INFORMATION TO USERS
This manuscript has been reproduced from the microfilm master. UMI films the text directly from the original or copy submitted. Thus, some thesis and dissertation copies are in typewriter face, while others may be from any type of computer printer.
The quality of this reproduction is dependent upon the quality of the copy submitted. Broken or indistinct print, colored or poor quality illustrations and photographs, print bleedthrough, substandard margins, and improper alignment can adversely affect reproduction.
ln the unlikely event that the author did not send UMI a complete manuscript and there are missing pages, these will be noted. Also, if unauthorized copyright material had to be removed, a note will indicate the deletion.
Oversize materials (e.g., maps, drawings, charts) are reproduced by sectioning the original, beginning at the upper left-hand corner and continuing from left to right in equal sections with small overtaps.
Photographs included in the original manuscript have been reproduced xerographically in this copy. Higher quality 6n x gn black and white photographie prints are available for any photographs or illustrations appearing in this copy for an additional charge. Contact UMI directly to order.
Bell & Howell Information and Leaming
300 North Zeeb Road, Ann Arbor, Ml 48106-1346 USA 800-521-0600
NOTE TO USERS
Page(s) missing in number only; text follows. Page(s) were
microfilmed as received.
107
Université de Sherbrooke
Mécanisme de déclenchement de l'apoptose par des lésions à l'ADN : protection par l'induction de l'expression de p2 l Wafl/Cipl.
Par Nathalie Bissonnette
Département de médecine nucléaire et de radiobiologie
Thèse présentée à la faculté de médecine en vue de l'obtention du grade de Philosophiae Doctor (Ph.D.) en radiobiologie
l+I
National Libraryof Canada Bibliothèque nationale du Canada Acquisitions and
Bibliographie Services Acquisitions et services bibliographiques
395 Wellington Street Ottawa ON K1 A ON4 Canada
395, rue Wellington Ottawa ON K1A ON4 Canada
The author has granted a
non-exclusive licence allowing the
National Library of Canada to
reproduce, loan, distribute or sell
copies of this thesis
inmicrofonn,
paper or electronic formats.
The author retains ownership of the
copyright
inthis thesis. Neither the
thesis nor substantial extracts from it
may be printed or otherwise
reproduced without the author' s
pernnss10n.
Our 6le Notre référence
L, auteur a accordé une licence non
exclusive permettant à la
Bibliothèque nationale du Canada de
reproduire, prêter, distribuer ou
vendre des copies de cette thèse sous
la forme de microfiche/film, de
reproduction sur papier ou sur format
électronique.
L, auteur conserve la propriété du
droit d'auteur qui protège cette thèse.
Ni la thèse
nides extraits substantiels
de celle-ci ne doivent être imprimés
ou autrement reproduits sans son
autorisation.
0-612-56989-6
À qui de droit,
Par la présente, nous autorisons Nathalie Bissonnette à inclure dans sa thèse l'article publié dans la revue EA1BO Journal ( 1996; 15: 1615-1624) et intitulé: .. Functional interactions between p53 and the TFIIH complex are affected by tumour-associated mutations".
Thierry Léveillard Ladislav Andera, PhD
c:r;;Jent
ScVa~,PhD
... )4' -Laurent Bracco, PhD / Bohdan Wasylyk,PhD-À qui de droit,
Par la présente. nous autorisons Nathalie Bissonnette à inclure dans sa thèse l'article publié dans la revue Carcinogenesis ( 1998; 19: 1545-1552) et intitulé: "Induction by estrogens of
methotrexate resistance in MCF-7 breast cancer cells".
~-
- \.
>==---Jau1 A. Thibodeau
C'·
/Suzanne Kocsis Bédard
"= ,/ -{ ( ' 1 <17'>
< ~ Darel Hunting, PhD
À qui de droit,
Par la présente, j"autorise ~athalie Bissonnette à inclure dans sa thèse l'article publié dan" la
revue Oncogene (1998; 16:3461-3469) et intitulé: ··p21-induced cycle arrest in Gl protects cells from apoptosis induced by UV-irradiation or RNA polymerase II blockage".
' -=::...,j
À qui de droit,
Par la présente, nous autorisons Nathalie Bissonnette à inclure dans sa thèse !"article publié dans la revue Biochem. Cel!. Bio!. (1997; 75:351-358) et intitulé: "The apoptotic and transcriptional transactivation activities of p53 can be dissociated''.
Je vis! Viennent la pluie et le temps! Insensible, Portant ma destinée et sachant mon délai, Je marchais en riant sous le pays horrible Des astres que traverse une route de lait. Séparons-nous ici, vous êtes arrivé.
Voici le lieu, voici l'arbre, voici la porte.
Compagnon, nous avons marché de bonne sorte, Coursant ainsi, depuis que le jours 'est levé. Eh bien, c'est là le toit qui.fut par vous trouvé. Dites: C'est moi! Criez, heurtez !'huis à mainforte! La femme et le repas vous attendent. Qu 'importe Le compagnon d'un jour ? Vous êtes arrivé. La veille mère est là; l'épouse douce et sage Est là ; prenez-les dans vos bras. Le reste est vain. L'enfant nouveau vous met les mains sur le visage. Adieu. Riez, soyez heureux jusqu'à la fin,
Mangez de votre pain, buvez à votre verre,
Pour moi un long chemin encore me reste à faire. Paul Claudel, 1891
Table des matières
Liste des abréviations···-··· ... V Résumé ... ···-... VII Chapitre I - Introduction
1- Généralités . . . . .. . . ... .. ... . .. .. . . ... ... . . ... . . .... ... .. .. . . .. . ... . . ... .. .. .. .. . . . .. . .. . .. ... . . .. . . .. . . .. . . 1
1.1 Protéine p53 ···-··· ... 2
1.2 Apoptose indépendante de p53 ···-··· 6
1.3 L'activation des caspases est essentielle à l'apoptose ... 6
1.4 Famille de protéines Bcl-2 ···-··· 7
2- Projets de recherche ···-··· 11
3- Observations reliées aux articles présentés··-··· 13
Chapitre II - Premier article The apoptotic and transcriptional transactivation. activities of p53 can be dissociated ... _ .. . . . .. .. . . .. ... . . .. . . .. . . .. . .. . . .. . . 15
Chapitre ill -2ième article DNA lesions repaired by the nucleotide excisioru. repair pathway trigger apoptosis through a blockage of RNA polymerase II ... 24
Chapitre IV - 3ième article p2 l-induced cycle arrest in G1 protects cells frorn apoptosis induced by UV-irradiation or RNA polymerase II blockage ... 58
Chapitre V - 4ième article Functional interactions between p53 and the TFilH complex are affected by tumour-associated mutations ... 68
Chapitre VI - 5ième article Induction by estrogens ofmethotrexate resistance in the MCF-7 breast cancer cells ···-··· 79
Chapitre VII - Discussions 1- Rôle de p53 dans l'apoptose···-···~·-··· 108
2- Apoptose faisant intervenir une signalisation membranaire ... 110
3- Induction de l'apoptose par des dommages à l'ADN inhibant la transcription et non par les dommages ox:ydatifs créés par les UVB, les UVC et le 4-nitroquinoline N-oxide ... 112
4- Induction de l'apoptose par des radiations ionisantes selon le type cellulaire ... 114
5- La céramide, un messager prioritaire de l'apoptose ... 114
6- Facteurs de croissance ou mitogéniques ... 116
7- p21 Wafl/Cipl : un agent régulant l'horloge apoptotique ... 118
8- Protéines Bcl-2 et Bax ... 120
9- Clivage de p53, pourquoi pas! ... ~ ... 121
10- Stabilisation de la protéine p53, la boîte noire ou la bête noire ... 126
11- Apigénine : faites vos jeux! ... 128
12- Induction de l'apoptose et clivage de p53 par les inhibiteurs des PPl et PP2A ... 133
13- Induction de l'apoptose par des inhibiteurs des topoisomérases d'ADN ... 134
14- Réparation de l'ADN et p53 ... 135
15- TFIIH : une pièce du puzzle reliée à p53 ... 136
Chapitre VIlI - Conclusions ... 142
Remerciements ... 143
Références ... 144
Annexe I - Induction de l'apoptose dans les cellules MCF-7 en fonction de la dose d'UVB et de 4-nitroquinoline N-oxide ... 156
Annexe II - Protection de l'apoptose par la cycloheximide et la mimosine dans les cellules MCF-7 ... 157
Annexe fil - Effet du sérum sur la cinétique d'induction de l'apoptose dans les cellules MCF-7 ... 158
Annexe IV - Absence de protection par la mimosine de l'apoptose induite par l'apigénine. Induction de l'apoptose par la cisplatine, la mitomycine C et l'inhibiteur de kinase wortmannin ... 159
Annexe V - Distribution des cellules A431 dans les différentes phases du cycle cellulaire après un relâchement des cellules de la phase G1/S en fonction du temps ... 160 Annexe VI -Effet de l'inhibition de l'apoptose par une préincubation à la mimosine
pour une période s'étendant jusqu'à 18 heures suivant un relâchement des
cellules A43 l de la phase G1/S ... 161 Annexe VII -Étude comparative de différents traitements sur la distribution des
cellules A43 l dans les différentes phases du cycle cellulaire ... 162 Annexe VIII - Induction de l'expression de la protéine Bax durant l'apoptose
.d. -dl .. 6
m mte par un arret e a transcnpt1on ... 1 3 Annexe IX - Clivage de la protéine p53 endogène des cellules A43 l lors
de l'apoptose ... 164 Annexe X - Clivage de la protéine p53 des cellules A43 l en apoptose induite
par différents agents ... 165 Annexe XI - Clivage in vitro de la protéine p53 par la caspase-3/CPP32 ... 166 Annexe XII - Hypothèse impliquant l'effet de l'induction de l'apoptose par
un blocage de la transcription ... 167 Annexe XIII - Relation entre le facteur de transcription TFIIH
et la protéine p53 ... 168 Annexe XIV - Effet de l'a.-amanitine dans les cellules A43 l ... 169 Annexe XV - Effet de la présence de la protéine p53 sur l'apoptose induite
Abréviations
AP0-1-2 -3: protéine de l'apoptose -1 -2 -3
ARN pol II : ARN polymérase II
Atm : protéine du gène ATM, dont la mutation est caractéristique du syndrome Ataxia telangiectasia
BER : réparation par excision de bases
CAD : désoxyribonucléase activée par une caspase (3)
CAK : kinase activant les cdks composée de cdk7, cycline H et MAT 1 CDK: kinase dépendante des cyclines
CED-3 : protéine homologue à ICE du nématode Caenorhabditis elegans du gène #3 "death" CPP32 : protéase à cystéine de 32 kDa
CRADD : caspase et adapteur RIP avec un "domaine de mort" ("death domain") DNA-PK: protéine kinase DNA-dépendante
DRB : 5 ,6-dichloro-l-f3-D-ribofuranosylbenzimidazole
EGTA: acide éthylène glycol-bis(f3-aminoéthyle éther)-N,N,N',N-tétraacétique EDTA : acide éthylène di-amine tétraacétique
FADD/MORTl : protéine associée à Fas avec un "death domain" (domaine de mort) HU : hydroxyurée
ICE : enzyme convertissant l'interleukine 1-j)
IKB-a. : protéine régulatrice a. de facteurs de transcription de la famille NF-KB
JNK : kinase du facteur jun
MAPKK : kinase phosphorylant la protéine-kinase activée par des facteurs mitogéniques NER : mécanisme de réparation par excision de nucléotides
NF-KB : facteur (de transcription) nucléaire K-B
4-NQO : 4-nitroquinoline N-oxide
p21/W AFl/CIPl : protéine de 21 kDa/ fragment 1 activé de la protéine p53 sauvage/ protéine inhibitrice 1 des CDKs
PARP : polymérase de poly(ADP-ribose)
RAIDD : protéine homologue à ICE et à CED-3 associée à RIP avec un "death domain"
pRb : protéine du gène du rétinoblastome
RIP : protéine interagissant avec le récepteur SAPK : protéine kinase activée par un stress
IFllH : facteur H de transcription par l'ARN polymérase II TNF : facteur de nécrose tumorale
TRADD: protéine associée au "death domain" (domaine de mort) de TNFR-1
Ul-70 kDa: protéine nucléaire de 70 kDa comprise dans la petite ribonucléoprotéine Ul
UV : ultraviolet
XP -A -B ou -C : xéroderma pigmentosum du groupe de complémentation -A -B ou -C Lignées cellulaires
A43 l : lignée de kératinocytes cancéreux de la wlve humaine HeLa : carcinome utérin humain
HL60 : leucémie promyélocytique humaine
Jurkat : leucémie lymphoblastique-T aigüe humaine
MCF-7: lignée de cellules épithéliales cancéreuses du sein humain
K562 : lignée de cellules hématopoïétiques positive pour la protéine Bcr-Abl U93 7 : lymphome histocytique humain
Inhibiteurs liant irréversiblement le site actif des protéases à cystéines Ccaspases)
'Zr V AD-fmk : N-benzyloxycarbonyl-V al-Ala-Asp-fluorométhylcétone, un inhibiteur universel des
caspases (l'apoptose induite par la céramide qui n'est pas distinguable de celle induite par
TNF-alpha est bloquée par cet inhibiteur Z-V AD)
'ZrFA-fmk: Z-Phe-Ala-fink, un inhibiteur de cathepsine G
'ZrAAD-cmk : Z-Ala-Ala-Asp-chloromethylketone, un inhibiteur de granzyme B ALLN : N-acetyl-Leu-Leu-norleucinal, un inhibiteur de calpaïne-cathepsine AcDEVD-cho: inhibiteur de la caspase-3/CPP32/Apopaïne
Résumé
Les agents endommageant l'ADN sont connus pour avoir des effets mutagènes. Ils influencent le passage des cellules dans les différentes phases du cycle cellulaire. Les agents alcoylants, les radiations, ainsi qu'un grand nombre d'agents chimiothérapeutiques peuvent provoquer un arrêt transitoire en phase G1 du cycle cellulaire. Cet arrêt est un moyen de défense important pour prévenir ia réplication de l'ADN endommagé. Le suppresseur de tumeur p53 est un médiateur de cet arrêt du cycle cellulaire en phase G1 _ La protéine p53 est stabilisée et activée en réponse aux dommages à l'ADN. Cela amène une augmentation du niveau de la protéine p21 Wafl!Cipl, laquelle est un inhibiteur des kinases dépendantes des cyclines, requises pour que la
cellule puisse entrer en phase de synthèse d'ADN du cycle cellulaire. De nombreuses études ont démontré le rôle essentiel que joue p53, le gardien du génome, dans la réponse cellulaire à
différents types de stress. L'apoptose induite par un stress génotoxique est parfois associée au rôle de la protéine p53 agissant -comme suppresseur de tumeur. En effet, la protéine p53 peut empêcher les cellules ayant subi un stress génotoxique de se dpliquer, soit de façon transitoire, par l'arrêt du cycle en G1, soit de façon définitive, par l'induction de l'apoptose. Cette protéine joue donc un rôle modulateur de la réponse cellulaire aux agents cytotoxiques. Des travaux de recherche ont démontré que l'activité de transactivation transcriptionnelle du p53 est requise pour l'arrêt en G1 après un stress génotoxique, mais n'est pas nécessaire pour l'induction de l'apoptose. Également, il a été démontré que le niveau d'expression de la protéine p53 est déterminant pour l'induction de l'apoptose. Par contre, peu d'informations sont disponibles quant à la nature des dommages induisant l'apoptose.
Utilisant les cellules A43 l et MCF-7, nous avons observé une très bonne corrélation entre l'induction de l'apoptose et l'incapacité de la protéine p53 à transactiver un gène en aval lors de la réponse au stress génotoxique, soit p21/WAFJICIPJ. Nous avons émis l'hypothèse qu'une inhibition de la transcription par l'ARN polymérase II serait l'élément déclencheur de la voie menant à l'apoptose. Nos observations supportent l'hypothèse selon laquelle _l'apoptose faisant intervenir la protéine p53 peut être indépendante de son activité de transactivation
transcriptionnelle. Ces résultats sont publiés dans la revue "Biochemisty and Cellular Biology" (1997, 75 : 351-358) et font l'objet du deuxième chapitre du présent document. Une étude plus élaborée quant à la nature des dommages pouvant induire l'apoptose nous a menée à émettre l'hypothèse suivante : les dommages à l'ADN qui sont de bons inhibiteurs de la transcription par l'ARN polymérase II et qui sont réparés par le mécanisme d'excision de nucléotides/resynthèse sont des précurseurs de l'apoptose. Ces résultats sont présentés au troisième chapitre et seront soumis à la revue "Oncogene". L'étude de la fonction de la protéine p53 comme facteur de transcription nous a révélé que les cellules A43 l comportent une protéine p53 mutée, inapte à la transactivation transcriptionnelle. Ce résultat a été confirmé par la caractérisation génomique d'une mutation en son domaine central (R273H). Les kératinocytes A43-I, comportant une protéine p53 mutée non fonctionnelle comme transactivateur de la transcription, n'induisent pas le gène p21/WAFJICIPI et ne présentent pas d'arrêt dans le cycle cellulaire en phase G1. En utilisant la mimosine comme inducteur de p21 Wafl/Cipl par une voie indépendante du p53, nous
avons démontré que l'arrêt en phase Gi, concomitant à l'expression de la protéine p21 Wafl!Cipl,
protège la cellule de l'apoptose. Ces résultats constituent le quatrième chapitre et sont sous presse dans la revue "Oncogene" (1998). Certaines études utilisant des fibroblastes porteurs d'une mutation homozygote dans le gène TP 5 3 (dérivés de patients atteints du syndrome de Li-Fraumeni) ont démontré que la protéine p53 jouerait un rôle dans le mécanisme de réparation par l'excision de nucléotides. En utilisant un système élaboré à partir d'extraits cellulaires, nous n'avons pu observer d'effet de la protéine p53 sauvage, ni de la protéine mutée, sur le mécanisme de réparation par l'excision de nucléotides/resynthèse in vitro. Une partie de ces résultats, qui ont été publiés dans la revue EMBO (1996, 15 : 1615-1624), est présentée dans le cinquième chapitre. Une autre partie de ces résultats est comprise dans le troisième chapitre.
La corrélation entre l'état sauvage de la protéine p53 et sa capacité à transactiver le gène p21 Wafl/Cipl après un stress génotoxique nous ont permis de développer un test biochimique pouvant déterminer l'état fonctionnel de la protéine p53 dans différents types cellulaires. En effet, l'induction de l'expression de la protéine p21 Wafl!Cipl suivant une irradiation (ionisante ou UV)
protéine p53 sauvage. Avec ce test, nous avons pu caractériser l'état fonctionnel de la protéine p53 contenue dans des clones de cellules épithéliales MCF-7 résistants au méthotrexate produits dans le laboratoire du Dr Benoît Paquette. Une partie de ces résultats constitue le sixième chapitre, a été accepté pour publication dans la revue "Carcinogenesis".
Chapitre 1
Introduction
!-Généralités
L'homéostasie cellulaire est contrôlée par un équilibre entre la prolifération, l'arrêt de croissance, la différentiation et la mort cellulaire programmée, appelée apoptose. L'identification des facteurs de contrôle négatif de la croissance, incluant l'arrêt transitoire lors du cycle cellulaire, ou définif lors de l'apoptose, est nécessaire pour comprendre comment un nombre défini de cellules est maintenu et comment une altération de l'équilibre homéostatique peut contribuer à une évolution maligne.
Le cancer est caractérisé par une prolifération cellulaire incontrôlée, résultant d'une accumulation d'événements mutationnels affectant les glnes mis en cause dans la régulation de la prolifération cellulaire. Plusieurs études ont démontré que les récurseurs cellulaires des oncogènes, les proto-oncogènes, codent pour des protéines qui sont étroitement reliées aux processus de régulation de la prolifération cellulaire et de différentiation. Ces oncoprotéines peuvent être des facteurs de croissance, des récepteurs de facteurs de croissance, des transducteurs de signaux ou des protéines associées à l'ADN qui modulent l'expression des gènes. Il est reconnu que l'activation constitutive des oncogènes contribue directement au processus de transformation maligne. Par contre, la prolifération cellulaire peut être inhibée par une autre classe de produits de gènes, appelés suppresseurs de tumeur. Or, les proto-oncogènes et les suppresseurs de tumeur, qui sont respectivement des régulateurs positifs et négatifs de la prolifération cellulaire, subissent tous deux des mutations dans les cancers humains. La mutation d'un proto-oncogène entraînera l'activation constitutive de la protéine (régulation positive de la croissance cellulaire), tandis que la mutation d'un suppresseur de tumeur annihilera sa capacité à
Les agents endommageant l'ADN sont connus comme ayant des effets mutagènes. Ils influencent le passage des cellules dans les différentes phases du cycle cellulaire. Les agents alcoylants (Plant et Roberts, 1991 ), les radiations (Kastan et al., 1991; Kuerbitz et al., 1992), et un grand nombre d'agents chimiothérapeutiques (Fritsche et al., 1993; Nelson et al., 1994) peuvent induire un arrêt transitoire en phase Gl du cycle cellulaire. Cet arrêt est un moyen de défense important pour prévenir la réplication de l'ADN endommagé (Smith et Fronace, 1996; Levine, 1997). Le suppresseur de tumeur p53 est impliqué dans différents aspects du cycle cellulaire, l'apoptose, le contrôle de l'intégrité génomique et la réparation de l'ADN. La protéine p53, qui est stabilisée et activée en réponse aux dommages à l'ADN, est un médiateur de l'arrêt du cycle cellulaire en phase G1 (Kastan et al., 1991 ). La molécule p53 activée amène une augmentation du niveau de la protéine p21Wafl/Cipl (El-Deiry et al., 1993; Xiong et al., 1993), un inhibiteur des kinases dépendantes des cyclines (Harper et al., 1993) qui sont requises pour que la cellules puisse entrer dans la phase de synthèse de l'ADN du cycle cellulaire.
L'apoptose induite suivant un stress génotoxique est souvent associée au rôle du suppresseur de tumeur p53. De nombreuses études ont démontré le rôle essentiel que joue p53, le gardien du génome, dans la réponse cellulaire à différents types de stress. La protéine p53 peut empêcher les cellules ayant subi un stress génotoxique de se répliquer, soit de façon transitoire, par l'arrêt du cycle en phase Gi, soit définitivement, par l'induction de l'apoptose. Cette protéine joue donc un rôle modulateur de la réponse cellulaire aux agents cytotoxiques.
1.1 Protéine p53
À partir d'une étude épidémiologique rétrospective menée en 1969, deux épidérniologistes, Frédérick Li et Joseph Fraumeni, ont pu caractériser une forme d'agrégation familiale de cancers associant des sarcomes de tissus mous et des carcinomes du sein. Cette maladie fut nommée le syndrome de Li-Fraumeni (Li et Fraumeni, 1969). En 1990, on documentait des mutations dans le gène p53 (TP53) de familles atteintes du syndrome de Li-Fraumeni (Malkin et al., 1990;
Srivastava et al., 1990) dont les tumeurs examinées présentaient la perte de l'allèle sauvage (Srivastava et al., 1992).
Cette perte de la fonction de la protéine p53 par une mutation du gène TP53 est un événement fréquent et important dans la genèse et la progression de plusieurs tumeurs malignes humaines. La perte de la fonction apoptotique de p53 et la perte de son activité comme facteur de transcription semblent être en cause dans plusieurs cas de carcinogenèse, puisque l'apoptose est envisagée comme un mécanisme d'élimination des cellules qui présentent un excès de lésions au génome ou un dérèglement de la croissance, et qui deviendraient des précurseurs de clones de cellules transformées.
La protéine humaine p53 est composée de 393 acides aminés et se subdivise en quatre parties. Le domaine amino-terminal (1-43) est responsable de l'activation de la transcription. Le domaine central (100-300), où réside la grande majorité des mutations ponctuelles, est responsable de la reconnaissance spécifique et de la liaison à l'ADN (Pavletich et al., 1993). Le domaine carbox:y-terminal (300-393) se divise en deux parties, soit le domaine de tétramérisation (320-362) et le domaine de régulation de la capacité de liaison à sa séquence spécifique (363-393). Le domaine pro-apoptotique serait situé dans une région riche en pralines (77-90) (Walker et Levine, 1996; Sakamuro et al., 1997).
Deux activités de la protéine p53 ont été clairement identifiés : son rôle apoptotique et sa fonction de facteur de transcription. La biochimie et la génétique moléculaire de p53 ont fait l'objet de récentes synthèses (Ko et Prives, 1996; Levine, 1997; Agarwal et al., 1998). À première vue, il semble qu'une augmentation du niveau de la protéine p53 (stabilisation) dans les cellules corresponde à une augmentation de son activité de transactivation transcriptionnelle. Toutefois, il a été démontré que l'induction de la transcription dépendante de p53 par irradiation à faibles doses d'UV ou par micro-injection d'un anticorps reconnaissant l'extrémité carbox:y-terminale, même en l'absence de dommage à l'ADN, ne requiert pas l'accumulation de la protéine p53 (Hupp et al., 1995). De leur côté, Chemov et Stark (1997) ont observé que le salicylate de sodium, qui inhibe
les protéines kinases, bloque l'activation transcriptionnelle de p53 san:s empêcher l'accumulation de la protéine. Chemov et Stark ont également mis en évidence qu'en u.itilisant un inhibiteur de la protéine kinase C, la protéine p53 s'accumule dans le noyau et n'a pas cl'activité transcriptionnelle, en dépit de sa capacité de liaison à sa séquence d'ADN consensus (Chemov et al., 1998). Or, différents processus de régulation de l'activité du p53 semblent avoilr un rôle à jouer, soit la phosphorylation (Lohrum et Scheidtmann, 1996), la glycosylation (Sha'-V et al., 1996), la liaison à
certaines protéines régulatrices (Hansen et al., 1996), l'épissage alternatif (Wu et al., 1997) et l'acétylation (Gu et Raeder, 1997).
La phosphorylation/déphosphorylation est un mécanisme de contrô·le modulant l'activité de plusieurs protéines régulatrices dans la cellule, et la protéine p53 ne fait pas exception. Plusieurs kinases de sérines induisent la phosphorylation de la protéine p53 humaine, soit PKC aux résidus 371, 376 et 378 (Hupp et Lane, 1994; Takenaka et al., 1995), CKII au résidu 392 (Hupp et al., 1992) et les cdks au résidu 315 (Wang et Prives, 1995) dans le clomaine carbosy-terminal. Également dans le domaine amino-terminal, p53 subit la phosphoryla"'tion par la DNA-PK aux résidus sérines 15 et 3 7 (Fiscella et al., 1993; Maya et al., 1997), par La CKI aux résidus sérines 4, 6 et 9 (Milne et al., 1992), par la MAP kinase (Milne et al., 1994), la JNKI -2 ou -3 (Milne et al., 1995; Hu et al., 1997), la Raf (Jama et
zm:
1995) et par la kinase CAK (Ko et al., 1997) au résidu sérine 33. Il a été démontré que la phosphorylation du p53 à l'extrémité arnino-terminale survient en réponse à des dommages à l'ADN (Knippschild et al., 1996;. Shieh et al., 1997; Meek et al., 1998). Les phosphatases PPl et PP2A sont des agents de déphosphorylation des domaines amino- et carboxy-terminaux de la protéine p53 (Scheidtmann et al., 1991; Helps et al., 1995; Takenaka et al., 1995).Le p53 est une phosphoprotéine qm existe normalement sc:»us forme latente en un complexe tétramérique et qui agit comme activateur de la transcription <le gènes cibles lorsqu'elle est activée. Ces derniers comportent dans leur région promotrice un élément de réponse défini comme étant une séquence consensus d'ADN reconnue par p53, pouvant contenir deux ou plusieurs copies du décamère 5'-PuPuPuC(A!I)(T/A)GPyPyPy-3' (El-Deiry et al., 1992). La
protéine p53 est un facteur de transcription qu'on retrouve retenue au cytoplasme. A la suite d'un stress génotoxique, la protéine est transloquée au noyau, puis elle transactive un groupe de gènes,
par exemple p21/WAF1/CIP 1, GADD45, MDM2, cyc/in G, PCNA, BAX, AP0-1/FAS, 1NF-a et
IGF-BF3 (Owen-Schaub et al., 1995; voir aussi le résumé de Ko et Prives, 1996). Plusieurs groupes ont observé une affinité différentielle de p53 pour sa séquence consensus, se traduisant par une transactivation différentielle des gènes cibles (par exemple, voir Lohrum et Scheidtmann, 1996). Il semblerait que l'activité de transactivation transcriptionnelle du p53 fasse intervenir l'interaction de son domaine amino-terminal (domaine de transactivation) avec certains polypeptides du mécanisme générale de la transcription, comme le facteur TBP ("Tata Box Binding Protein") (Seto et al., 1992), les facteurs TAFs (Transcription Activating Factor) comme TAF3 l ou TAF70 (Lu et Levine, 1995), et les coactivateurs cellulaires p300/CBP (Avantaggiati et al., 1997; Gu et al., 1997; Lill et al., 1997). La protéine p53 peut également réprimer certains gènes ne comportant pas la séquence consensus reconnue par p53, tel que le gène de la topoisomérase Ila (Wang et al., 1997). Le mécanisme de répression n'est pas encore bien compris
à ce jour et ne ferait pas intervenir l'interaction avec TBP (Farmer et al., 1996).
Bien qu'il soit généralement accepté que le p53 induit l'apoptose via la transactivation transcriptionnelle de BAX, il a été démontré que l'apoptose dépendante de p53 ne nécessite pas
Mdm2, p21 Wafl/Cipl, Fas/AP0-1 ni Bax après une exposition aux radiations ionisantes (Knudson
et al., 1995; Reinke et Lozano, 1997). Un modèle d'induction de l'apoptose dépendante du p53 a été proposé. En fait, ce modèle a été élaboré à partir de l'observation d'une augmentation de certains ARNm au cours de l'apoptose induite par l'expression de p53. Les protéines codantes prédites de ces ARNm se sont avérées faire partie de la réponse au stress oxydatif (Polyak et al., 1997). Toutefois, il a été démontré que la protéine p53 peut induire l'apoptose sans faire intervenir son activité de transactivation transcriptionnelle (Caelles et al., 1994).
1.2 Apoptose indépendante de p53
Un mécanisme d'apoptose indépendant de p53 implique la voie de signalisation par les récepteurs appartenant à la famille des récepteurs de facteurs de nécrose tumorale (TNF), dont le FAS (APO/CD95), le TNF-Rl, le DR3 (TRAMP/wsl-l/AP0-3/LARD/AIR), et le DR4ffRAIL-Rl/AP0-2 (voir pour résumé: Nicholson et Thornbeny, 1997; Peter et al., 1997; Cohen, 1997). Le phénomène apoptotique s'amorce lorsque ces facteurs se lient aux récepteurs de surface cellulaire, provoquant leur agrégation (Nagata et Golstein, 1995). La partie cytoplasmique du récepteur s'associe à des protéines contenant le domaine de mort ("death domain") inclus dans les molécules cytoplasmiques adaptatrices, telles que MORTl/FADD (Boldin et al., 1995; Chinnaiyan et al., 1995), TRADD (Hsu et al., 1995), MADO (Schievella et al., 1997), RAIDD/CRADD (Duan et Dixit, 1997) ou RIP (Stanger et al., 1995). De cette interaction protéine-protéine via le domaine de mort, découle une cascade d'événements de phosphorylation/déphosphorylation qui ultimement entraîne le recrutement et l'activation des caspases-8/FLICE/MACH/MchS et -10 de la famille des cystéine-protéases de l'enzyme convertissant l'interleukine-1-P (ICE). À son extrémité amino-terminale, la molécule F ADD contient un domaine effecteur de mort (DED : "death effector domain") qui est également retrouvé dans la caspase-8/FLICE (Medema et al., 1997).
1.3 L'activation des caspases est essentielle
à
l'apoptose
Que le signal menant au phénotype apoptotique fasse intervenir p53 ou non, des caspases (çystéinyl aspartate-spécifiques protéinases) seront ultimement activées et seront responsables collectivement du phénotype apoptotique (Sabbatini et al., 1997). Ces caspases sont des protéases ubiquitaires, qui sont produites sous une forme inactive (pro enzymes ou procaspases ), qui sont activées par le clivage du précurseur protéique dans un domaine de liaison entre les deux domaines catalytiques, et se transforment en hétérodimères actifs. Après leur activation, les
caspases procèdent au clivage d'une variété de substrats cellulaires initiant un démantèlement ordonné de la cellule. La clivage a lieu après le résidu aspartate d'une séquence peptidique consensus de quatre acides aminés. Le clivage de la molécule se fait en deux temps : d'abord, la reconnaissance du site (S4-S l) qui est adapté pour recevoir la chaîne aspartique du site (S l) commune à toutes les caspases (puisqu'étant elles-mêmes des substrats de caspases), puis la différenciation des caspases en fonction de leur spécificité par la reconnaissance des résidus S2-S4 du substrat (Thornberry et al., 1997). La découverte de la caspase-3 (CPP32/Yama/apopaïne) et l'étude de son activité ont permis de mieux comprendre les transformations morphologiques et physiologiques qui surviennent au cours de l'apoptose. Certains de ces substrats incluent le facteur de réplication C (REF-C) (Rhéaume et al., 1997), le facteur de fragmentation de l'ADN (DFF) (Liu et al., 1997), l'inhibiteur de la désoxyribonucléase activée par la caspase (ICAD) (Enari et al., 1998; Sakahira et al., 1998), la kinase dépendante de l'ADN (DNA-PK) (Song et al., 1996),
la polymérase d'ADP-ribose (PARP) (Lazebnik et al., 1994), la protéine associée au métabolisme
des riboprotéines (70 kDa-Ul) (Casciola-Rosen et al., 1994), la protéine du rétinoblastome (pRB) (Jânicke et al., 1996), la protéine de liaison de l'élément de régulation du stérol (SREBP) (Wang X. et al., 1996), l'huntingtine (Goldberg et al., 1996), la 1ŒKK (Cardone et al., 1997), le D4-GDI (Na et al., 1996), la kinase PKCô (Emoto et al., 1995) et l'oncogène Mdm2 (Chenet al., 1997). Pour de récentes synthèses, voir Salvesen et Dixit, 1997; Peter et al., 1997. La caspase-3 participe également à un processus non-apoptotique, à savoir la maturation de la cytokine pro-interleukine-16 (Zhang et al., 1998). La caspase-6, appelée laminase, est aussi activée au cours de l'apoptose et procède au clivage de la lamine, qui est essentielle à l'intégrité de l'enveloppe nucléaire (Takahashi et al., 1996). Ces caspases sont généralement activées en aval de la famille des protéines Bcl-2.
1.4 Famille des protéines Bcl-2
Il est connu que les membres de la famille des protéines Bcl-2 jouent ~n rôle important
Bad, d'autres sont anti-apoptotiques, tels que Bcl-2 et Bel-xi.. et tous semblent être associés à des points de contrôle entre le signal pro-apoptotique (émanant de la membrane ou du stress génotoxique) et l'activation des caspases (pour un résumé, voir Brown, 1997). Il est généralement accepté que Bcl-2 protège de l'apoptose tandis que Bax s'oppose à cet effet protecteur. Bax est un produit de l'activité de transactivation transcriptionnelle de p53, mais il a été démontré qu'il n'est pas indispensable dans l'apoptose dépendante de p53 induite par les radiations ionisantes dans les thymocytes (Knudson et al., 1995), suggérant ainsi que d'autres facteurs importants entrent en jeu. Les mécanismes par lesquels les membres de la famille Bcl-2 affectent la survie cellulaire ne sont pas très bien élucidés. Ainsi, le gène BCL-2 code pour une protéine de 25-26 kDa et ne révèle aucune structure particulière pouvant suggérer qu'il ait un rôle à jouer dans l'apoptose. Cependant, les protéines Bcl-2 comportent une extrémité carboxy-terminale hydrophobe essentielle à son ancrage membranaire des mitochondries, du réticulum endoplasmique et de l'enveloppe nucléaire. Il a été démontré que les 21 acides aminés hydrophobes sont nécessaires pour permettre à Bcl-2 de contrôler l'apoptose (Tanaka et al., 1993). Une hypothèse a été émise selon laquelle Bcl-2 exercercit son action dans les pores membranaires et les canaux de perméabilité, en contrôlant le transport de molécules dans ces organelles (Minn et al., 1997; Antonsson et al., 1997; Schendel et al., 1997). Il a été démontré qu'une fuite des mitochondries suffit à induire l'apoptose (Zamzami et al., 1996). Certains prétendent que Bcl-2 contrôle le potentiel membranaire ainsi que le volume homéostatique des mitochondries (Vartder Heiden et al., 1997). D'autres ont constaté que Bcl-2 contribuait à la régulation des intermédiaires réactifs de l'oxygène au niveau cellulaire (Hockenbery et al., 1993; Melkova et al., 1997). L'hypothèse la plus plausible, qui rassemble ces observations, confère à
Bcl-2 ou à Bcl-xL un rôle de protection contre le relâchement de cytochrome c de l'espace intermembranaire des mitochondries au cytosol {Adachi et al., 1997, pour un résumé, voir Read, 1997). Le cytochrome c est une protéine soluble, chargée positivement, dont le rôle dans la chaîne respiratoire est d'interagir avec des partenaires rédox du complexe ID et du complexe IV (Mathews, 1985). Le mécanisme conduisant au relâchement des cytochromes c des mitochondries dans le cytoplasme n'est pas connu. Les cytochromes c micro-injectés dans la cellule induisent l'apoptose (Li F. et al., 1997; Zhivotovsky et al., 1998; Hengartner, 1998). Des facteurs ont été
isolés et apparaissent nécessaires pour l'activation des caspases, à savoir Apaf-1, Apaf-2 et Apaf-3 (pour 11Apoptosis protease activating factor"). Il a été établi que le facteur Apaf-2 était le
cytochrome c (Zhivotovsky et al., 1998; Hengartner, 1998) et que le facteur Apaf-3 correspondait à la caspase-9 (Li P. et al., 1997). Le facteur Apaf-1 forme un complexe ternaire avec Bel-XL et la caspase-9 (Pan et al., 1998). L'activité anti-apoptotique de Bel-XL est renversée par l'expression de Bax ou de Bak qui empêchent l'interaction de Bel-XL avec Apaf-1 et la caspase-9 (Pan et al., 1998). En l'absence du partenaire anti-apoptotique de la famille Bcl-2, la caspase-9 et le facteur Apaf-1 se lient via leur domaine amino-terminal en présence du cytochrome c et de dATP. Cet événement mène à l'activation de la caspase-9 qui active alors la caspase-3 (Li P. et al., 1997). Le relâchement des cytochromes c des mitochondries survient avant ou au même moment que l'activation des caspases-3/CPP32/Apopaïne. En effet, malgré que l'inhibiteur peptidique irréversible général des caspases, en l'occurence z-V AD-fink (voir la liste des abréviations, p. V) inhibe l'apoptose, il n'a pas d'effet sur la translocation des cytochromes. Cette inhibition de l'apoptose s'observe par l'empêchement du clivage de P ARP, de la pro-caspase-3 et de la formation de corps apoptotiques (Bossy-Wetzel et al., 1998). L'activité des caspases n'est donc pas requise pour la translocation des cytochromes c au cytosol, mais cette translocation semble nécessaire pour l'activation des caspases. En revanche, on a relevé un cas d'induction de l'apoptose n'impliquant pas les cytochromes c, ni la fonction protectrice de Bcl-2. Ce dernier fait intervenir la granzyme B (Vaux et al., 1992), qui transforme directement la pro-caspase-3 en pro-caspase-3 active (Darmon et al., 1995).
L'augmentation du calcium cytosolique est un phénomène fréquemment observé durant l'apoptose (pour un résumé sur le rôle du calcium, voir Malviya et Rogue, 1998). Le rôle du calcium (Ca2+) nucléaire s'est révélé important de par son association à la maladie d'Alzheimer. La démence associée à une dégénérescence neuronale qui caractérise cette maladie découlerait d'une accumulation de fragments de préséniline-2. Ceux-ci sont produits par le clivage effectué par des caspases, en l'occurrence la caspase-3/CPP32Nama (Li J. et al., 1997; Vito et al., 1997), et seraient dépendantes du Ca2+. Une autre indication du rôle du Ca2+ nucléaire dans l'apoptose concerne les membres de la famille de Bcl-2 qui sont également situés dans la membrane nucléaire
externe. Les membres de la famille Bcl-2 localisés dans les membranes du réticulum endoplasmique et des mitochondries servent aussi de tampon et de régulateur du Ca2+ cytosolique. Il a été démontré que la suppression de l'apoptose par Bcl-2 se produit parallèlement à l'exclusion du Ca2+ nucléaire (Marin et al., 1996). Dans le même ordre d'idées, les membres de la famille Bcl-2 nucléaires pourraient jouer un rôle de régulation du Ca2+, c'est-à-dire en contrôlant la régulation du ca2+ nucléaire.
Quoiqu'il en soit, il semble que Bcl-2 soit placé à un point de contrôle où convergent les signaux apoptiques provenant de la membrane et qu'il protège également de l'apoptose en présence d'une variété d'agents endommageant l'ADN. Cette classe d'inducteurs de l'apoptose inclut les radiations ionisantes, la campthotécine (inhibiteur des topoisomérases d'ADN), la moutarde nitrogénée (dont la lésion simple est un dommage mimant les UV et réparée par le NER) (Walton et al., 1993; Miyashita et Reed, 1993). De plus, Bcl-2 protège de l'apoptose induite par la céramide, issue de l'activation des récepteurs membranaires de type F AS-APO (Geley et al., 1997). De même, Bcl-2 empêche l'induction de l'apoptose par p53, lequel est un détecteur des dommages à l'ADN (Chiou et al., 1994). Le mécanisme de protection de l'apoptose induite par un stress génotoxique implique que la protéine Bcl-2 empêche la translocation de p53 vers le noyau, malgré l'augmentation de son niveau d'expression (Beham et al., 1997). Cette observation est fort intéressante puisque l'apoptose induite par p53 suppose que la protéine soit transloquée au noyau où elle y jouerait un rôle.
Les lecteurs sont invités à consulter les introductions des chapitres II à IV pour obtenir des informations supplémentaires concernant ces travaux de recherche.
2 - Projets de recherche
Le scénario actuellement accepté reliant protéine p53, stress génotoxique et apoptose se décrit comme suit : p53 cause un arrêt de croissance à certaines phases du cycle cellulaire (points de contrôle), dont le mieux identifié est l'arrêt en phase G1 tardive. À la suite d'un stress cytotoxique, la protéine p53 induit cet arrêt par la transactivation transcriptionelle du gène p21/Wafl/Cipl, dont le produit est un inhibiteur des kinases dépendantes des cyclines (CDK) (El-Deiry et al., 1993). L'arrêt du cycle cellulaire induit par l'expression de p21Wafl/Cipl mettrait en cause l'interaction avec BRCAl (Somasundaram et al., 1997). Cette hypothèse est supportée par l'observation que BRCAl interagit directement avec p53 et régule positivement s<>n activité transcriptionnelle (Ouchi et al., 1998). Il a également été proposé que l'arrêt du cycle cellulaire, principalement en phase S, fasse intervenir PCNA (Flores-Rozas et al., 1994; Wood et Blow, 1994; Jackson et al., 1995; Chen J.J. et al., 1996), mais cette dernière hypothèse demeure contreversée (Nakanishi et al., 1995). Dans certaines circonstances, la protéine p53 peut déclencher l'apoptose par la transactivation transcriptionndle du gène BAX (Miyashita et Reed, 1995; Yonish-Rouach et al., 1996). Ces fonctions en tant que facteur de transcription requièrent une protéine non-mutée, c'est-à-dire pouvant lier via son domaine protéique central la séquence d'ADN consensus de reconnaissance du gène à transactiver. Il est proposé que l'arrêt en G1 alloue à la cellule le temps de réparer ses dommages et, dans le cas où ceux-ci sont trop importants, la protéine p53 déclenchera la voie apoptotique. Par contre, ce concept ne précise pas la nature des lésions et l'étendue des dommages pouvant provoquer un arrêt transitoire du cycle cellulaire ou un arrêt définitif: c'est-à-dire l'apoptose. Un des buts premiers de cette thèse a été d'identifier la nature des lésions pouvant être à l'origine de l'apoptose induite par p53.
En réponse à des signaux de stress très divers (radiations, agents toxiques, carence en facteurs de croissance, diminution de la concentration de nucléotides, etc.), la protéine p53 se stabilise et s'accumule dans le noyau cellulaire (Kastan et al., 1991; Zhan et al., 1993; Nelson et Kastan, 1994). Dans les cellules normales non endommagées, la protéine pS? est présente en quantité infime, sous forme latente rapidement dégradée (t'Y2 -30 min), probablement par un
processus d'ubiquitination (Chowdary et al., 1994; Brown et Pagano, 1997) faisant appel à la protéine Mdm2 responsable de la boucle de rétrocontrôle (Haupt et al., 1997; Kubbutat et al., 1997; Piette et al., 1997). De même, l'état de phosphorylation (via la DNA-PK) influence la capacité de p53 de se lier à Mdm2 et se répercute sur son activité de transactivation et sur sa stabilité (Shieh et al., 1997). L'activité de p53 et sa stabilisation sont contrôlées post-transcriptionnellement et post-traductionnellement, en dépit de sa capacité à transactiver son propre gène TP53 ( Deffie et al., 1993) et de s'associer à son propre ARN messager (Mosner et al., 1995). Les facteurs influant sur la stabilité de la protéine p53 incluent la phosphorylation/déphosphorylation, le changement de conformation, l'association protéine-protéine et le signal de localisation nucléaire. Les inhibiteurs de la transcription n'empêchent pas l'augmentation du niveau de la protéine p53 dans la cellule (Fritsche et al., 1993), mais bien au contraire, l'inhibition de la transcription peut, à elle seule stabiliser la protéine sans que le moindre dommage soit perçu dans la cellule (Kastan et al., 1991 ). Pour un résumé faisant état de l'effet de la phosphorylation de p53 sur sa conformation, sur sa capacité de liaison à sa séquence consensus et sur sa capacité de transactivation, les lecteurs sont invités à consulter Milczarek et al. (1997).
Il persiste dans la littérature deux hypothèses concernant l'activité de p53 dans l'induction
de l'apoptose. Il a été démontré hors de tout doute que p53 peut transactiver le gène BAX, dont
le produit du gène forme un hétérodimère avec la protéine anti-apoptotique Bcl-2. Lorsque le niveau de la protéine pro-apoptotique Bax est supérieur au niveau de Bcl-2, l'apoptose survient (Miyashita et al., 1994; Zhan et al., 1994; Miyashita et Reed, 1995). Par contre, il a également été démontré que p53 entraîne l'apoptose par un mécanisme indépendant de son activité de transactivation transcriptionnelle (Caelles et al., 1994; Haupt et al., 1995; Wang X.W. et al.,
1996; Chen X.B. et al., 1996). Un des objectifs de cette recherche était de définir l'état fonctionnel de la protéine p53 dans les cellules A43 l, en tant que facteur de transcription et/ou d'apoptose.
3 - Observations reliées aux articles présentés
Dans ce projet de recherche, nous avons principalement utilisé une lignée de cellules épithéliales cancéreuses (MCF-7) et de kératinocytes cancéreux (A43 l). Ces cellules sont sensibles aux rayons ultraviolets et nous avons observé que cette sensibilité se traduit par l'induction de l'apoptose. Nous avons pu constater une très grande corrélation entre l'induction de l'apoptose et l'incapacité du p53 à transactiver les gènes en aval lors de la réponse au stress
génotoxique, en l'occurrencep21/WAFJICJPJ. Nous avons émis l'hypothèse qu'une inlu'bition de
la transcription par l'ARN polymérase II (ARN pol Il) enclencherait la voie menant à l'apoptose. Nos observations traduisent l'hypothèse selon laquelle l'apoptose provoquée par un stress génotoxique, qui fait intervenir la protéine p53, peut être dissociée de son activité de transactivation transcriptionnelle. Les résultats de ces observations ont déjà été publiés dans le journal "Biochemistry and Cell Biology" (1997, 75: 351-358; voir chapitre II, p. 15).
Une étude plus élaborée quant à la nature des dommages pouvant conduire à l'apoptose nous a mené à émettre l'hypothèse suivante : les dommages à l'ADN qui sont de bons inhibiteurs de la transcription par l'ARN pol II et qui sont réparés par excision/resynthèse sont des précurseurs initiaux de l'apoptose. Ces résultats sont en voie d'être soumis (voir chapitre III, p. 24).
L'étude de la fonction du p53 comme facteur de transcription nous a révélé que les cellules A43 l comportent une protéine mutée (Harlow et al., 1985), inapte à la transactivation transcriptionnelle, fait confirmé par la caractérisation génomique d'une mutation en son domaine central (R273H). Ces kératinocytes A431 comportant une protéine p53 mutée (R273H) non fonctionnelle comme transactivateur de la transcription (Rolley et al., 1995; Wang et al., 1996), compte tenu de l'abolition de l'interaction de l'histidine avec la double hélice de l'ADN (Cho et al., 1994). Ces cellules A431 ne présentent pas d'arrêt dans le cycle cellulaire en phase G1 après une exposition à un stress génotoxique, car elles n'induisent pas le gène WAFJIC/PJISDIJ. En utilisant la mimosine comme inducteur de p21 Wafl!Cipl par une voie indépendante du p53, nous
avons démontré que l'arrêt en phase Gi concomitant à l'expression de la protéine p21 protège de l'apoptose (voir chapitre IV, p. 58).
Des études utilisant des fibroblastes homozygotes pour le gène 1P53 muté dérivés de patients ayant le syndrome de Li-Fraumeni (Ford et Hanawalt, 1995; 1997) ont démontré que la protéine p53 serait impliquée dans la réparation par excision de nucléotides. Utilisant un système élaboré à partir d'extraits cellulaires, nous n'avons pu observer aucun effet de la protéine p53 sauvage, non plus que de la protéine mutée sur le mécanisme de réparation par excision/resynthèse in vitro (voir les chapitres ill, p. 24, et V, p. 68).
La corrélation entre l'état sauvage de la protéine p53 et sa capacité à transactiver le gène p21 Wafl/Cipl après un stress génotoxique nous ont permis de développer un test biochimique pour déterminer l'état fonctionnel de la protéine p53 dans différents types cellulaires. En effet, l'induction de l'expression de la protéine p2 l Wafl/Cipl après irradiation est garante de l'activité
fonctionnelle de transactivation de p53, donc signifie la présence d'une protéine p53 sauvage. Avec ce test, nous avons pu caractériser l'état fonctionnel de la protéine p53 contenue dans les clones de cellules épithéliales MCF-7 résistants au méthotrexate, produits dans le laboratoire du Dr Benoît Paquette. Une partie des résultats a été soumise et l'article est accepté pour publication (voir chapitre VI, p. 79).
Chapitre
II
The apoptotic and transcriptional transactivation activities of p53 can be dissociated. Nathalie Bissonnette. Bohdan Wasylyk and D.J. Hunting
Biochem. Cel/ Bio/. 75: 351-358 (1997).
Dans cet article, l'auteure de la thèse a produit la totalité des résultats, réalisé les illustrations et rédigé l'article. Elle est également responsable de l'hypothèse selon laquelle la protéine p53 mutée (R273H) qui est présente dans la lignée cellulaire A43 l, qui est inactive comme activateur de la transcription et qui subit un clivage lors de l'apoptose, pourrait avoir des propriétés apoptotiques. Le fragment clivé de p53 observé au cours de l'apoptose pourrait interagir avec TFIIH pour ainsi promouvoir l'apoptose.
The apoptotic and transcriptional
transactivation activities of p53 can be·
dissociated
N. Bissonnette, B. Wasylyk, and D.J. Hunting
351
Abstract: Previous studies have shown that the apopcotic response of cells following DNA damage requires p53 expression. Wild-type p53 procein levels increase in response to DNA damage and its growth-suppressive action is thoughc co be mediaced by cranscriptional activation of the p21/WAF l ICIP l gene, the product of which is a pocent inhibitor of
cyclin-dependent kinases. The mechanism by which ele,·aced p53 levels lead co apoptosis is not known, bue is believed co involve transcriptional activation of apoptotic genes. such as BAX. We have studied transformed human cells that constirutively express high levels of the R273H mutant p53. which has been reponed to Iack cranscriptional activation activity. We used the inability to induce the p21/Wafl/Cip l procein as a marker co verify the lack of cranscriptional activation accivity. Cells expressing the R273H mutant of p53 do not show an increase in p2I/Wafl/Cipl following irradiation with ionizing or UVB radiation. Surprisingly, these cells are very susceptible to induction of apoptosis by UVB radiation, as seen by the formation of a nucleosomal ladder and the proceolytic cleavage of poly(ADP-ribose) polymerase. This suggests that the R273 mutant p53 can fonction normally in apoptosis but not in cranscriptional activation following DNA damage. Furthennore, an inhibitor of RNA polymerase II is a potent inducer of apoptosis in these cells, demonstrating that transcription is not required for apoptosis and suggesting that stalled RNA polymerase II complexes can initiate apoptosis. Incerestingly, proceolytic cleavage of p53 occurs during apoptosis in these cells, generating a 45-kDa fragment and liberating the DNA repair helicase binding domain of p53. We propose that the peptide liberated from the carboxy terminus of p53 may contribute to its apoptotic activity, possibly through interaction with the XPB and XPD DNA helicases.
Key words: DNA damage, p21/WAFl/CIPI, proteolytic cleavage.. p53. RNA polymerase II inhibition. apoptosis.
Résumé : Des études antérieures ont montré que la réponse apoptocique des cellules après une lésion de r ADN nécessite l'expression de la protéine p53. Le taux de protéine p53 sauvage augmente à la suite d'une lésion de l'ADN et l'arrêt de croissance qu'elle entraîne serait attribuable à l'activation de la transcription du gènep21/WAFJICIPI, dont le produit est un
puissant inhibiteur des kinases cycline-dépendantes. Le mécanisme par lequel un taux élevé de p53 entraîne l'apoptose est inconnu. mais il nécessiterait l'activation de la transcription de gènes d'apoptose, tel BAX. Nous avons étudié des cellules humaines transformées exprimant la R273H. une p53 mutante ne pouvant activer la transcription de gènes, de façon constitutive et à des eaux élevés. Nous avons vérifié l'absence d'activité activatrice de la transcription de gènes par l'incapacité d'induire la protéine p21/Wafl/CipL Il n'y a pas d'augmentation de p21/Wafl/Cipl à la suite d'une irradiation des cellules exprimant la protéine p53 mutante, R273H. par des rayons UV-8 ou des radiations ionisantes. Étrangement, les rayons UV-8 induisent une forte apoptose dans ces cellules. tel qu'indiqué par la formation d'une échelle de nucléosomes et par la protéolyse de la poly(ADP-ribose) polymérase. Cela suggère que la protéine p53 mutante. R273H, fonctionne normalement au cours de l'apoptose, mais non durant l'activation de la transcription à la suite d'une lésion del' ADN. De plus, un inhibiteur del' ARN polymérase II est un puissant inducteur de l'apoptose dans ces cellules, ce qi..:i démontre que la transcription n ·est pas requise pour l'apoptose et suggère que les complexes d' ARN polymérase II stationnaires peuvent amorcer l'apoptose. Il est intéressant de noter que la p53 est clivée dans ces cellules au cours de l'apoptose. ce qui génère un fragment de 45 kDa et libère son domaine de liaison aux hélicases de réparation de l' ADN. Nous proposons que le peptide libéré de l'extrémité carboxyle de p53 contribuerait à son activité d'amorçage de l'apoptose, peut-être grâce à une interaction avec les hélicases XPB et XPD.
Mors clés: lésion del' ADN, p21/WAFl/C/P/, protéolyse. p53. ARN polymérase II. inhibition. apoptose.
[Traduit par la rédaction]
Received March 21, 1997. Revised August 6, 1997. Accepted August 11, 1997.
Abbreviations: RNA pol IL RNA polymerase II; PARP, poly(ADP-ribose) polymerase; TFDH. transcription factor IIH.
N- Bissonnette and D.J. Hunting.1 MRC Group in the Radiation Sciences, Faculté de médecine. Université de Sherbrooke,
Sherbrooke, QC JlH 5N4, Canada.
B. Wasylyk. Institut de génétique et de biologie moléculaire et cellulaire. 67404 Illkirch Cédex. France.
1 Author to whom ail correspondence should be addressed.. e-mail: dhunting@counier.usherb.ca
352
Introduction
The p53 tumor suppressor phosphopretein has critical roles in cellular stress responses and the maintenance of genome sta-bility (for reviews, see Hartwell and Kastan 1994; Smith and Fomace 1995). Following genotoxic stress, such as chat pro-duced by UV or ionizing irradiation, chemotherapeutic agents. or conditions of hypoxia, induction of p53 results in cell-cycle arrest or apoptosis (for a review, sec Ko and Prives 1996).
p53 has been demonstrated bath to bind DNA in a sequence-specific manner via its central do main (Kern et al. 1991) and to contain a strong transcriptional activation sequence nea.r its amino terminus (Fields and Jang 1990; Raycroft et al. 1990). One of its transcriptional targets is the gene for the cyclin- dependent kinase inhibitor p2//WAF/ICIPI (El-Deiry
et al. 1993), which inhibits the protein kinase activities of G1 - cyclin-dependent kinase complexes, chus preventing
entrance into S phase.
It is Iess clear how p53 stimulates apoptosis; however, there are several downstream genes linked to apoptosis including BAX. These are trans-activated following p53 expression in some cell types, but whether the apoptotic activicy of p53 is dependent on its sequence-specific transcriptional activation propenies is controversial (Caelles et al. 1994; Haupt et al. 1995; Miyashita and Reed 1995; Owen-Schaub et al. 1995; Sabbatini et al. 1995; Chenet al. 1996; Attardi et al. 1996; Hansen and Braithwaite 1996). If indeed p53 can stimulate apoptosis in theabsenceofcranscriptional cransactivation, then
it will be important to identify the domains or regions of the
protein responsible for this function.
Extensive analysis bas thus far revealed three functional domains in the p53 protein (Ko and Prives 1996). The p53 polypeptide consists of an activation domain located within the 43 amino-terminal amino acids, a sequence-specific DNA binding domain located within the central, conserved portion of the protein, and within the carboxyl terminus resides a tetramerization do main (Stürzbecher et al. I 992; Clore et al. 1995; Jeffrey et al. 1995) as well as a regulatory region that controls the ability of the protein to allosterically switch from a latent form to one that is active for sequence-specific DNA binding (Hupp et al. 1992, 1995; Jayaraman and Prives 1995). The vast majority of the missense mutations in p53 from hu-man tumors reside within the central DNA-binding domain and impair or abolish sequence-specific DNA binding by p53 (Vogelstein and Kinzler 1992). The C-terminal region is in-volved in many fonctions. It binds to damaged DNA (Lee et al. 1995; Reed et al. 1995), reanneals DNA and RNA (Bakalkin et al. 1994; Wu et al. 1995), inhibits transcription in transfec-tion assays (Shaulian et al. I 995), has transforming potential (Shaulian et al. 1992; Reed et al. 1993), restores the apoptocic response when directly microinjected into primary normal hu-man fibroblasts (Wang et al. 1996), and most incerestingly. restores DNA binding activity to a subset of p53 mutants both in vivo and in vitro (Abarzua et al. 1996). Moreover. de-Ietion of the carboxy-terminal 30 amino acids of p53, binding of monoclonal PAb421 whose epitope maps to amino acids 370-378, phosphorylation of the C-terminal domain, or addi-tion of the C-terminal p53 peptide were all shown to enhance the specific DNA binding function of wild-type p53 in vitro (Hupp et al. 1992, 1995).
Furthermore, the C-cerminal domain inhibits bath helicases
Biochem. Cell Biol. Vol. 75, 1997
of the transcription factor IIH (TFIIH) complex (Léveillard et al. 1996). The TFIIH complex phosphorylates RNA polym-erase II (pol II) and bas a -role in RNA pol II transcription (Gerard etaL 1991; Aoreset al. 1992). DNA repair(van Vuuren et al. 1994 ). and possibly cell cycle control (Seraz et al. I 995). In this study. we show that DNA-damaging agents as well as an inhibitorofRNA pol II induce a rapid apoptotic response in keratinocytes expressing high levels of the mutant fonn (R273H) of p53. which is defective in transcriptional activation. This death response is concomitant with a proteolytic cleavage of the C-terminal domain of p53, suggesting a raie for the release ofthis peptide in the p53-dependent apoptotic pathway.
Materials and methods
Cell culture
Primary mouse fibroblasts were established from the skin of p53 null BALBc mice (-/-)or wild-type BALBc mice (+/+)and kept in cul-ture for four passages. The status ofboth p53 alleles was determined by PCR analysis of DNA extracted from mouse skin using three primers in a single reaction. The common primer was specific for ex on 7 (GT ACTfGT AGTGGA TGGTG) whereas the second primer was either forexon 6 (CGTGGTGGT ACCTf A TGAG) or for the neo gene (CCTCGTGCITTACGGTATC). Normal human primary
fi-broblast cells (Coriell Institute for Medical Research: AG01519A) and epidenn:il carcinoma cells (American Type Culture Collection: CRLl555) were grown as monolayers in Dulbecco's modified Eagle's medium (DMEM: Life Technologies, Inc, Grand Island, N.Y.) sup-plemented with 9% fec.il bovine serum (FCS) at 37°C in a humidified atmosphere of 7% CO:? in air.
Irradiation and cellular lysate preparation
Cells were irradiated with a <ioco source ac 15 cGy/s (gamma radia-tion) or with UVB (302 nm) at a dose rate of 20 W/s. Cells were washed and scr:iped in buffered saline. Total cellular protein was extracted with lysis buffer (25 mM Hepes (pH 7.8). 2 mM ethylene-diaminetetraacetic acid (EDTA), 1 % NP40. 15% glycerol, 1 mglmL phenylmethanesulfonyl fluoride, IO glmL of peptide inhibitor cocktail (leupeptin. chymostatin, and pepstatin A). and 2 mM dithiothreitol).
Electrophoresis and immunoblotting
SOS-PAGE was perfonned in minigels using the buffer system de-scribed by Laemmli (1970). A 5% stacking gel and a 12% separating gel were used. Equal amouncs of protein from differenc samples were placed in boiling water for 4 min in the presence of SDS gel sample buffer (125 mM Tris-HCI (pH 6.8), 4% SDS, 0.01% bromophenol blue, 10% 2-mercaptoethanol, and 15% glycerol) and electrophore-sed for 90 min at 160 V. After the transfer onto nitrocellulose, the membrane was firsc blocked with 8% powdered skimmed milk in TBS (10 mM Tris-HCI (pH 8.0) and 150 mM NaCI) for l-2 h and incubated with the appropriate first antibody ovemight. The second antibodv was visualized with a chemiluminescence detection
proced~re (PIERCE) according co the manufaccurer's protocol
(Chromatographie Specialities Inc.). Antibodies
The anti-p53 antibody. monoclonal DO-!, was provided by Dr. Bohdan Wasylyk (Institut de génétique et de biologie moléculaire et cellu-laire, Illkirch, France): anti·p2 l antibody was purchased from Onco-gene Science (Cambridge, Mass.) and anti-PARP (poly(ADP-ribose) pc;ilymerase) antibody was kindly provided by Dr. Gilbert deMurcia (Ecole supérieure de biotechnologie de Strasbourg. Illkirch., France).
Bissonnette et al.
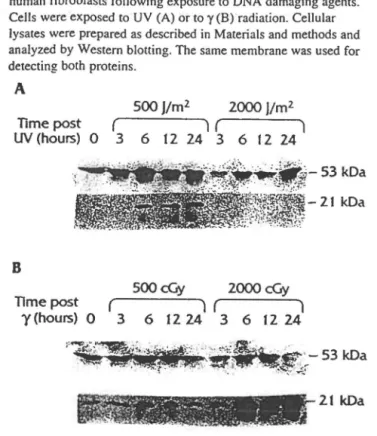
Fig. 1. Time course of p53 and p2 l protein expression in normal human fibroblasts following exposure to DNA damaging agents. Cells were exposed to UV (A) or to y (B) radiation. Cellular lysates were prepared as described in Materials and methods and analyzed by Western blotting. The same membrane was used for detecting both proteins.
A Time post UV (hours) 0 500 J/m2 2000 j/m2 ( H ) 3 6 1 l 14 3 6 1 2 24
·-~ ;~·:_;
..-.·-,~~~
.•·~
·•.r.,--~
..i:~~-~~r~-
53 kDa B llme post y(hours) 0 ( 3 SOOcGy 2000cGy ) ( ) 6 12 24 3 6 12 24 Characterization of apoptosis -21 kDa 21 kDaCells (100-mm Petri dish) were incubated for 20 min in 700 µL lysis buffer (IO mM EDTA and 0.6% SDS) and NaCI solution was added to give a final concentration of 1 M. After an overnight incubation at 4°C, the lysate was centrifuged in a microcentrifuge at 4°C for 30 min. The supernatant was incubated at 50°C for 2 h with 100 µg/mL pro-teinase K and brought to 1.3 M NaCI prior to isopropanol precipita-tion. Pellets were resuspended in TE ( 10 mM Tris (pH 7 .5) and 2 mM EDTA) and incubated for 3 h at 37°C with 80 µg/mL of RNase A. The DNA samples were electrophoresed through a 1.2% agarose gel. Results
p53 expression and p21 induction do not correlate at the protein level
Using nonnal human fibroblasts, we studied the dose response of p53 and p21 expression after exposure to ionizing (y) and UV radiation. We found that UV radiation was more efficient than y radiation for induction of p53 (Fig. IA); however, p2 l induction was much higher following ionizing radiation (Fig. l B ). The kinetics of induction of p53 and p2 l are shown in Fig. 1. Interestingly, the level of p53 diminished at early times (3 h) after a high dose of UVB (2000 J/m2), returned to normal levels at 6 h, and was elevated 24 h after damage (Fig. lA).
Furthermore, there is a total discordance between p53 and p2I expression. p21 was clearly expressed following a low dose of UV (500 J/m2) at 6 h up to 24 h after irradiation whereas no induction was seen after a high dose (2000 J/m2). This decrease in p2 l expression was dose dependent for doses above 1000 J/m2 (data not shown).
We also studied the level ofp53 protein in cells exposed to ionizing radiation and found that the kinetics of accumulation
353 Fig. 2. Expression of p21/Wafl/Cipl protein in normal and p53 knockout cells as a function of the dose of damaging agent. Primary fibroblasts from wild type p53 (+/+)(A) or knockout p53 (-/-) mice (B) were exposed to different doses of UV or
y radiation as indicated and the cells were harvested 7 h after exposure. Cellular extracts were prepared as described in Materials and methods and analyzed by Western blouing.
A (p53+/+ cells) 8 (p53-/- cells) UV (j/m2) y (cGy) E __ ( )( l~ Treatment
g
C ~ c8 8
N 11'1§ §
8 8
N 11'1§ §
a_ OO - N N -U -U 'ô_ "- ~~~ :1 ... :, ': r.r--,_,__~..•
'•!,,"(;• ••:.·.
·. _.
,
'~~':f--~.,...
21 kDaand disappearance of the p53 wi>re very similar following exposure to 500 and 2000 cGy. Following y irradiation, p53 expression was only slightly elevated over that in untreated control ce lis (Fig. lB ). With a dose of 500 cGy, the p2 l/W AF 1 protein levels peaked between 6 and 12 h and retumed to control levels by 24 h. However, p2 l expression markedly increased following y irradiation at higher doses (2000 cGy) and this elevated level persisted at 24 h. These differences in response were not attributable to differences in p53 ex-pression levels.
Induction of p21 following DNA damage requires p53
expression
To deterrnine if p53 is required for induction of p2 l expression following DNA damage, we investigated the dose-response for the induction of p2 I in primary cultures of fibroblasts from normal Balb C mice (p53 +/+) and p53 (-/-) mice (Fig. 2). p2 I was induced in a dose-dependent manner up to 500 J/m2
and then decreased at doses over 1000 J/m2 (Fig. 2A). As
with normal human fibroblasts (Fig. IA), 2000 J/m2 reduced
protein levels below those of unirradiated control cells. This p53 protein level reduction persisted over 12 h (data not shown). Gamma irradiation increased p21 expression in a dose-dependent manner. In agreement with previous studies, p2 l induction was not seen following DNA damage in knockout p53 (-/-) primary mouse fibroblasts (Fig. 2B), arguing that p53 is required for p21 induction after DNA damage (Zhan et al. 1996). p2 I cannot be induced in a mutant p53 (R273H) cell line following UV or y-irradiation
We ha\"e studied a tumor-derived line of transformed human keratinocytes (A43 l cells) containing one allele of p53, mu-tated in its central core at the residue 273 (Arg His) (Harlow