HAL Id: hal-03043844
https://hal.archives-ouvertes.fr/hal-03043844
Submitted on 7 Dec 2020HAL is a multi-disciplinary open access archive for the deposit and dissemination of sci-entific research documents, whether they are pub-lished or not. The documents may come from teaching and research institutions in France or abroad, or from public or private research centers.
L’archive ouverte pluridisciplinaire HAL, est destinée au dépôt et à la diffusion de documents scientifiques de niveau recherche, publiés ou non, émanant des établissements d’enseignement et de recherche français ou étrangers, des laboratoires publics ou privés.
Theoretical insights on acceptor–donor dyads for organic
photovoltaics
Michele Turelli, Domenico Alberga, Gianluca Lattanzi, Ilaria Ciofini, Carlo
Adamo
To cite this version:
Michele Turelli, Domenico Alberga, Gianluca Lattanzi, Ilaria Ciofini, Carlo Adamo. Theoretical insights on acceptor–donor dyads for organic photovoltaics. Physical Chemistry Chemical Physics, Royal Society of Chemistry, 2020, �10.1039/D0CP03038D�. �hal-03043844�
Theoretical Insights on Acceptor-Donor dyads for Organic Photovoltaics
Michele Turelli1,2,3, Domenico Alberga4, Gianluca Lattanzi2,3*, Ilaria Ciofini1 and Carlo Adamo1,5*
Chimie ParisTech, PSL University, CNRS, Institute of Chemistry for Life and Health Sciences, Paris, France; Physics Department of Trento University, Via Sommarive 14, Povo (Trento), 38123, Italy; Trento Institute for Fundamental Physics and Applications (INFN-TIFPA), Via Sommarive 23, Povo (Trento), 38123, Italy; Dipartimento di - , Via E. Orabona, 4, I-70126 Bari; and Institut Universitaire de France, 103 Boulevard Saint Michel, F-75005 Paris, France
Abstract
The field of organic photovoltaics has witnessed a steady growth in the last decades and a recent renewal with the blossom of single-material organic solar cells (SMOSCs). However, due to the intrinsic complexity of these devices (both in terms of their size and of the condensed phases involved), computational approaches enabling to accurately predict their geometrical and electronic structure and to link their microscopic properties to the observed macroscopic behaviour are still lacking.
In this work, we have focused on the rationalization of transport dynamics and we have setup a computational approach that makes a combined use of classical simulations and Density Functional Theory with the aim of disclosing the most relevant electronic and structural features of dyads used for SMOSC applications. As prototype dyad we have considered a molecule that consists in a dithiafulvalene-functionalized diketopyrrolopyrrole (DPP), acting as electron donor, covalently linked to a fulleropyrrolidine (Ful), the electron acceptor. Our results, beside a quantitative agreement with experiments, show that the overall observed mobilities results from the competing packing mechanisms of the constituting units within the dyad both in the case of crystalline and amorphous phases. As a consequence, not all stable polymorphs have the same efficiency in transporting holes or electrons which often results in a highly directional carrier transport that is not, in general, a desirable feature for polycrystalline thin-films. The present work, linking microscopic packing to observed transport, thus opens the route for the in-silico design of new dyads with enhanced and controlled structural and electronic features.
1)Chimie ParisTech; 2) Universityof Trento; 3) INFN-TIFPA; 4) University of Bari; 5) IUF *) corresponding authors: [email protected]; [email protected]
1. Introduction
The field of organic photovoltaics has been steadily growing for the past two decades. A wide and concerted research effort has been successful in improving the efficiency of organic solar cells so as to reach commercially viable values, even if their efficiency still lags behind the conventional inorganic technologies1. This research area remains nonetheless extremely appealing thanks to the versatile chemistry of organic compounds that promises virtually unlimited possibility for fine-tuning and purpose-driven design.
However, these benefits come hand in hand with intrinsic difficulties. The physics of organic solar cells is more complex and less understood compared to that of their inorganic counterpart2-4. The persistence of gaps in our understanding is partly due to the vast chemical space that these compounds occupy which results in a variety of possible behaviours that need to be traced back to more general principles. The coming maturity of the field is signalled by the increasing necessity to rationalise the wealth of experimental data already collected into a theoretical framework capable of directing the investigation and synthesis of novel materials through a list of design principles5–9. Indeed, theoretical approaches are becoming increasingly widespread as tools to guide the search for new molecules10-15, the majority of which, however, are still being discovered and tested experimentally16.
The need for a deep theoretical understanding aimed at establishing a series of sound design principles is even more pressing for the class of materials employed in the so-called single-material organic solar cells (SMOSCs). The domain of SMOSCs has recently seen a boost due to the ability of these materials to circumvent the disadvantages of the more established donor-acceptor bulk heterojunction architecture17-19, such as thermodynamic instability20,21 and the recent identification of molecules with higher conversion efficiencies22–32. The treatment of these systems brings an additional layer of complexity both in terms of the physical process involved and their molecular size. Indeed, their size is often too large to allow the application of the quantum chemical methods needed to accurately predict the relevant properties33 and forces treatments based on less accurate semi-empirical methods34,35. In this regard we present here a computational study of one of these materials employed in a SMOSC active layers that has been shown experimentally to have a remarkable performance22. For this system, a full characterisation is difficult and leaves the interpretation of the mechanisms underlying its efficiency unsatisfactory. This makes it an ideal test-case to prove the value and efficacy of theoretical methods aimed at complementing and directing the experimental work.
The molecule belongs to a particular class of materials employed in SMOSCs called dyads36,37. A dyad is a system combining an electron donor with an acceptor group by covalently linking them in a single molecular architecture, thus realising a molecular
heterojunction38. In the majority of cases, dyads employed in organic photovoltaics are obtained via the covalent bonding of a fullerene (C60), playing the role of acceptor, with a
π-conjugated monodisperse oligomer, acting as donor39. The dyad we focus on consists in a dithiafulvalene-functionalized diketopyrrolopyrrole (DPP) acting as donor and a [60]-fulleropyrrolidine (Ful) as acceptor (see Figure 1). Experimentally22, the two moieties were selected on the basis of reported performances of the separated molecules in the literature rather than through the application of more rigorous purpose-oriented design rules.
In dyads the first step of light-to-energy conversion common to organic active materials, i.e. the dissociation of the exciton into separate charge carriers, is achieved on the molecule itself16,32. However, differently from inorganic semiconductors, in dyads the mechanism is more complex and nuanced, involving a series of intermediate steps and transitions that influence the overall performance4. Following charge separation, charges move in the active layer towards the electrodes: the efficiency of this transport is fundamental in determining the performance of the solar cell40,41. In this work, we will focus on transport dynamics with the aim of pointing out the electronic and structural features of the dyads that are most relevant to enhance solar cell efficiencies. It is worth to notice that, at the best of our knowledge, this is the first time that this kind of theoretical study has been carried out on dyads for SMOSC applications.
2. Computational details
The modelling of DPP-Ful dyad poses specific challenges due to its nature of ambipolar conductor. Carriers of both signs can be transported independently over morphologies whose microscopic details are not entirely known22. From a theoretical point of view this requires the prediction of both ordered and disordered morphologies and to simulate transport in both cases.
The first mandatory steps consist in the gas-phase calculations on the isolated molecule. These calculations were performed at density functional theory (DFT) level applying the B3LYP functional in conjunction with the triple- Pople’s basis set (6-311G(d,p) )48,49. Dispersion interactions were accounted for through the Grimme’s empirical potential (D3)50. All optimisations, including those carried out to compute reorganisation energy λ for both
anionic and cationic states, were performed at the same level of theory via the Gaussian09 package51.
Ordered morphologies were obtained following the approach adopted in reference 52. We sampled the thermodynamically most relevant crystal structures, i.e. crystal morphologies with a non-trivial probability of being occupied at experimental conditions, via the polymorph predictor module of Materials Studio53. The prediction of crystal structures is carried out by arranging the packing motifs within a given space group and then by simultaneously optimizing the molecular packing inside the cell and the unit cell parameters. These steps were carried out using the Dreiding force field54, largely used for molecular crystal prediction10, 52. We limit our search to the most probable space groups reported for similar organic compounds i.e. P212121, P21/c, P , P21, C2/c and Pna2155. A number of independent
searches have been performed until no new low-energy structures, significantly different from those already obtained, are found. The resulting structures are then clustered and ranked according to their energy and density and those with the highest rank are selected. This ranking is then refined according to the results of full DFT geometry optimizations of the unit cells and the atomic positions performed including periodic boundary conditions (PBC) using the CRYSTAL17 code56. In this case, the same exchange correlation functional (B3LYP-D3) is used together with an optimised basis set, tailored to organic crystals, available in the SI. Amorphous configurations are extracted following the same computational procedure proposed in reference 52. We obtain the disordered configurations for a system of 500 molecules by taking 150 uncorrelated snapshots from a molecular dynamics (MD) trajectory. The initial conditions are chosen as to accurately reproduce the disordered phase. DPP-Ful molecules are initially placed at random positions in a cubic cell 270 Å wide. This configuration is evolved through a simulation in the NPT ensemble at high temperature T=800 K and P=1 atm until the volume of the cell reaches convergence. Another NPT simulation is then performed at lower temperature, T=300 K. The system is further evolved through a third NPT simulation that produces the trajectory from which configurations are extracted. All simulations are terminated at convergence of the volume. The final trajectory is 30 ns long. Simulations were carried out via the NAMD2.12 package57 with CGenFF parameters58,59. Van der Waals cut-off is set at 12 Å and electrostatic interactions are treated according to the particle mesh Ewald method.
Charge dynamics is simulated, for both crystalline and amorphous phases, using the non-adiabatic high-temperature limit of semiclassical Marcus charge transfer theory42. The approximation is justified by the fact that charge carriers in organic materials are mostly
localised on single molecules that weakly interact with their neighbours40,41. The formula for the evaluation of the rate constants reads:
The two main parameters in equation 1, encoding microscopic information, are the reorganisation energy λ, a molecular level property evaluating the energy cost of exchanging a charge carrier, and the transfer integral Jab, that, computed for each pair of exchanging
molecules, quantifies the propensity of the carrier to be transferred mediated by the interaction Hamiltonian of the pair.
We note that reorganisation energy in our calculations neglects the contribution due to the change in polarisation of the surrounding environment before and after the exchange and only accounts for intramolecular geometry reorganisation, according to:
where the subscripts indicate molecular state (c or n, charged or neutral) and molecular geometry (C or N, charged optimised or neutral optimised). For sake of coherence, the same DFT approach, B3LYP-D3, has been used for the calculations of reorganization energies. Our previous experience52,60,61 showed that mobilities computed the B3LYP level are in (very) good agreement with the experiments, even if, generally speaking, theirs values could be affected by the choice of the DFT approach62.
Transfer integrals are computed treating the molecular pair as an isolated dimer under the assumption that its orbitals arise from the interaction of monomer frontier orbitals – HOMO for hole transport and LUMO for electron transport – φiH/L and φj H/L 63:
This approximation is justified by the small coupling, between monomers in the pair34. All the orbitals needed to estimate the electronic coupling are obtained through the semi–empirical ZINDO method35, which has been proven to provide accurate enough estimates31,32,51.
Rates are evaluated for all pairs specified in this list. The final step to estimate mobility is to perform kinetic Monte Carlo simulations using the computed rate constants. The mobility is given by the ratio of〈v〉, the average velocity in the direction of the electric field E and the magnitude of the latter.
Owing to the fact that the molecule carries both a DPP electron donor moiety and a fullerene electron acceptor group and that the two act as separate transport channels, the two distinct
conducting moieties were defined in order to simulate the charge-separated (CS) state. The separation of the two moieties is performed accordingly to the localisation of the HOMO and LUMO orbitals (see Figure 2). The separation is carried out by cutting the bond between the pyrrolidine in the fullerene moiety and the proximal thiophene ring, as sketched in Figure 1. The bonds at the cut are saturated with hydrogen atoms and the computational procedure to simulate transport is applied to the morphologies of the separated moieties.
3. Results and discussion
The most widely applied theoretical framework to describe dynamics in such systems is Marcus theory40–42. An approach based on the non-adiabatic high-temperature limit of this theory is well justified since the working conditions and components, i.e. high temperature (300 K) and highly localised charge carriers, largely fit into the range of validity of the theory. The convenience of Marcus theory lies in its ability to describe dynamics in molecular solids as a set of charge transfer reactions with associated rates. Rates depend critically on two parameters (see Eq.1) associated with molecular electronic properties and, more importantly, to bulk structural properties (i.e. crystal structure packing). Their prediction is essential in giving an informed estimate of charge transport performance.
Bulk properties are linked to solid phase morphologies that are the result of the self-organisation of large numbers of molecules. The prediction of solid phase packings, either crystalline or amorphous, remains to this day a challenging task47. This is especially true for relatively large organic compounds like dyads that might exhibit polymorphism43,44. The reason lies in the heavy computational cost of an accurate treatment of intermolecular interactions that require a high-level description of the electronic structure that includes dispersion forces, a key driver for aggregation in molecular solids43,45. In order to overcome this issue, crystal packings are guessed via a less-demanding computational procedure, that employs a classical force field coupled with a Monte Carlo sampling46. The output is a set of the most thermodynamic viable crystal polymorphs whose ranking is refined through higher level calculations. The procedure still overlooks other contributions to the ranking that might change it significantly47 however, the comparison between properties evaluated for these packings and the available experimental data may lead to the reliable identification of the molecular packing responsible for the reported performance. This is of course extremely relevant since it provides direct knowledge of microscopic details that either boost or inhibit transport efficiency of charge carriers. Moreover, the availability of crystal morphologies not directly accessible in the experiments due to their unfavourable thermodynamic rank allows a
direct comparison between them. Starting from this, a more general picture can be drawn, thus paving the road towards a set of synthetic principles to be followed in the design of dyads with enhanced performance.
3.1 Isolated molecules and crystalline polymorph
A first step has been the characterisation of molecular properties relevant to charge transfer in gas-phase. Previous works22 have largely stressed how, in dyads, properties of the isolated donor and acceptor moieties should be retained in order to ensure an optimal transport of the charge carriers, meaning that they should remain sufficiently decoupled in terms of their respective electronic structure. A qualitative indication of this decoupling is given by the localisation of the frontier orbitals of the molecule reported in Figure 2, whose energies are reported in Table S1. The strong localisation of frontier orbitals in different parts of the molecule is clear evidence that the moieties are kept largely independent and can efficiently produce a charge-separated state48. Indeed, the HOMO is highly localised on the donor moiety, DPP, while the probability amplitude of the LUMO is markedly centred on the Ful moiety. This effect can be traced back to structural features of the dyad. In particular, the possibility of weak interactions of the fullerene with the closest aliphatic chain reduces the planarity of the thiophene rings linking the fullerene moiety to the diketopyrrolopyrrole group. As a consequence, the π-conjugation is constrained to the remaining part of the donor moiety, producing a certain degree of insulation between the two parts, as can be seen in Figure 2. This effect is further modulated by the interactions proper to each polymorph packing where it is generally amplified.
Starting from the gas-phase data, reorganisation energies were also estimated according to equation 2. The ambipolar character of the molecule implies that both anionic and cationic states should be investigated. Despite the moieties being virtually independent in terms of transport after charge separation, we consider the response of the whole molecular structure to the presence of a charge carrier. Computed values for the hole (λ(h) = 361 meV) are compatible with those reported for other diketopyrrolopyrrole derivatives, λ(h)~ 300-400 meV64,65. At the same time, the energy cost for the exchange of an electron (λ(e) = 212 meV) is close to the reported reorganisation energies for similar fullerene derivatives, λ(e)~130-150 meV for PCBM66. This agreement is another strong indication of the efficient decoupling of the two moieties in the dyad.
As already mentioned in the introduction, polymorphs can exhibit dramatically different transport properties. Thin-films employed as active layers usually have a pronounced
polycrystalline character, so that it could be expected that different crystal domains, with distinct orientations and possibly packing, will coexist with less ordered regions67. In order to provide a reasonable description of the structures present in DPP-Ful thin-films we predicted and analysed the 5 lowest energy crystal packings obtained following the procedure described in the computational details.
In Table 1 we report the unit cell data for the crystal structures identified after full geometry optimization at the DFT level following a preliminary optimization using an empirical force field. Structural parameters for these latter are reported in Table S2, while their structures are reported in Figure S1-S5. The structures belong to the P 212121 symmetry group (structure 0
-i.e. the most stable-, 1, 3, 4) and P n a 21(structure 2). At this level, all the extracted
polymorphs are within a range of about 20 kcal/mol from the most stable structure taken as reference. It is interesting to notice that when moving from empirical to DFT calculations a significant change of the ranking is observed, with variations affecting both the energies values and the relative stabilities. It is nonetheless reassuring, that the two most stable polymorphs are still the same according to both approaches, even if their structural parameters are significantly changed (see Table 1 and Table S3). Since we expect the DFT structures to be more accurate than those obtained via the molecular mechanics approach, only the former will be discussed in detail.
The DFT energy differences between the polymorphs considered are rather pronounced. Based on these values in fact, the probability to populate any of the structures beyond polymorph 1 is absolutely negligible. Nonetheless, one has to consider that in actual thin-films several polymorphs could co-exist due to the experimental conditions of preparation which could favour the nucleation of a particular structure over another and achieve stability thanks to large kinetic barriers. At the same time, an incertitude of about 3 kcal/mol in the stabilities should be expected68,69. As a consequence, all these polymorphs will be discussed in some details, starting from their structural features.
Given the character of ambipolar conductor of the molecule, the most desirable packing should arise from a balance between donor and acceptor competing arrangements, both considerably impacting the magnitude of carrier mobilities. The main factor affecting these latter is the value of transfer integrals, Jij, which depends on the orbital overlap between the
two moieties involved in the charge transfer. The relative distance between exchanging moieties, dij, is the most natural index in term of structure, but other, more complex,
geometrical features emerge, among these the angle between conjugation planes in donor moieties and their relative shift.
A detailed analysis of the polymorphs obtained can give more precise examples of these geometric features in the particular case of DPP-Ful, but results can easily be generalised since many characteristics are common to other dyads and, more in general, to organic photovoltaic materials.
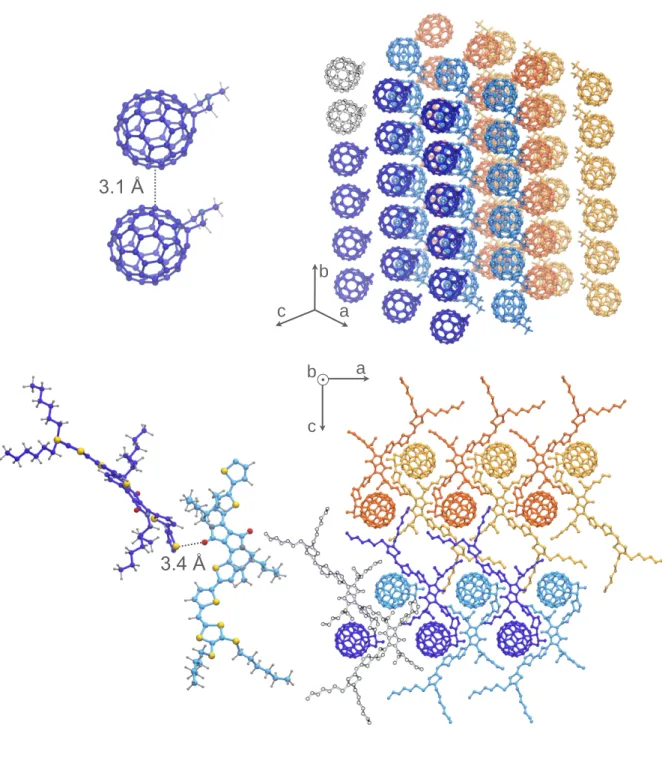
The optimized structure of the most stable polymorph (0), belonging to the orthorhombic
P212121 space group, features four dyad molecules in the asymmetric unit. In the supercell
these molecules are arranged according to a double herringbone in the ac-plane of the crystal, as shown in Figure 3. These two herringbones are identical but have opposite orientation, i.e. along the positive and negative directions of crystal axis a. The minimum approach distance for donor moiety dimers is dij = 3.4 Å and it is achieved for consecutive molecules belonging
to the same herringbone (Figure 3 lower-left). Layers are then stacked along b, where fullerenes form columns acting as conducting channels for electrons. The distance between fullerenes pairs in consecutive stacking planes is of 3.1 Å (10.3 Å centre-to-centre). The closest contacts are achieved in the stacking direction for fullerenes, as well as in the orthogonal plane (bc) for DPP molecules. The optimized structure of this polymorph is also the one showing, among those investigated, the best agreement with the limited structural data available. In particular, a comparison with the X-ray diffractogram reported in ref. 22 shows that many of the peaks are in reasonable agreement with the simulated XRD pattern for crystal structure 0, as shown in Figure S6. We also remark that the most stable polymorph is not the one with the highest density (see Table 1) suggesting dispersion-driven interactions between DPP-moieties are able to confer a higher stability and that these moieties play a key role in stabilising the packing from a thermodynamic point of view.
The optimized motif of the next most-stable packing, polymorph 1, features a structure in which fullerenes are distributed in a regular lattice in the bc-plane, with the space in between occupied by a close-knitted donor moiety matrix (Figure 4, right). Similar planes are then stacked in the a direction where the closest approach between fullerenes belonging to stacked planes is achieved with a distance of dij= 2.9 Å allowing rather large values of Jij. The donor
structure across the planes is composed of a network of DPP moieties with their backbones arranged almost perpendicularly so that no π-π stacking is observed for DPP moieties in close proximity (see figure 4, left). The packing is rather efficient and confers to the polymorph a density of 1.443 g/cm3. The closest approach distance between DPP pairs is rather large, dij =
4.9 Å, and does not allow efficient transport of holes within the bc-plane nor along a.
Crystal motif of polymorph 2, presents one of the most efficient packings in the set. In the a direction fullerene moieties are arranged again in separated columns that constitute the
backbone of two repeated herringbone structures in the ab-plane (Figure 5, right). The minimum distance between a pair of DPP moieties is realised between molecules belonging to neighbouring herringbone structures along c, with a minimum distance dij of 3.45 Å. The
approximate conjugation planes of the pair at the point of closest approach are almost parallel and consequently the orbital overlap is large. DPP pairs in the other directions have reduced contact due to the hindrance of lateral aliphatic chains that keeps them distanced. Fullerene moieties in the backbone of the herringbone structures and stacked along a also have a minimum approach distance dij of 3.2 Å.
In polymorph 3 we have again a motif composed by two herringbone structures, evident in the
bc-plane that are then stacked along crystal axis a. In this case the two rows of molecules
making up each distinct herringbone structures are more intertwined (see Figure 6) with respect to other similar structures in the other polymorphs. In this configuration fullerenes are packed close to another fullerene belonging to a neighboring herringbone structure. These pairs are then stacked along a where we have again column-like stacking. All elements in the column reach a minimum distance of 3.4 Å, ensuring rather large values for transfer integrals. The fact that rows belonging to the same herringbone motif are very intertwined in the bc-plane means that donor moieties remain more distant to accommodate for the fullerenes so that DPP moieties cannot reach efficient packing. Minimum distance in the bc-plane is around 4.3 Å and the approach configuration is edge-on, detrimental to transfer efficiency. In the stacking direction a, the hindrance of the aliphatic chains brings the minimum approach distance at over 9 Å, making transport along this direction completely inefficient.
The structure of polymorph 4, is characterised by two herringbone motifs wedged into each other in the bc-plane and then stacked along a, as shown in Figure 7. In this herringbone structure the closest approach for DPP moieties is realised between elements of different herringbone motifs (Figure 7). The minimum distance is dij =3.5 Å but the approach is
edge-on, i.e. the approximate conjugation planes of the DPP backbones are almost perpendicular to each other. This arrangement lowers considerably the orbital overlap. The stacking along a of the motif in the bc-plane results in the formation of fullerene columns or channels similar to what already observed for polymorph 1, see Figure 4. The minimum approach distance for acceptor moieties in this direction is 3.0 Å, a value small enough to allow large rates of electron transfer between fullerenes.
Polymorphs 2 and 3 boast the largest densities in the set with 1.454 and 1.463 g/cm3 respectively.
As mentioned, the analysis of the localized structures provides valuable insight about the main features boosting or hampering transport at supramolecular level and relating packing to transport efficiency through the assessment of carrier mobilities will help identify and possibly generalise the main mechanisms at play. Results obtained for the theoretical mobilities for all DFT structures are reported in Tables 2 and 3. Data computed using the optimized molecular mechanics structures are reported in Tables S4 and S5 for sake of completeness, but they will be not discussed, since they are less reliable as supported by the worse agreement with the experimental mobilities (see below).
The most stable polymorph (0) is also the one with the best overall performances in terms of both electron and hole transport. Transport takes place mainly along the a-direction for holes while transport across different planes, i.e. along c, is very limited. In the ac-plane the configuration of the closest exchanging pair allows a rather large coupling element, Jij=4.11⋅10
−3 eV, yielding a Marcus rate of 2.04⋅1011
s-1. The rather large orbital overlap is ensured by the proximity of the conjugated backbone of one exchanging moiety in to the diketopyrrolopyrrole unit of the second. The overall mobility along a reaches μa(h)=4.86⋅10−4
cm2/V⋅s, with the herringbone structures acting as main transport channels. Along crystal axis
c on the same plane transport is significantly less efficient due to a smaller coupling between
DPP moieties belonging to different herringbone structures, Jij = 4.11⋅10−4 eV giving μc(h)= 2.84 ⋅10−7 cm2/V⋅s. In the stacking direction, corresponding to crystal axis b, the hole mobility is very low μb(h)= 4.8 ⋅10−11 cm2/V⋅s. Conversely, along this direction we see the
largest value for electron mobility, that takes place along the fullerene channels, μb(e)=4.99
⋅10−4
cm2/V⋅s, while being negligible in the other directions.
As remarked above, this packing is not the most dense, yet its structure is able to maximize dispersion interactions by allowing a pronounced interdigitation of DPP moieties that ultimately gives it the highest stability. Comparison with the experimental data, (μ(e)=1.1⋅10−4
cm2/V⋅s, μ(h)=1.3⋅10−4 cm2/V⋅s), reveals that our estimates (μavg(e)=2.88⋅10-4 cm2/V⋅s, μavg(h)=2.81⋅10-4 cm2/V⋅s) are able to reproduce the behaviour of the material experimentally observed. Our estimates for both charge carriers are roughly 2.8 times larger than experimental mobilities but the order of magnitude and the relative values between holes and electrons are compatible with the available data22. The difference in the absolute values is to be expected given the many effects normally present in a real-life device that can lower the value of mobility with respect to an ideal periodic structure. Real-life devices operate in the centimetre-scale, this means that charges are required to be transported over large distances
where other effects (e.g. electrodes, interfaces, higher-scale morphology of the active layer) that lie outside the range of our treatment may affect the overall transport efficiency.
Polymorph 1 exhibits a much more unbalanced transport behaviour. Electron transport is more efficient than polymorph 0 given the closer packing in the a direction, and again only takes place along this axis. The minimum distance between two fullerenes belonging to different bc-planes is only 2.6 Å. This allows a large transfer integral, Jij=1.68⋅10−2 eV and a
large exchange rate giving a mobility of μa (e) = 1.58⋅10−2 cm2/V⋅s. Despite the proximity
of DPP moieties in the close-knitted donor structure the backbones in the closest exchanging pair cross almost orthogonally, resulting in a limited overlap of orbitals and low transport efficiency across the bc-plane with mobilities μb (h) = 1.28⋅10−7
cm2/V⋅s and μc (h) = 8.18⋅10−8 cm2/V⋅s, several orders of magnitude lower than the
electronic one.
The crystal motif of polymorph 2 reaches the best compromise between the packings of the two moieties and shows very large transport efficiencies for both carriers (μa(e)=2.16 ⋅ 10−1
cm2/V⋅s and μc (h)=2.84 ⋅10−1 cm2/V⋅s). Optimal transport occurs in orthogonal directions for
holes and electrons, respectively along c and a. This means that electrons move preferentially along the fullerene channels that are formed along a, while holes move preferentially within the plane perpendicularly to the direction of the herringbone structures (see Figure 5).
Polymorph 3 offers an example of another limiting case. Fullerenes are optimally packed and can transport electrons efficiently reaching very large mobilities compared to the standard of organic photovoltaics materials, reaching a value of 1.34 cm2/V⋅s in the a direction, while in the other two directions transport is absolutely negligible . Donor moieties on the other hand have reduced orbital overlap, with the largest value of the transfer integral being Jij
= 4.64⋅10−3
eV and largest hole mobilities in the order of 10−6 cm2/V⋅s.
Polymorph 4 structure shows low transport efficiencies for both carriers. While also in this case the stacking along a gives rise to channels of fullerenes for electron transport, the larger distance compared to polymorph 1 and the configuration of the pairs with the closest approach translate in a smaller electron mobility, μa(e) =
1.58⋅10−5
cm2/V⋅s along the direction of the fullerene stacking that is negligible in the other orthogonal directions. DPP pairs in closest proximity have a shortest distance of 3.5 Å which is comparable to the one found in polymorph 0 but the particular configuration of the exchanging pair makes the orbital overlap sensibly smaller (Jij
=3.56⋅10−4
eV) and results in very small mobilities along all directions, the largest being μc (h) = 6.88⋅10
−11
cm2/V⋅s
The comparison we carried out offers an overview of the strongly different behaviours that polymorphs of a same molecule can have and more in general of the fundamental role played by supramolecular effects. Nonetheless, the main goal remains to prove the effectiveness of computational approaches in complementing and interpreting experimental data. From our analysis DPP-Ful reveals a dominant crystalline form that is thermodynamically favoured over the others. This is consistent with data reported in reference 22, suggesting the presence of “long-range ordering” in active layers made with this material.
Our results suggest that the computational approach we implemented is able to reproduce the main microscopic features of the crystal form observed experimentally providing a direct microscopic picture of the molecular packing responsible for the large reported efficiency of DPP-Ful-based devices.
3.2 Amorphous Phase
Having discussed crystal polymorphs, let us consider the amorphous phase, which remains interesting for two main reasons. Firstly, amorphous regions may coexist with more ordered ones and contribute to the overall performance of the device. Secondly, completely amorphous films are generally more convenient to produce compared to polycrystalline or crystalline ones66,70. The opposite is true in simulations, where the amorphous phase is more challenging to model compared to the ordered one. In fact, the absence of long-range order, makes it formally inaccurate to apply PBC. This requires the production of a significant number of configurations with relatively large amounts of molecules in order to have reliable estimates of the structural effects affecting the mobility (no border effects). Following the approach detailed in the computational details we produced configurations of 500 molecules in cells whose linear size is 106 Å. For all sufficiently close molecular pairs we computed the transfer integrals and the associated rates. This step of the calculation is more expensive in this case. Generally, in ordered phases periodicity limits the number of possible pair configurations and only a small set of rates repeated over the lattice is sufficient. Mobility estimates are computed for every configuration extracted. Disordered systems are to a good extent isotropic, so different directions of the applied electric field can be considered in order to collect some statistics on mobility, results are reported in Table 2. Amorphous mobilities show a marked difference between the two charge carriers.
The reason can be deduced from the analysis of the pair distribution functions g(r) for acceptor and donor moieties. The pair distribution function is defined according to the following:
Where N(r) is the number of molecules with distance r from a given molecule and the denominator is the linear density of an ideal gas with the same total density ρI of the cell. The
features of the distribution signal deviations with respect to the ideal gas behaviour and reveal how the geometry and chemical character of the material may introduce some degree of order at supramolecular scale. We calculated g(r) for both moieties separately.
For each moiety we chose a point from which to compute distances according to their respective characteristics. Fullerene moieties are symmetric enough for the centroid to be a sensible choice while diketopyrrolopyrrole moieties have a more complex geometry. For this reason, we considered as relevant point the centre of charge of the donor moiety in its positively charged state. The pair distribution functions for both moieties are reported in the plot in Figure 8.
By complementing the discussion of Figure 8 with data for the amorphous phase reported in Table 2 can give us useful insight in the behaviour of DPP-Ful in disordered morphologies. The first peak at around 7.5 Å in Figure 8 is an indication that fullerenes tend to attract each other and form small clusters with a rather efficient packing and high exchange rates reaching 1011 s-1. The depletion zone following this first peak is a sign that the donor moieties attached to the fullerenes have more hindrance and require a certain volume around the cluster to be accommodated. This more or less regular alternation of clusters and regions occupied by donor moieties causes the amorphous material to have a poor performance in terms of electron transport as electrons are very unlikely to be able to move across neighbouring clusters and hence remain trapped.
The difficulty of donor moieties in assembling due to the hindrance of aliphatic chains and their linear geometry results instead in a more favourable morphology for hole transport. The structure is sufficiently intertwined and homogeneous to achieve non-negligible transport performances. Electronic couplings between neighbouring molecules have an average magnitude of 10-3 eV (see Table 2) and the homogeneous structures allows an average transport rate of 1011 s-1. These structural features are reflected in the values of mobilities. Thanks to conformational disorder, holes can achieve mobilities of μ(h)=2.29⋅10-6 cm2/V⋅s on
and 4 (see Table 2). Conversely, due to the relative ease with which fullerene moieties are able to assemble, clusters severely affect the electron transport efficiency of the amorphous medium with a very low value of μ(e)=2.32⋅10−9 cm2/V⋅s, not comparable with any of the
crystal polymorphs studied exhibiting, on average, far larger mobilities for electrons.
4. Conclusions
Our computational study has allowed us to appropriately characterise, at the molecular level, the packing that is responsible for the transport properties of DPP-Ful active layers reported in ref. 22. The comparison of transport efficiencies for both carriers in the different polymorphs extracted enabled us to point out some general features that might be well applicable to other dyads and hence prove useful in directing future experimental work.
Linking aliphatic chains to donor moieties has been shown to improve solubility and processability of the material71 but it also makes more difficult for DPP moieties to achieve efficient packing. Despite the possibility for these chains to promote order at ambient temperature due to steric hindrance, our analysis of the predicted structures suggests that they introduce stringent constraints to the packing of donor moieties, considerably hindering the π-π stacking and hence transfer efficiencies between molecules. At the same time, their presence on the diketopyrrolopyrrole unit seems to be promoting the interaction with the fullerene moiety so as to enhance the relative insulation of donor and acceptor moieties by diminishing the π conjugation of the linker. Clearly, these effects might be overestimated by the method used to predict crystal structures. However, even assuming a substantial geometric change of aliphatic chains in the experimental crystal structure their effect could remain important. Indeed, a modulation of their length may very well lead to dyads with better stacking properties of donor moieties while maintaining good stability of the crystal structure at ambient temperature.
In addition, we found that in general fullerene moieties and donor moieties do compete for optimal packing. This means that not all stable polymorphs have the same efficiency in transporting holes or electrons unless an equilibrium is reached against these mechanisms. This fact results crucially in a highly directional carrier transport that is not, in general, a desirable feature for polycrystalline thin-films, with many randomly oriented ordered regions coexisting between which carriers should be able to cross as swiftly as possible. Moreover, holes and electrons in packings exhibiting high transport efficiencies tend to be optimally conducted in directions orthogonal to each other. This is possibly due to the fact that
fullerenes are rather large molecules and, when stacked in columns determine orthogonal planes with considerable spacing between them.
Competing clustering of fullerene and donor moieties emerges also in the amorphous phase. An indication that the typical hurdles linked to fullerene-derivatives in bulk heterojunction blends are also transferred to molecular heterojunctions.
A number of these features can be extrapolated to a substantial degree to the larger class of organic photovoltaics dyads. This demonstrates the validity of our computational approach that is able to estimate molecular level properties and crucially to complement it with information about packing at the supramolecular level. The latter task remains one of the most delicate steps, in an ideal implementation an appropriate description including disorder and effects of morphologies at higher scales, would allow the simulation of more realistic structures fundamental in providing more accurate predictions of photovoltaic performance.
Acknowledgements
We thank Dr Frédéric Labat for his help with the CRYSTAL code. We The authors acknowledge the use of computational resources at Tr s rand entre de alcul (TGCC) provided by GENCI through project A0050810135 and the CINECA computing facility for the availability of high-performance computing resources and support.
This work has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme ( o STRI ES; grant agreement No 648558)
References
1) N. R. E. L. (NREL), Best Research-Cell Efficiency Chart, 2019.
2) .- . r das, D. Beljonne, V. Coropceanu and J. Cornil, Chem. Rev., 2004, 104, 4971– 5004.
3) . ippelen and .- . r das, Energy Environ. Sci., 2009, 2, 251–261.
4) V. Coropceanu, X. -K. Chen, T. Wang, Z. Zheng and J. -L. r das, Nat. Rev. Mater., 2019, 1–19.
5) A. Wadsworth, M. Moser, A. Marks, M. S. Little, N. Gasparini, C. J. Brabec, D. Baran and I. McCulloch, Chem. Soc. Rev., 2019, 48, 1596–1625.
6) H. Li, T. Earmme, G. Ren, A. Saeki, S. Yoshikawa, N. M. Murari, S. Subramaniyan, M. J. Crane, S. Seki and S. A. Jenekhe, J. Am. Chem. Soc., 2014, 136, 14589– 14597.
7) B. M. Wong and J. G. Cordaro, J. Phys. Chem. C, 2011, 115, 18333–18341.
8) N. M. O’ oyle, . M. ampbell and . R. Hutchison, J. Phys. Chem. C, 2011, 115, 16200–16210.
9) I. Y. Kanal, S. G. Owens, J. S. Bechtel and G. R. Hutchison, J. Phys. Chem. Lett., 2013, 4, 1613–1623.
10) . N. Sokolov, S. tahan-Evrenk, R. Mondal, H. . kker- man, R. S. S nchez-Carrera, S. Granados-Focil, J. Schrier, S. C. Mannsfeld, A. P. Zoombelt, Z. Bao et al., Nat. Commun., 2011, 2, 437.
11) . Hachmann, R. Olivares- maya, S. tahan-Evrenk, . mador- edolla, R. S. S nchez-Carrera, A. Gold-Parker, L. Vogt, A. M. Brockway and A. Aspuru-Guzik, J. Phys. Chem.
Lett., 2011, 2, 2241–2251.
12) J. Xi, M. Long, L. Tang, D. Wang and Z. Shuai, Nanoscale, 2012, 4, 4348–4369.
13) P. Raccuglia, K. C. Elbert, P. D. Adler, C. Falk, M. B. Wenny, A. Mollo, M. Zeller, S. A. Friedler, J. Schrier and A. J. Norquist, Nature, 2016, 533, 73–76.
14) A. Jain, Y. Shin and K. A. Persson, Nat. Rev. Mater., 2016, 1, 1–13.
15) D. P. Tabor, L. M. Roch, S. K. Saikin, C. Kreisbeck, D. Sheberla, J. H. Montoya, S. Dwaraknath, M. Aykol, C. Ortiz, H. Tribukait et al., Nat. Rev. Mater., 2018, 3, 5–20.
16) J. Roncali and I. Grosu, Adv. Sci., 2019, 6, 1801026.
17) P. Peumans, S. Uchida and S. R. Forrest, Materials for Sustainable Energy: A Collection
of Peer-Reviewed Research and Review Articles from Nature Publishing Group, World
Scientific, 2011, pp. 94–98.
18) G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789– 1791.
19) J. Halls, C. Walsh, N. C. Greenham, E. Marseglia, R. H. Friend, S. Moratti and A. Holmes, Nature, 1995, 376, 498–500.
20) P. Cheng and X. Zhan, Chem. Soc. Rev., 2016, 45, 2544–2582.
21) N. i, . D. Perea, T. assar, M. Richter, T. Heumueller, . . Matt, . Hou, N. S. ldal, H. Chen, S. Chen et al., Nat. Commun., 2017, 8, 1–9.
22) K. Narayanaswamy, A. Venkateswararao, P. Nagarjuna, S. Bishnoi, V. Gupta, S. Chand and S. P. Singh, Angew. Chem., Int. Ed. Engl., 2016, 55, 12334–12337.
23) M. Marinelli, M. Lanzi, A. Zanelli, M. Zangoli, F. Di Maria, E. Salatelli et al., J. Mater.
Chem. C, 2020.
24) T. L. Nguyen, T. H. Lee, B. Gautam, S. Y. Park, K. Gundogdu, J. Y. Kim and H. Y. Woo,
Adv. Funct. Mater., 2017, 27, 1702474.
25) T. Nishizawa, H. K. Lim, K. Tajima and K. Hashimoto, Chem. Commun., 2009, 2469– 2471.
26) S. Izawa, K. Hashimoto and K. Tajima, Phys. Chem. Chem. Phys., 2012, 14, 16138– 16142.
27) J. H. Lee, C. G. Park, A. Kim, H. J. Kim, Y. Kim, S. Park, M. J. Cho and D. H. Choi, ACS
Appl. Mater. Interfaces, 2018, 10, 18974–18983.
28) G. Feng, J. Li, F. J. Colberts, M. Li, J. Zhang, F. Yang, Y. Jin, F. Zhang, R. A. Janssen, C. Li et al., J. Am. Chem. Soc., 2017, 139, 18647–18656.
29) F. Pierini, M. anzi, P. Nakielski, S. Pawłowska, O. Urbanek, . Zembrzycki and T. . Kowalewski, Macromolecules, 2017, 50, 4972–4981.
30) A. Labrunie, J. Gorenflot, M. Babics, O. Aleveque, S. Dabos- Seignon, A. H. Balawi, Z. Kan, M. Wohlfahrt, E. Levillain, P. Hudhomme et al., Chem. Mater., 2018, 30, 3474– 3485. 31) A. P. Diac, L. Szolga, C. Cabanetos, A. Bogdan, A. Terec, I. Grosu and J. Roncali, Dyes
Pigm., 2019, 171, 107748.
32) S. ucas, T. eydecker, P. Samor , E. Mena-Osteritz and P. uerle, Chem. Commun., 2019, 55, 14202– 14205.
33) H. Oberhofer, K. Reuter and J. Blumberger, Chem. Rev., 2017, 117, 10319–10357.
34) B. Baumeier, J. Kirkpatrick and D. Andrienko, Phys. Chem. Chem. Phys., 2010, 12, 11103–11113.
35) M. C. Zerner, Rev. Comput. Chem.,1991,2,313– 365.
3 ) N. Mart n, . S nchez, . Illescas and I. P rez, Chem. Rev., 1998, 98, 2527–2548. 37) J. Roncali, Adv. Eng. Mater., 2011, 1, 147–160.
39) . . Segura, N. Mart n and D. M. Guldi, Chem. Soc. Rev., 2005, 34, 31–47.
40) V. Coropceanu, J. Cornil, D. A. da Silva Filho, Y. Olivier, R. Silbey and J.- . r das,
Chem. Rev., 2007, 107, 926–952.
41) .- . r das, J. P. Calbert, D. da Silva Filho and J. Cornil, Proc. Natl. Acad. Sci. U.S.A., 2002, 99, 5804–5809.
42) R. A. Marcus, Rev. Mod. Phys., 1993, 65, 599. 43) G. J. Beran, Chem. Rev., 2016, 116, 5567–5613.
44) J. G. Brandenburg and S. Grimme, Acta Crystallogr., Sect. B, 2016, 72, 502–513.
45) S. L. Price and J. G. Brandenburg, Non-covalent Interactions in Quantum Chemistry and
Physics: Theory and Applications, Elsevier, 2017, pp. 333–336.
46) R. L. Akkermans, N. A. Spenley and S. H. Robertson, Mol. Simul., 2013, 39, 1153–1164. 47) J. Hoja, H.-Y. Ko, M. A. Neumann, R. Car, R. A. DiStasio and A. Tkatchenko, Sci. Adv., 2019, 5.
48) A. Becke, J. Chem. Phys., 1993, 98, 648–5.
49) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785. 50) S. Grimme, R. Huenerbein and S. Ehrlich, ChemPhysChem, 2011, 12, 1258–1261.
51) M. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. Petersson, 2009.
52) D. Alberga, I. Ciofini, G. F. Mangiatordi, A. Pedone, G. Lattanzi, J. Roncali and C. Adamo, Chem. Mater., 2017, 29, 673–681.
53) BIOVIA, Materials Studio, 2020, v. 2018, San Diego, Dassault Syst mes.
54) Mayo, S. L.; Olafson, B. D.; Goddard, W. A., J. Phys. Chem. 1990, 94, 8897-8909.
55) A. D. Mighell, V. L. Himes and J. Rodgers, Acta Crystallogr., Sect. A, 1983, 39, 737–740. 56) R. Dovesi, V. Saunders, C. Roetti, R. Orlando, C. Zicovich-Wilson, F. Pascale, B. Civalleri, K. Doll, N. Harrison, I. Bush et al., 2017.
57) J. C. Phillips, R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kale and K. Schulten, J. Comput. Chem., 2005, 26, 1781–1802.
58) K. Vanommeslaeghe, E. Hatcher, C. Acharya, S. Kundu, S. Zhong, J. Shim, E. Darian, O. Guvench, P. Lopes, I. Vorobyov et al., J. Comput. Chem., 2010, 31, 671–690.
59) W. Yu, X. He, K. Vanommeslaeghe and A. D. MacKerell Jr, J. Comput. Chem., 2012, 33, 2451–2468.
60) D. Alberga, G.F. Mangiatordi, F. Labat, I. Ciofini, O. Nicolotti, G. Lattanzi, C. Adamo J.
61) Y. Jiang, C. Cabanetos, S. Jungsuttiwong, D. Alberga, C. Adamo, J. Roncali Chem. Sel. 2017, 2, 6296.
62) . Sancho- arc a, Chem, Phys., 2007, 331, 321.
63) J. Kirkpatrick, Int. J. Quantum Chem., 2008, 108, 51–56.
64) M. V. Makarova, S. G. Semenov and O. A. Guskova, Int. J. Quantum Chem., 2016, 116, 1459–1466.
65) F. Günther, S. Gemming and G. Seifert, J. Phys. Chem. C, 2016,120, 9581–9587.
66) F. Gajdos, H. Oberhofer, M. Dupuis and J. Blumberger, J. Phys. Chem. Lett., 2013, 4, 1012–1017.
67) J. Rivnay, S. C. Mannsfeld, C. E. Miller, A. Salleo and M. F. Toney, Chem. Rev., 2012,
112, 5488–5519.
68) M. Cutini, B. Civalleri, M. Corno, R. Orlando, J. G. Branden burg, L. Maschio and P. Ugliengo, J. Chem. Theory Comput., 2016, 12, 3340–3352.
69) K. Hongo, M. A. Watson, R. S. Sanchez-Carrera, T. Iitaka and A. Aspuru-Guzik, J. Phys.
Chem. Lett., 2010, 1, 1789–1794.
70) M. Baldo, Z. Soos and S. Forrest, Chem. Phys. Lett., 2001, 347, 297–303. 71) I. Meager, R. S. Ashraf, S. Mollinger, B. C. Schroeder, H. Bronstein, D. Beatrup, M. S. Vezie, T. Kirchartz, A. Salleo, J. Nelson et al., J. Am. Chem. Soc., 2013, 135, 11537–11540.
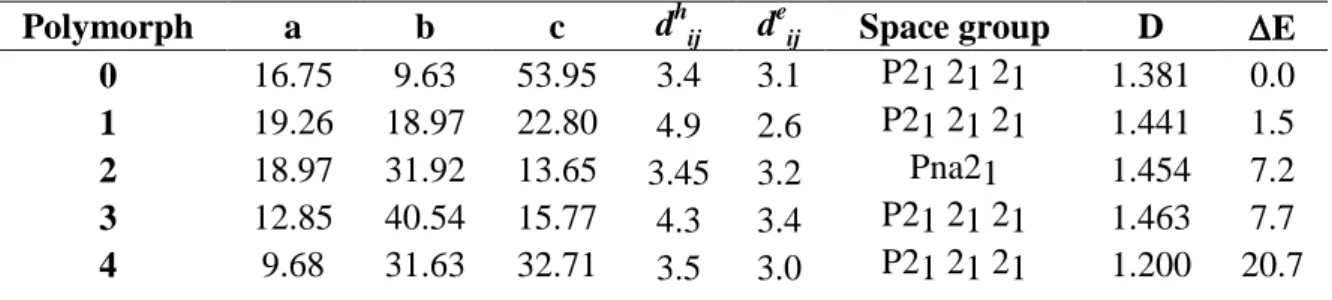
Table 1. Computed cell parameters (a,b, c, Å ), shortest distance between donor (dhab, Å) and acceptor groups (deab, Å), densities (D, g/cm3) and relative stabilities (E, kcal/mol) for the five different crystalline polymorphs. All reported values refer to polymorphs fully optimized at the B3LYP-D3 level.
Polymorph a b c dhij deij Space group D E
0 16.75 9.63 53.95 3.4 3.1 P21 21 21 1.381 0.0
1 19.26 18.97 22.80 4.9 2.6 P21 21 21 1.441 1.5
2 18.97 31.92 13.65 3.45 3.2 Pna21 1.454 7.2
3 12.85 40.54 15.77 4.3 3.4 P21 21 21 1.463 7.7
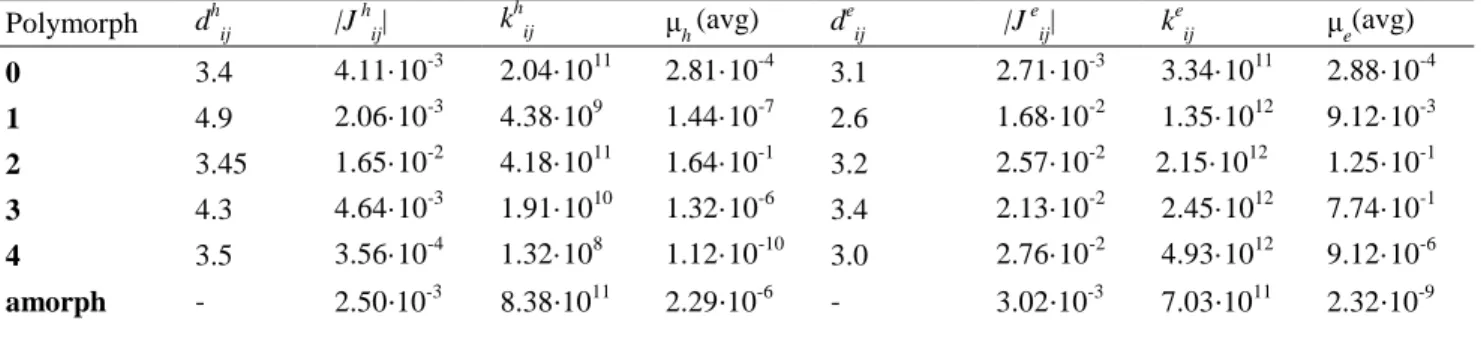
Table 2. Data on hole transport and electron transport dij in Å, Jij in eV, kij in s-1, μ in cm 2
/V⋅s. All reported values are obtained using B3LYP-D3 optimized structures.
Polymorph dhij |J hij| khij μh(avg) deij |J eij| keij μe(avg)
0 3.4 4.11·10-3 2.04·1011 2.81·10-4 3.1 2.71·10-3 3.34·1011 2.88·10-4 1 4.9 2.06·10-3 4.38·109 1.44·10-7 2.6 1.68·10-2 1.35·1012 9.12·10-3 2 3.45 1.65·10-2 4.18·1011 1.64·10-1 3.2 2.57·10-2 2.15·1012 1.25·10-1 3 4.3 4.64·10-3 1.91·1010 1.32·10-6 3.4 2.13·10-2 2.45·1012 7.74·10-1 4 3.5 3.56·10-4 1.32·108 1.12·10-10 3.0 2.76·10-2 4.93·1012 9.12·10-6 amorph - 2.50⋅10-3 8.38⋅1011 2.29⋅10-6 - 3.02⋅10-3 7.03⋅1011 2.32⋅10-9
Table 3. Electron and hole mobilities along all crystal axes a,b and c. All reported values are
in cm2/V⋅s and are obtained using the B3LYP-D3 optimized structures.
Polymorph μa(e) μb(e) μc(e) μa(h) μb(h) μc(h)
0 1.00⋅10-6 4.99⋅10-4 1.17⋅10-11 4.86⋅10-4 4.80⋅10-11 2.84⋅10-7
1 1.58⋅10-2 7.56⋅10-11 1.46⋅10-10 1.98⋅10-7 1.28⋅10-7 8.18⋅10-8
2 2.16⋅10-1 2.15⋅10-11 1.11⋅10-4 3.80⋅10-7 7.04⋅10-10 2.84⋅10-1
3 1.34⋅100 8.90⋅10-13 1.64⋅10-9 8.47⋅10-10 9.09⋅10-10 2.28⋅10-6
Figure captions
Figure 1. Chemical structure of DPP-Ful with [60]-fulleropyrrolidine (Ful) acceptor moiety
in blue and the diathiafulvalene-functionalised diketopyrrolopyrrole (DPP) donor moiety in red.
Figure 2. Isosurface (values +/-0.005) representation of frontier orbitals. (left: HOMO; right :
LUMO).
Figure 3. Structural features of the most stable polymorph (0) obtained after a full DFT
optimization: the shortest distance between fullerene pairs and the distribution of acceptor moieties (top), the shortest distance between DPP pair and a view of the herringbone structures in the ac-plane (bottom).
Figure 4. Structural features of the polymorph 1 obtained after a full DFT optimization: the
shortest distance between DPP pairs moieties with the largest orbital overlap for the structure (left) and a view of the crystal motif along the bc-plane (right).
Figure 5. Structural features of the polymorph 2 obtained after a full DFT optimization: the
shortest distance between for a DPP pair (left) and a view of the bc-plane (right).
Figure 6. Structural features of the polymorph 3 obtained after a full DFT optimization: the
shortest distance between for a DPP pair (left) and a view of the bc-plane (right).
Figure 7. Structural features of the polymorph 4 obtained after a full DFT optimization: the
shortest distance between for a DPP pair (left) and a view of the bc-plane (right).
Figure 8. Radial Pair distribution functions for the donor (red) and acceptor (blue) moieties of
Figure 3 a c b a c b 3.4 Å 3.1 Å
Figure 5
3.4 Å
b
a c
Figure 7
3.45 Å
b
c a