tests
–
n-hexadecane hydroisomerization ... 49
3.1 Catalyst shape ... 49
3.2 Chemical products... 49
3.3 Experimental apparatus ... 50
3.4 Catalytic test conditions ... 53
3.5 Reactor Filling... 53
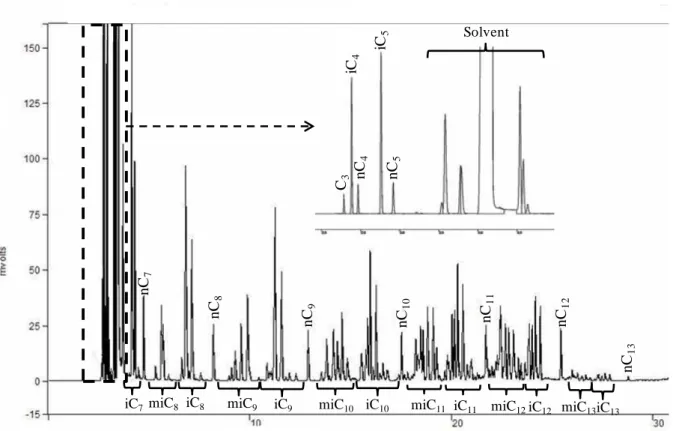
3.6 Chromatographic analysis ... 54
Chapter III
– Spilt-over diffusion and interaction with hydrocarbons
61
1
Objective ... 64
2
Active site characterization ... 64
3
Toluene hydrogenation ... 65
3.1 Summary ... 68
4
Pyridine hydrogenation ... 68
4.1 Pyridine hydrogenation reaction mechanism ... 69
4.2 Pyridine hydrogenation: HBEA ... 71
4.3 Pyridine hydrogenation: PtHBEA series ... 73
Optimization of the balance between activity and selectivity on a hydroisomerization catalyst
viii
4.5 Pyridine hydrogenation: PtA+HBEA (S2) series... 77
4.6 Pyridine hydrogenation: PtA+HBEA (S1) series... 78
4.7 Discussion ... 80
4.8 Summary ... 81
5
Hydrogen chemisorption ... 82
5.1 Summary ... 83
6
Conclusion ... 83
Chapter IV
– Impact of the proximity and balance of the active sites on
hydroisomerization
85
1
Objective... 88
2
Catalysts characterization... 88
3
Catalysts activity... 89
3.1 Summary ... 934
Catalysts selectivity... 94
4.1 Total isomers yield ... 95
4.2 Monobranched (M) and multibranched (B) isomers selectivity ... 99
4.3 Cracking products selectivity ... 100
4.4 Summary ... 101
5
Conclusion ... 102
P
ART
II:
IMPROVEMENT OF THE HYDROISOMERIZATION PERFORMANCE OF BEA NANOCRYSTALSChapter I
– Literature Review:
Hydroisomerization catalysts
105
Optimization of the balance between activity and selectivity on a hydroisomerization catalyst
ix
1.1 Hydrogenation/dehydrogenation function ... 108
1.2 Acid function... 108
2
Hierarchical zeolites ... 117
2.1 Hierarchical zeolite single crystals ... 118
2.2 Zeolite nanocrystals ... 119
2.3 Supported zeolite crystals ... 120
2.4 Hierarchical zeolites for hydroisomerization ... 120
3
Problematic ... 121
Chapter II
– Experimental techniques: Morphological, textural and diffusion
properties
123
1
X-Ray diffraction (XRD) ... 127
1.1 Experimental procedure ... 127
1.2 Scherrer method ... 127
1.3 Zeolite loading quantification ... 128
2
Nitrogen physisorption ... 129
2.1 Experimental procedure ... 129
2.2 Isotherm classification ... 130
2.3 Specific Surface – BET surface ... 131
2.4 External surface ... 132
2.5 Pore Volume ... 133
2.6 Mesopore size distribution ... 137
3
Scanning Electron Microscopy (SEM) ... 137
3.1 Experimental procedure ... 138
3.2 Particle size distribution ... 138
3.3 Average particle size ... 138
4
Transmission Electron Microscopy (TEM) ... 139
Optimization of the balance between activity and selectivity on a hydroisomerization catalyst
x
4.2 Platinum particles average size and distribution ... 139
5
Infrared spectroscopy ... 139
5.1 Hydroxyl (OH) stretching vibration bands ... 140
5.2 Zeolite structure bands ... 141
6
Reverse Flow - Gas Chromatography (RF-GC) ... 143
6.1 Experimental procedure ... 143
6.2 Physicochemical parameters quantification ... 145
Chapter III
– Impact of the BEA nanocrystals agglomeration on
hydroisomerization
151
1
Objective ... 155
2
BEA samples ... 155
2.1 BEA (PQ) ... 155 2.2 BEA (S) ... 155 2.3 BEA (M) ... 1563
Samples physical properties ... 157
3.1 Structural analysis ... 157
3.2 Morphological properties ... 158
3.3 Textural properties ... 161
3.4 Summary ... 163
4
Active function characterization ... 163
5
n-hexadecane hydroisomerization ... 165
5.2 Summary ... 169
6
Reverse-Flow Gas Chromatography (RF-GC) ... 169
6.1 Experimental data ... 170
Optimization of the balance between activity and selectivity on a hydroisomerization catalyst
xi
6.3 i-butane / BEA system adsorption and diffusion rates ... 175
6.4 Summary ... 178
7
Discussion ... 178
8
Conclusion ... 180
Chapter IV
– BEA nanocrystals germination on alumina: Synthesis,
characterization and catalytic tests
181
1
Objective ... 184
2
Catalysts synthesis ... 184
2.1 BEA/Al2O3 ... 184
3
Synthesis of the coated samples ... 185
4
Physical characterization ... 185
4.1 Structural properties ... 185
4.2 Composite samples zeolite loading ... 186
4.3 Textural properties ... 187
4.4 Morphological properties ... 189
4.5 Summary ... 197
5
Active sites characterization ... 197
5.1 Acid sites ... 198 5.2 Platinum sites ... 200 5.3 Summary ... 202
6
n-Hexadecane hydroisomerization ... 203
6.1 Activity... 203 6.2 Isomers Selectivity ... 2046.3 Cracking products distribution ... 206
6.4 Composite catalyst synthesis repeatability ... 208
Optimization of the balance between activity and selectivity on a hydroisomerization catalyst
xii
7
Conclusion ... 209
Chapter V
– Impact of the alumina phase on the BEA germination and
hydroisomerization
211
1
Objective ... 214
2
Catalyst synthesis ... 214
3
Physical characterization ... 214
3.1 X-ray Diffraction ... 215 3.2 Textural properties ... 216 3.3 Summary ... 2194
Active sites characterization ... 220
4.1 Samples acidity ... 220 4.2 Metallic function ... 221 4.3 Summary ... 223
5
n-hexadecane hydroisomerization ... 223
5.1 Activity... 224 5.2 Isomers selectivity... 2255.3 Cracking products distribution ... 226
5.4 Summary ... 227
6
Conclusion ... 228
Chapter VI
– Comparison of BEA and TON zeolites on hydroisomerization
229
1
Objective ... 232
2
Catalyst samples ... 232
Optimization of the balance between activity and selectivity on a hydroisomerization catalyst xiii 2.2 BEA(Z)... 232 2.3 BEA/Al2O3 ... 233 2.4 ZSM-22 ... 233
3
Catalyst characterization ... 233
3.1 Textural properties ... 233 3.2 Active sites ... 235 3.3 Summary ... 2374
n-hexadecane hydroisomerization ... 237
4.1 Activity... 238 4.2 Isomers selectivity... 2394.3 Cracking products distribution ... 241
4.4 Global catalyst evaluation ... 242
5
Conclusion ... 243
General conclusions
245
Perspectives
251
Bibliographic references
259
Optimization of the balance between the activity and selectivity on a hydroisomerization catalyst
3
Index
1
Diesel Fuel Emissions ... 4
2
Fischer-Tropsch to diesel... 6
2.1 Fischer-Tropsch process ... 6
2.2 Diesel fuel specifications ... 7
2.2.1 Cetane number ... 8
2.2.2 Density, volatility and viscosity ... 8
2.2.3 Cold properties ... 9
2.3 Upgrading LTFT products ... 9
Context
4
1
Diesel Fuel Emissions
Diesel is among one of the most used fuels for transport application. Indeed, in the European Union (27) diesel is not only the most used fuel for transportation, but also its consumption has been increasing on the last decade (Fig 1).
Fig 1. Transports fuel consumption on the European Union (27) [1].
Diesel engines are known both for their durability, robustness and for their combustion efficiency which means a lower consumption and less CO2 generation. Nonetheless, diesel motors produce a
high amount of nitrogen oxides (NOx) gases and particles, which constitute a serious threat to the
environment and human health. Taking awareness of the harmful effects of pollution to the environment the governmental authorities were obliged to adopt emission standards. These standards set the maximum pollutants allowed in automobile exhaust and are becoming more and more restrictive. Since the 90’s significant progress has been made to reduce lead (Pb), carbon monoxide (CO), particle matter (PM), sulfur (S) and unburned hydrocarbon (HC) emissions.
“Euro” standards, created in the framework of the European Auto-Oil EPEFE4, have been
established every 4 to 5 years since 1992, each time increasing the pollution constraints severity, as depicted in Fig 2. The Euro 5 standard is in operation since 2009 and a new Euro 6 standard is predicted for 2014 [2]. 0 50 100 150 200 250 300 350 400 2001 2002 2003 2004 2005 2006 2007 2008 2009 2010 F u el co n su m p tio n ( h u n d re d s o f to n ) LPG Gasoline Jet Fuel Diesel
Optimization of the balance between the activity and selectivity on a hydroisomerization catalyst
5
Fig 2. Diesel vehicles pollution emission restrictions evolution according to the “Euro” standards. Adapted from [3].
Among all the restrictions imposed over the pollution emissions, sulfur is the most defying for the refining industry. As sulfur is present in the crude oil, the final fuel contamination depends on the crude source and on the treatments performed during refining. Moreover, the sulfur content of a diesel fuel has been proven to enhance the emission of other pollutants, in particular CO, NOx, PM,
Volatile Organic Compounds (VOC’s), sulfur dioxide (SO2), sulfate particles and benzene [4].
The need to comply with the sulfur restrictions and the high impact of the crude oil sulfur content on the final product has renewed the interest on the Fisher-Trospch process. This process enables the production of sulfur free fuels from a variety of raw materials, such as natural gas, coal or biomass [5, 6]. The synthetic fuels obtained, using this method, have high purity (no sulfur or aromatics) and a very high cetane number. The sulfur absence in the diesel fuels would then decrease the exhaust pollution emissions when compared with a standard diesel [7, 8], as shown in Table 1.
Table 1. Comparison between an Ultra Low Sulfur Diesel (ULSD) and Fischer Tropsch Diesel (FTD) pollutant emissions (g/km). Adapted from [9].
Fuel HC CO CO2 NOX HC+NOX PM
ULSD 0.059 0.439 0.101 152 0.475 0.534 0.037 FTD 0.016 146 0.464 0.480 0.026 PM CO SDiesel S Gasoline HC+NOx
Context
6
2
Fischer-Tropsch to diesel
2.1 Fischer-Tropsch process
The Fisher-Tropsch reaction transforms CO and H2 (Syngas) into hydrocarbon molecules with a
wide carbon number range. The syngas used as the reaction raw material may result from a variety of sources (natural gas, coal, biomass, etc.) increasing the versatility of this process. Indeed, a wide range of raw materials are suited for the Fischer-Tropsch, however their economical viability highly depends on the crude oil barrel price. Studies have been made, showing that the most economically viable raw material is natural gas followed by coal and biomass, which attain the break-even point when the crude oil attains the price of $36, $60 and $75 per bbl, respectively [5]. Still, the use of biomass as a raw material highly contributes to a reduction of the greenhouse gas emissions, at least 50%, when compared with the remaining possibilities and crude itself [5].
The performance of the Fischer-Tropsch synthesis can be affected by a variety of parameters, such as gas composition, catalyst formulation and operating temperature, which change the final products yield [6]. Two completely different reactor conditions are then often found: High Temperature Fischer-Tropsch (HTFT) and Low Temperature Fischer-Tropsch (LTFT). The HTFT process works at higher temperatures (320 to 350°C [10]) using iron based catalysts and generates a higher olefin yield with a narrower carbon number range (Table 2). On the other hand, the LTFT process works at lower temperatures (220 to 250°C [10]) using iron or cobalt based catalysts and generates a wide carbon number range hydrocarbons, mainly paraffins (Table 2).
Optimization of the balance between the activity and selectivity on a hydroisomerization catalyst
7
Table 2 HTFT and LTFT product yield distribution, excluding C1-C2 hydrocarbons. Adapted from
[11].
HTFT
(Synthol process)
LTFT (Arge process) Carbon number distribution (wt.%)
C3-C4 30 10 C5-C10 40 19 C11-C22 16 22 C22 and heavier 6 46 Aqueous products 8 3 Compound classes (wt.%)
Paraffins >10% Major product Olefins Major product >10%
Aromatics 5-10% <1%
Oxygenates 5-15% 5-15%
The paraffinic content of the LTFT synthesis conditions makes it preferential for the production of middle distillates, including diesel fuels [12]. Nonetheless, in order to maximize both the diesel cut and to obtain the required fuel properties the products resulting from the LTFT process must be upgraded. Indeed, the amount of high carbon number linear paraffins on the LTFT outlet stream needs to be corrected to maximize amount products on the middle distillate range. In addition, since the paraffins resulting from the LTFT are mainly linear an extra-requirement concerning the fuel properties under cold conditions, which must be respected for further commercialization, must be made.
2.2 Diesel fuel specifications
The physical and chemical properties of a diesel fuel are used to describe its quality. Several parameters must be taken into account for a fuel to be acceptable for commercial application. Parameters like cetane number, volatility, viscosity, and the so called cold properties, are of high importance and need to be balanced according to the engine and climatic conditions before a diesel fraction to be commercialized.
Context
8
2.2.1 Cetane number
The cetane number measures the capability of a fuel towards self-ignition on a scale of 0 to 100, where 0 has the -methylnaphthalene as reference and 100 to cetane (n-hexadecane). A high cetane number enables better performances, regarding emissions and noise [13]. This property depends upon the fuel chemical hydrocarbon composition. In general, linear paraffins (unbranched saturated hydrocarbons) have high cetane numbers. On the other hand, isoparaffins (branched saturated hydrocarbons) and aromatic hydrocarbons have low cetane numbers, while the olefins (unsaturated hydrocarbons (alkenes)) and naphthenes have intermediate cetane numbers [14].
2.2.2 Density, volatility and viscosity
These three characteristics, which often depend one upon the other, are important for the fuel injection and for good self-ignition air/fuel mixture.
Density: The fuel density is important as the pump and injectors are set to deliver a precise flow.
Moreover, this parameter also controls the weight ratio between air and fuel. In Europe, according to the norm 590 (EN 590), a diesel fuel must have a density between 0.820 and 0.845 kg/dm3.
Volatility: This property is expressed through two characteristics: the flash point and the distillation
curve. The flash point has no direct influence on the combustion or motor performances, but it is of high importance for the storage and distribution security criteria [14]. On the other hand, the distillation curve is directly related with combustion properties. In Europe, the norm EN 590 establishes three criteria for the distillation curve fractions: less than 65% at 250°C; more than 85% at 350°C; and more than 95% at 360°C. A good balance between high and low bowling point must be achieved, whereas low boiling point compounds facilitate cold starting and high boiling point molecules contribute for power [15].
Viscosity: It represents the resistance of a fluid to flow when submitted to external forces. The
viscosity of a fuel must be balanced to enable both an adequate spray to be obtained, so that a good air-fuel mixture will be produced and therefore a clean and efficient combustion (favored by low viscosity), and to provide lubrication for the fuel pump and injectors (favored by high viscosity).
Optimization of the balance between the activity and selectivity on a hydroisomerization catalyst
9
2.2.3 Cold properties
The properties of a diesel at low temperatures are mostly important for the proper functioning of the mechanic parts. In particular, before entering the motor itself the diesel fuel must traverse a very tight filter (several micrometers), to avoid the entrance of impurities and particles into the injection pump [14]. Depending on the diesel composition, which is mainly composed by linear paraffins that can go from C10 to C35, particles may be formed at low temperatures through paraffin
crystallization. This phenomenon may lead to the filter obstruction, problems in the injection pump,
etc…
Like before, several specifications are imposed concerning the diesel cold properties. The European Norm 590 specifies three different parameters: cloud point (CP), pour point (PP) and cold filter plugging point (CFPP). The restrictions, however, change from region to region and are related to the climate conditions. Each one of the referred properties represents a different scenario: CP represents the temperature at which dissolved solids are no longer completely soluble, precipitating as a second phase giving the fluid a cloudy appearance; PP the temperature after which the fuel may no longer flow; and CFPP correspond to the temperature after which the fuel may no longer traverse the filter.
2.3 Upgrading LTFT products
The LTFT products, which are mainly linear paraffins, need to be upgraded so that at least part of these products can be used as a diesel fuel. Moreover, the products distribution in the FT process itself needs to be corrected in order to maximize the fuel production, since the Fischer-Tropsch synthesis product distribution is very wide (Table 2). The paraffins of high molecular weight have very poor cold properties, so in order to include them into a fuel pool they must be transformed. The selective cracking of the Fischer-Tropsch effluent stream is the most common procedure (hydrocracking). This upgrading technique uses the hydrocracking reaction to transform the large carbon number paraffins into cracked fragments of the desired carbon number range. Nonetheless, the reaction conditions must be so that the products which are already in the desired range should not suffer cracking. Moreover, the formation of light hydrocarbon gases, with less commercial value, should be minimized.
The catalytic dewaxing of the long chain linear paraffins presents itself as an economical alternative for the LTFT products upgrading, since it enables to produce high quality diesel and base oils used
Context
10
on high-performance lubricants [12, 16]. This process consists on n-alkanes isomerization, as isomer paraffins have much lower melting points, as shown over Fig 3.
Fig 3. Difference between the fusion points of the monobranched isomers and the corresponding n -alkanes [17].
The alkane isomerization of the diesel fraction C15 to C22, derived from the LTFT process, is of
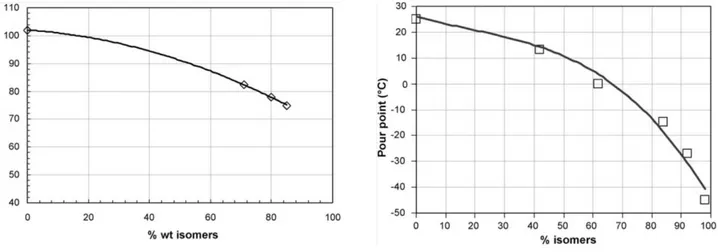
growing interest for the transport fuel due to the high cetane number and the good cold properties that can be obtained [9]. Even though the isomer paraffins cetane number is somewhat lower than that of the linear ones, due to the high carbon number and the absence of aromatics and naphthenes the diesel specifications can be easily achieved. This is clearly seen in Fig 4 where the cetane number and the pour point of a C15 to C22 diesel fraction are evaluated as a function of the cuts
isomer composition.
Fig 4. Cetane number (a) and pour point (PP) (b) evolution with the isomer content on a C15 to C22
diesel fraction [9].
Both hydrocracking and catalytic dewaxing/hydroisomerization occur through the same bifunctional mechanism, however, the catalytic dewaxing process requires the reaction to stop before the paraffins to undergo cracking (hydrocracking). In the case of the transformation of large
Optimization of the balance between the activity and selectivity on a hydroisomerization catalyst
11
carbon number paraffins this issue may become a major problem, since the isomers can undergo cracking quite easily. Therefore, it is important to use an adequate catalyst with high selectivity towards isomerization products.
3
Objective
One of the main challenges when developing adequate catalysts for the catalytic dewaxing process (hydroisomerization) is to maximize the isomerization products yield all while maintaining the selectivity towards light cracking products low. This is a significant challenge since the cracking reactions rate increases with the paraffin isomerization degree. As a consequence, the main objective of this Ph. D thesis was to obtain, study and compare high performance hydroisomerization catalysts. In order to do this two parallel studies were performed: the first concerning the analysis of the hydroisomerization reaction mechanism (Part I); and the second concerning the development of high performance hydroisomerization catalysts using the large pore zeolite BEA as an acid support (Part II).
Indeed, for a proper development of an adequate catalyst a full knowledge of the reaction mechanism is required, namely what are the active sites and what is their role during the reaction. Even if in literature it is undeniable that a metallic and an acidic function are the base of the hydroisomerization reaction mechanism, in the last decades several authors have discussed the role of each one of these active sites on the reaction mechanism (further information on Part I – Chapter I). Therefore, in order to bring some light into this subject, a full study of the hydroisomerization reaction mechanism was made. In particular this study will be focused on the impact of the proximity between the acidic and metallic functions. This study will be presented on the first part of this Ph. D. manuscript (Part I).
In parallel to the study of the hydroisomerization reaction mechanism, the development of high performance hydroisomerization catalyst using, as acid support, the large pore zeolite BEA was made (Part II). This work followed the Ph. D. conclusions of R. Merabti [15], who proposed that BEA zeolite nanocrystals could be used on the hydroisomerization of large carbon number paraffins with a good selectivity towards isomerization products and a high activity. In fact, R. Merabti showed that the BEA zeolites could be almost equally selective and several times more active than the medium pore zeolite ZSM-22 (the zeolite with the highest selectivity towards isomerization products) (further information on Part II – Chapter I). Nonetheless, R. Merabti also stated that the hydroisomerization performance of the BEA zeolite nanocrystals was limited by their typical agglomeration into large micrometric clusters. As a consequence, a detailed study of the impact of
Context
12
the BEA nanocrystals agglomeration on the hydroisomerization of large carbon number paraffins was made (Part II – Chapter III). On the other hand, to confirm the R. Merabti Ph. D. conclusions, the reduction of the BEA nanocrystals agglomeration was performed by directly germinating the nanocrystals on a support (alumina) (Part II – Chapters IV and V). Finally, the best BEA catalysts will be compared to the ZSM-22 zeolite (Part II – Chapter VI).
P
ART
I
Hydroisomerization:
I
Literature Review
Part I – Hydroisomerization: Impact of the proximity between the active sites
17
Index
1
Introduction ... 18
2
Reactions involved in hydroisomerization ... 18
2.1 Acidic function reactions ... 19
2.1.1 Carbocation chemistry ... 19 2.1.2 Isomerization ... 20 2.1.3 Cracking ... 22 2.1.4 Oligomerization ... 24 2.1.5 Other acid function reactions... 25
2.2 Metallic function reactions... 26
2.2.1 Hydrogenation (Dehydrogenation) ... 26
2.3 Bifunctional mechanism ... 27
2.3.1 Weisz’s mechanism (Classical mechanism) ... 27 2.3.2 Spilt-over hydrogen (Hsp) mechanisms ... 29
3
Problematic ... 31
Chapter I – Literature Review: Reaction Mechanism
18
1
Introduction
The research and development of new and more suitable catalysts requires a full and profound knowledge of the catalytic reaction for which the catalyst will be designed. Indeed, the full understanding of the reaction mechanism is necessary, since this can make the difference between a
suitable catalyst and the “ideal” catalyst. In fact, no “ideal” catalyst exists for a given reaction, in
particular for hydrocarbon conversion, as, depending on the final products application and raw materials, different properties may be considered suitable.
As referred before, the main objective of this Ph. D. is to find a suitable bifunctional catalyst for the hydroisomerization of long chain paraffins. Consequently, this first volume will be devoted to a full study of the bifunctional mechanism participating in hydroisomerization. The most widely accepted
reaction mechanism in literature for this reaction was proposed in the 50’s by P. Weisz [18-20].
However, in several occasions this reaction mechanism was proven to be insufficient for the explanation of all the experimental results [16, 21-29]. In those cases spilt-over species (hydrogen) were used to explain the catalytic performances that the mechanism proposed by Weisz could not. The impact of these spilt-over species on the reaction is still in great discussion within the scientific community where there is no consensual opinion. With this work we intend to uncover a little bit more about the impact of these spilt-over species on the linear paraffins hydroisomerization. The present chapter consists in a literature review about all the relevant data concerning the bifunctional mechanism required for the hydroisomerization reaction mechanism study.
2
Reactions involved in hydroisomerization
In order for hydroisomerization to take place two different types of active sites are required: acid function (AF) and hydrogenating / dehydrogenating function (HDF) [16, 30-33]. Each of these functions enables the occurrence of separate elementary reactions, which when combined together form the bifunctional mechanism responsible for the linear paraffins hydroisomerization. The comprehension of these reactions is, thereby, important for the study of the global reaction mechanism.
Part I – Hydroisomerization: Impact of the proximity between the active sites
19
2.1 Acidic function reactions
Several reactions can occur in the presence of an acid site, e.g. cracking, isomerization, oligomerization, hydrogen transfer, alkylation, etc [16, 29]. All these reactions happen through the activation of the hydrocarbon by addition or subtraction of a proton or a hydride / halogen, respectively. The protonation of the hydrocarbon is then performed on a Brønsted acid site [16, 30, 34-36], while the hydride / halogen subtraction is done on a Lewis acid site [16, 29, 37-39]. Regardless of the type of acid site used the hydrocarbon activation over acid sites always results from the formation of a carbocation.
2.1.1 Carbocation chemistry
The hydrocarbon transformation over acid function catalysts occurs through the formation of carbocations as reaction intermediates [30, 40, 41]. These can be formed by three different ways: protonation, hydride / halogen abstraction and as a result of another reaction. Two different types of carbocations exist: the carbenium being the most common consists in a positively charged ion containing a three-coordinated carbon atom (Fig. I.1 A); on the other hand, the carbonium equally represents a positively charge ion, this time with a five-coordinated carbon atom (Fig. I.1 B) [16, 29, 42].
Fig. I.1 Schematic representation of the carbenium (A) and carbonium (B) ions. Three different configurations are presented for the carbonium ion. Adapted from [16].
The carbocation stability is also a parameter to take into account when considering the formation of a possible reaction intermediate, as the hydrocarbon transformation via highly unstable carbocation is very unlikely. Several factors, both intramolecular and originated from the external environment, can affect the stability of carbocations. For instance the presence of a double bond near the charge,
e.g. benzenium, or if an electron attractor group exists on the molecule, e.g. Cl, highly stabilize the carbocation [16]. On the other hand, the environment near the molecule, as a counter ion or
R
R
ʹR
ʹ ʹC
+R
R
ʹR
ʹ ʹC
R
ʹ ʹ ʹH
+R
R
ʹR
ʹ ʹC
R
ʹ ʹ ʹH
+R
R
ʹR
ʹ ʹC
R
ʹ ʹ ʹH
+A
B
i
ii
iii
Chapter I – Literature Review: Reaction Mechanism
20
polarizing solid (e.g. zeolite), may also play a role on the carbocation stabilization [16]. Concerning the formation of non cyclic saturated carbocations, the most relevant in the bifunctional mechanism under study, one rule must always be clear: the higher substitution degree of the carbon containing the positive charge the higher the stability of the carbocation. Indeed, the occurrence of a carbocation on a carbon with three substitution groups (tertiary carbocation) is more likely than on a carbon with only one substitution group (primary carbocation) (Fig. I.2).
Fig. I.2 Carbocations stability. Formation heat values taken from [34].
The stability of the carbocations is intimately related to the reactivity of some hydrocarbons in the presence of acid sites, since a certain reaction is more likely to happen by passing through a more stable intermediate. The reactions involving acid sites will be detailed next with a major focus on cracking and isomerization, given that these reactions are the ones that occur more often during the linear paraffins hydrogenation. Moreover, only the reactions involving alkanes and alkenes will be described here, as only these are of concern towards the mechanism under study.
2.1.2 Isomerization
Once in the form of a carbocation a hydrocarbon can be transformed through isomerization. This reaction occurs through an intra-molecular mechanism and can occur both in cyclic and acyclic molecules. As it was said before, in this work only the acyclic reaction mechanism will be described. Still, two different types of isomerization can occur [16]:
Part I – Hydroisomerization: Impact of the proximity between the active sites
21
Type A: The displacement of an alky group is in the origin of an isomerization of type A. This type
of isomerization mechanism modifies the structure of the carbocation without changing the molecule branching degree.
Type B: This second isomerization type is a rearrangement of the ion structure with a modification
of the branching degree.
2.1.2.1 Type A isomerization
The migration of a negatively charged alkyl group (R-) is generally possible when the destination carbocation is one or two positions way [43]. The type A isomerization mechanism is described on Fig. I.3.
Fig. I.3 Isomerization type A: migration of an R group (R, e.g. methyl). Adapted from [16]. There is no real consensus about the impact of the alkyl group size on the migration rate, since some studies indicate that the migration of an ethyl group is faster than that of a methyl [44-46] while other indicate that the rate decreases with the alkyl group size [17, 47]. Still, the migration of a methyl group is quite easy, as its energy barrier is 5 kJ.mol-1 [48]. It is important to notice that the alkyl group migration rate highly depends on the nature of the carbocation from depart and arrival,
e.g. Tertiary Tertiary >> Tertiary Secondary.
2.1.2.2 Type B isomerization
The direct isomerization of a linear into a branched carbocation by the migration of terminal alkyl group is very unlikely, since this process requires the formation of a primary carbocation as an intermediate. Thereby, it is generally considered that the transformation occurs through the formation of cyclic carbocation. The most well known cyclic carbocation is the Protonated CycloPropane (PCP) shown in the type B isomerization mechanism presented in Fig. II.1.
H R Rʹ C+ C Rʹ ʹ H H R Rʹ C + C Rʹ ʹ H H R Rʹ C + C Rʹ ʹ H H R Rʹ C +C Rʹ ʹ H
Chapter I – Literature Review: Reaction Mechanism
22
Fig. I.4 Type B isomerization using a PCP as a reaction intermediate (R, e.g. methyl). Adapted from [16].
It is important to notice that the isomerization through PCP on small carbon chain carbocations, e.g.
4, is highly unlikely, since it requires the formation of a primary carbocation. For the same reason, the formation of a PCP on the chain extremities is disadvantaged. According to this fact the occurrence of alkyl groups through PCP is more likely to happen in the middle of the carbon chain,
e.g. n-methyl > (n-1)-methyl >> 2-methyl [17]. The formation rate for the formation of alkyl branches on a carbocation chain is also dependent on the size of the alkyl group. Indeed, the formation, for instance, of ethyl and propyl branching requires a four and five carbon ring intermediate, respectively, these being much less likely to be formed [17, 29]. This fact explains the much higher occurrence of methyl groups on isomerization products.
2.1.3 Cracking
The cracking reaction consists in the scission of a carbon chain C-C bond. Due to the increase of entropy and to the endothermic nature of this reaction, from a thermodynamic point of view, it is favored by high temperatures and low pressures [30, 49].
Two different mechanisms exist for the cracking of hydrocarbons on acid catalysts: -scission and protolytic cracking. Both mechanisms require the formation of carbocation species and will be described further [16, 29, 30, 48, 50-54]. H R Rʹ C1 H+ C3 C2 Rʹ ʹ H H H R Rʹ C1 C3 C2 Rʹ ʹ H H H + H R Rʹ C1 C3 C2 Rʹ ʹ H H H + H R Rʹ C1 C3 C2 Rʹ ʹ H H H + A B C
Part I – Hydroisomerization: Impact of the proximity between the active sites
23
2.1.3.1 -scission
The -scission mechanism promotes the scission of a C-H or a C-C bond in a carbenium. The C-H or C-C bonds to be broken are located on the position of the positively charged carbon (Fig. I.5).
Fig. I.5 C-H and C-C susceptible to -scission (R: H, CH3, C2H5…). Adapted from [16].
The cracking reaction mechanism through -scission corresponds to the electrophilic attack by the C-C bond located in position of the positive charge (Fig. I.6). This leads to the formation of a smaller carbocation and an olefin.
Fig. I.6 Cracking reaction mechanism through -scission (Rn: H, CH3, C2H5…). Adapted from [16].
Like in isomerization, the nature of the carbocation before and after the scission is directly related to the reaction rate. As an example the relative reaction rates of the different types of -scission for the n-decane cracking on a bifunctional catalyst Pt/HUSY are presented on Table I.1.
C
C
+
R
R
R
C
R
R
R
R1 R 5 C1 C3 C2 R6 H + R2 R 3 R4 R1 R 5 C1 C3 C2 R6 H + R2 R 3 R4 R1 C1+ R2 R3 +Chapter I – Literature Review: Reaction Mechanism
24
Table I.1. Relative -scission rates for the n-decane transformation on Pt/HUSY at 405 K [55]. -scission type B2 considered as reference.
Transformation Relative rate
Type A (Tertiary Tertiary) 1050
Type B1 (Secondary Tertiary) 2.8
Type B2 (Tertiary Secondary) 1.0
Type C (Secondary Secondary) 0.4
Type D (Secondary Secondary) ≈ 0
2.1.3.2 Protolytic cracking
The protolytic cracking occurs through the Haag-Dessau mechanism, which consists in the direct protonation of an alkane with the formation of a carbonium [29, 53]. The unstable intermediate will then collapse forming a smaller alkane (or hydrogen molecule) and a carbenium [49, 53]. This mechanism only becomes evident at temperatures around 800 K or higher [29, 53].
The high temperature required for the occurrence of this reaction makes it unlike to happen during hydroisomerization, since this reaction requires milder conditions to avoid cracking. Therefore, no further details will be provided about it.
2.1.4 Oligomerization
Oligomerization is a reaction that promotes C-C bonds formation, leading to higher molecular weight hydrocarbons. This reaction is exothermic and causes a reduction in the number of molecules. Accordingly, it is thermodynamically favoured at low temperatures and high pressures [49].
The oligomerization reaction mechanism is the inverse of -scission (Fig. I.6) and therefore not shown here. In general, oligomerization reactions on acid catalysts are followed by the cracking of the oligomers formed, principally at high reaction temperatures, thereby changing the cracking products distribution [48, 56]. The oligomerizaton-cracking mechanism occurs for an alkane as shown in Fig. I.7. It is important to notice that the mechanism occurring on alkene is the same without the hydride transfer step.
Part I – Hydroisomerization: Impact of the proximity between the active sites
25
Fig. I.7 Oligomerization cracking route for an alkane (RmH) involving the oligomerization of an
alkene (An) with a carbenium (Rm+) and subsequent -scission and hydride transfer [53].
The oligomerization reaction on the acid sites of the catalyst takes place via a carbenium ion (Rm+).
Once the carbenium ion is formed, for example in the initiation step of the catalytic cracking mechanism, it can react with a free olefin (An=) to form a heavier carbenium ion (Rm+n+), which can
subsequently suffer β-scission originating an alkene (Ap=) and a shorter carbenium ion (Rm+n-p+).
This carbenium ion can then by hydride transfer receive a proton from a reactant alkane molecule (RmH), leading to the formation of another carbenium ion (Rm+) and a desorbed alkane (Rm+n-pH)
different from the first alkane (RmH) [53].
The desorption of the high molecular weight carbenium produced through alkylation (dimerization)
occurs before the β-scission, the oligomerization reactions can be responsible for the formation of
products with long carbon chains that can be larger than the feed.
It is important to notice that, due to the bimolecular mechanism responsible for the oligomerization, this reaction is favoured on acid catalysts with high acid sites density and large pores [56-58].
2.1.5 Other acid function reactions
In addition to isomerization, cracking and oligomerization, other reactions may occur through acid catalysis. Yet, their relevance for the present work is small and for this reason they will not be detailed. Indeed, reactions like hydride transfer or coke formation have no impact on hydroisomerization due to the presence of a strong hydrogenation function. Yet, it is important to
An= Rm+ n+ Ap= Rm+ n-p+ Rm+ Rm+ n-pH RmH Oligomerization H-transfer -scission
Chapter I – Literature Review: Reaction Mechanism
26
notice than in the absence of an adequate hydrogenation function these reactions may occur, though this will be discussed further.
2.2 Metallic function reactions
The reactions occurring on the metallic function are of high importance for the bifunctional mechanism responsible for the hydroisomerization reaction, since, as it will be discussed latter, these are responsible for the first reaction step. The dehydrogenation and the hydrogenation reactions consist in the transformation of an alkane into an alkene and vice-versa. In addition to the hydrogenation and dehydrogenation, other reactions like the skeletal isomerization of hydrocarbons and hydrogenolysis (cleavage of a C-C bond) can occur on the metallic sites [29]. However, in the present work, the impact of these reactions was minimal. Therefore, they will not be further detailed.
2.2.1 Hydrogenation (Dehydrogenation)
As the hydrogenation and the dehydrogenation are in fact inverse reactions, the description will be focused only on the hydrogenation reaction mechanism.
The hydrogenation reaction consists in the saturation of a double bond with hydrogen. This reaction is highly exothermic and causes a reduction in the number of molecules [59]. Therefore, it is thermodynamically favoured at low temperatures and high pressures. On the other hand, in order for the reaction to take place a metallic catalyst is required, e.g. platinum. The main role of the catalyst is to activate the hydrogen so that the reaction can take place, as shown on Fig. I.8, since the thermal rupture of the H-H bound is energetically unapproachable. The hydrogenation of the ethylene molecule is given as example, on Fig. I.8, for the hydrogenation reaction mechanism demonstration.
Part I – Hydroisomerization: Impact of the proximity between the active sites
27
Fig. I.8 Ethylene hydrogenation reaction mechanism [59].
2.3 Bifunctional mechanism
The catalytic synergy resulting from the association of a hydrogenating / dehydrogenating function (HDF) and an acid function (AF) was since long proved [16]. The reaction mechanism explaining
this synergy was proposed by Weisz in the 50’s [18-20].
2.3.1 Weisz’s mechanism (Classical mechanism)
The bifunctional reaction mechanism proposed by Weisz considers that each one of the catalyst functions, HDF and AF, has an independent catalytic action that is, however, complementary of the opposite function. According to the classical reaction mechanism the hydrocarbon dehydrogenates through the HDF action into an alkene, diffusing then to the AF where it is transformed in a carbocation undergoing several possible transformations, i.e. skeletal rearrangement, -scission, cyclization, etc. After deprotonation, the resulting alkenes diffuse back to the HDF to be hydrogenated into saturated hydrocarbons. The schematic representation of this reaction mechanism can be seen in Fig. I.9. It is important to notice that under the reaction conditions (low temperature and hydrogen atmosphere), the dehydrogenation reaction is not thermodynamically favored. Still, the trace amounts of alkenes, at equilibrium, are enough to feed the acid sites and make the olefin transformation primary to the alkane [16].
Chapter I – Literature Review: Reaction Mechanism
28
Fig. I.9 Schematic representation of the classical mechanism. Adapted from [32].
In his studies, Weisz also observed that the olefin transfer between the active sites could be the limiting reaction step, reducing the activity and selectivity of the bifunctional catalyst [20].
Therefore, the “intimacy criterion” was proposed considering that the FA and HDF should be close
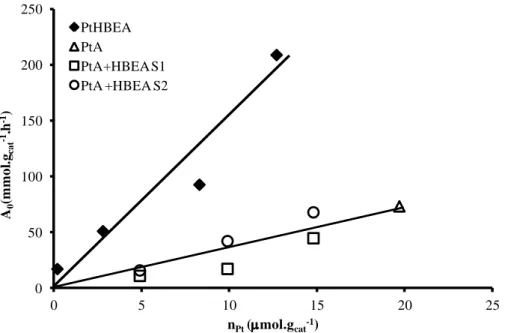
enough to avoid the olefin transfer limitations. Nonetheless, the criterion proved to be insufficient to obtain the ideal bifunctional catalyst. Therefore, the concept of balance between the FA and HDF was introduced, considering that on the ideal catalyst the HDF should be active enough to feed the AF with olefins [60].
Guisnet et coll. used the nMe/nH+ ratio (nMe– number of accessible metal sites; nH+ - Brønsted acid
site density) to quantify the balance between the two active functions [30, 46, 61]. Typically, for a given catalyst the reaction rate is limited by the HDF for low values of this ratio (Fig. I.10 A). Thus on a specific acid support, the catalyst activity increases when nMe/nH+ increases until a plateau is
reached, corresponding to a reaction rate limited by the number of protonic sites [46, 62, 63] (Fig. I.10 A). The value for which the plateau is observed depends, nonetheless, on several factors such as: the nature of the metal [64-67], of the hydrocarbon [32, 68] and of the zeolite [32, 69-72]. In addition to the reaction rate, Guisnet et coll. also made a relation between the nMe/nH+ ratio and the
catalysts stability and selectivity [30, 62]. Indeed, when the HDF is low, small nMe/nH+, the catalyst
deactivation by coke formation is possible. Likewise, at low HDF the hydrogenation rate of the olefin intermediates is slow, causing the appearance of multibranched isomers and cracking products at low reaction conversions (apparent primary products). The results obtained by Guisnet
et coll. on the n-decane hydroisomerization over Pt/HY are resumed in (Fig. I.10 B).
Diffusion Diffusion n-Cn + H2 - H2 n-C= n + H+ - H+ n-C= n n-C + n + H+ - H+ i-C+ n i-C= n - H2 + H2 i-C= n i-Cn n-/ iC+ n-m + H+ - H+ n-/ iC+ m n-/ iC= m + n-/ iC= n-m - H2 + H2 n-/ iC= m n-/ iC= n-m n-/ iCm n-/ iCn-m De / Hydrogenating Function Metallic Site: Noble metal
Acid Function
Brønsted acid site: H+
Part I – Hydroisomerization: Impact of the proximity between the active sites
29
Fig. I.10 Impact of the AF and HDF balance on hydroisomerization: A) Activity versus nMe/nH+; B)
Summary of the nPt/nH+ ratio variation on the n-decane hydroisomerization on PtHY catalysts (250
°C, Patm, H2/C10 =9). Adapted from [73].
2.3.2 Spilt-over hydrogen (Hsp) mechanisms
Despite the mechanism proposed by Weisz being the most well accepted for bifunctional catalysts and, therefore, for the hydroisomerization reaction, other mechanisms have been proposed in literature. These take into consideration the effect of spilt-over hydrogen species (Hsp) on the
bifunctional reaction.
2.3.2.1 Spilt-over definition
Spilt-over involves the transport of an active species (spilt-over species) sorbed or formed on a first phase (donor), onto another phase (acceptor) that does not under the same conditions, adsorb or form these species [29, 74, 75] (Fig. I.11). The result may be a reaction of this species on a second phase (acceptor) with other adsorbed molecules or the acceptor phase [29]. This phenomenon does not require, however, the direct deposition of the donor, since the spilt-over species have the capacity to diffuse between supports [76, 77] (Fig. I.11). The most widely discussed species in terms of spilt-over is atomic hydrogen. Yet, the diffusion of the hydrogen atoms on the acceptor phase is considered to be quite slow when compared to the di-hydrogen dissociation rate [74, 78-80]. Therefore, the rate limiting step for the spilt-over hydrogen phenomenon is the diffusion on the acceptor support. It is important to note that the diffusion rate of the hydrogen species on the acceptor support depends on several factors, such as temperature, hydrogen pressure and acceptor support nature.
Reaction rate limited by: AF HDF Ac ti v it y nMe/nH+ A nCn M B C nCn (M,B) C nCn M B C
nPt/nH+ High (> 0.10) Medium Low (< 0.03)
Sta bility Activity
Selectivity (i-C10)
Appa rent rea ction Scheme Perfect High High Medium Medium High Low Low Low B
![Table I.1. Relative -scission rates for the n -decane transformation on Pt/HUSY at 405 K [55]](https://thumb-eu.123doks.com/thumbv2/123doknet/8038531.269474/41.892.85.840.183.367/table-relative-scission-rates-decane-transformation-pt-husy.webp)
![Fig. III.3 Schematic representation of Yang et al . sulfur resistant hydrogenation catalyst [112]](https://thumb-eu.123doks.com/thumbv2/123doknet/8038531.269474/84.892.214.683.109.394/fig-schematic-representation-yang-sulfur-resistant-hydrogenation-catalyst.webp)

![Risiko- & [und] Schutzfaktoren der psychischen Gesundheit humanitärer Einsatzhelfer : eine systematische Literaturübersicht](data:image/gif;base64,R0lGODlhAQABAIAAAP///wAAACH5BAEAAAAALAAAAAABAAEAAAICRAEAOw==)