Two silencing
sub-domains of
v-erbA synergize with each
other,
but
not
with RXR
Bernd
Martin, Rainer
Renkawitz and Marc Muller*
Genetisches Institut, Justus-Liebig-Universitat, Heinrich-Buff-Ring 58-62,
D-35392
Giessen, Germany
Received September 19, 1994; Accepted October 12, 1994
ABSTRACT
Thethyroidhormonereceptor(TR)andthe retinoic acid
receptor(RAR)induce geneexpressioninthe presence of specific ligand and repress transcription in the absence of hormone. This repression is mediated
by
an active
silencing
mechanism rather thenby
interference with DNA binding activators.
V-erbA,
a variant form of TR which is unable to bindhormone,
represents a constitutive repressor. Here weshow,
using fusion
proteins
with the GAL4 DNAbinding
domain, that the minimal
silencing
domain ofv-erbA extendsfrom amino acids389 to632 andthatinternal deletions within this domain retain at least some repression function. Co-transfectionexperiments
of different deletion mutants indicate that thesilencing
domainis
composed
ofatleasttwosub-domainswhichare non-functional when tested
individually.
When combined ina heterodimericcomplex,
they synergize
such thatsilencing
activity
isregained.
In contrast tothe retinoic acidreceptorthe retinoidX receptor does
notcontainasilencingdomain. In addition it isunable
tocooperate with the
repression
function of TR orv-erbA in a heterodimer. INTRODUCTION
Themembers of thethyroid hormonereceptor (TR) and of the
retinoicacidreceptor(RAR)familyact onvertebratedevelopment andhomeostasisbybindingtospecific DNAsequences(1-3), thereby regulating transcription of target genes (4,5).
TRandRARareligand inducibletranscription factors which
repress transcription in the absence of hormone and induce
expression in the presence ofspecific ligand (6). Dependingon the promoter context or type of response element ligand
dependentrepressionhas beendescribedaswell (7-10). Here
weareanalyzing the repressionin the absence of ligand which ismediatedby an active silencing mechanism even acting on a
minimalpromoter composed of only a TATA box(11). Variant forms of these factors which are unable to bindhormonerepresent
constitutive repressors (6,12). Recently the analysis of thyroid
hormoneresistance syndromepatients illustrated the importance ofthis repression function. A dominantnegative TR mutant is
causinga severeimpairmentofphysicaland mental development,
which isnot seen uponTR genedeletion (13).Similarly,amajor
role for silencing is demonstrated by the chicken oncogene
product v-erbA.It does notbind T3 duetomutations inits ligand binding domain (14), it functionsas aconstitutive silencerprotein (6,11,12,15) and it interferes with normal erythropoiesis (16-20). A natural mutant of v-erbA (Pro 399 to Arg) lacks the oncogenic and the silencing activity (21,22).
Deletionanalysisofv-erbAassigned thesilencing functionto
about 300 C-terminal amino acids (a.a.) of the protein (23). Corresponding domains ofTRandRARwere ableto mediate both, repression andligand dependent induction. Incontrast to
otherrepression mechanisms (7-9,24-27), nothing is known
about the structural requirement for silencing domains, a
prerequisite forthemechanisticunderstanding of this effect(24).
Thefinding that bothTRandRARaswellasother members of thegenefamilyformheterodimers with RXRs addseven more
complexity to the transcription regulation by these factors (28 -34). The natural ligand for RXR was shownto be 9-cis retinoic acid(9-cis-RA),astereoisomer ofall-trans-retinoicacid, theligand forRAR(35,36).Heterodimerization ofTRwithRXR
stronglyenhancesboth, specificDNAbindingmeasuredinvitro
(28,30,37,38) and transcriptional induction in transient transfection experiments(31,32,34,39) orin vitro (40). Itwas
notknownwhetherRXRexhibitsorcontributestotherepression function in the ligand free heterodimer with TR orRAR.
Here weshow that thev-erbAsilencing domain iscomposed of atleasttworegions, both of whichare nonfunctionalontheir
own.Repression activity is restored when both regionsare present
inoneprotein, butinterestingly also whenpresent on twodifferent proteins which are ableto form heterodimers.
Wefindthat aregion of the RXRdomain, correspondingto the minimal silencing domain of v-erbA, does not mediate
transcriptional repression, although itreadily displays hormonal
induction. In addition it does not contribute to TR or v-erbA
silencing when present in a heterodimer. RESULTS
V-erbA contains a large silencing domain
Based onpreviousresults (23) wewanted to define more precisely
regions of v-erbA required for transcriptional repression. Therefore we fused the C-terminal part of v-erbA to the
heterologousDNAbinding domain (DBD) of the yeast activator GAL4. Aminoacids 1-147 of GAL4aresufficient formediating
several functions suchasnucleartranslocation,dimerization and binding to the specific upstream activator sequence (UAS), whereas the major trans-activation functions are deleted.
Transcriptional regulation mediated by GAL4-DBD orby the
chimericproteinswasmeasuredusingareporterplasmidcarrying
the bacterial chloramphenicol acetyl transferase (CAT) gene. CATexpressionwasunderthe control ofaGAL4DNAbinding
site upstream of the thymidine kinase (tk) promoter. This approach allows to study the function of v-erbA domains independentlyofendogenous members ofthethyroid hormone receptorfamily whichbind to the T3 responseelements, butnot to anUAS. Expressionand DNAbinding ability wastestedby
Reporter plasmid GAL 4 tk CAT UAS_ _ UAS Expression Plasmid 1 _ 147 346 v-erbA 639 362 639 389 639 409 639
b
N C L 1 % 2 3 4 Repression 1 1 100 140 190 1 5 6 7 8 so-U"4
It$
111 9 10 1 1 12 13 14 15 16Figure 1. Amino-terminal deletion mapping of the v-erbA silencing domain. Expression vectors coding for the indicated fusion proteins were transfected into Ltk- cellstogether with the indicator plasmid UAS-tkCAT (top line). Fold repression was calculated from CAT activities obtained in triplicate transfections
relativetothe CAT activity seen after co-transfection of a non-coding expression
vectorandthe reporter plasmid (23 2 %CAT conversion). Experiments were repeatedatleast twice.
s L I 1 ,fi,:4
lf,
input GAL-erb 409-639 (C) inputluciferase (L) GST-GAL-erb 508-639 GAL-erb 409-639 + GST luciferase input GAL-erb 362-508 (N) GST-GAL-erb 508-639 GST GAL-erb 362-508 + luciferase GST-erb 362-508 Bacterial in vitro 35S-labelled input luciferase (L)input GAL-erb 362-468/508-639 (A)
GST-GAL-erb 508-639 GAL-erb 362-468/ GST 508-639+luciferase input GAL-erb 508-639 (.) GST-GAL-erb 508-639 GST GAL-erb 508-639 + luciferase GST-erb 508-639 Bacterial in vitro N
35S-labelled
Expression plasmid 346 S 346 389 434 639 639 346 434 468 639 362 468 508 639 362 515 - . ,15 639 6: Repression 104 l 6 1 6 1Figure2.Internal deletions of the v-erbAsilencingdomain. Expressionvectors
codingfor the indicated fusionproteinsweretransfected intoLtk- cellstogether with the indicatorplasmidUAS-tkCAT (seelegendtoFig. 1). Expressionof UAS-tkCATtogetherwithanon-coding expression vectorwas40 X 3% CAT conversion.
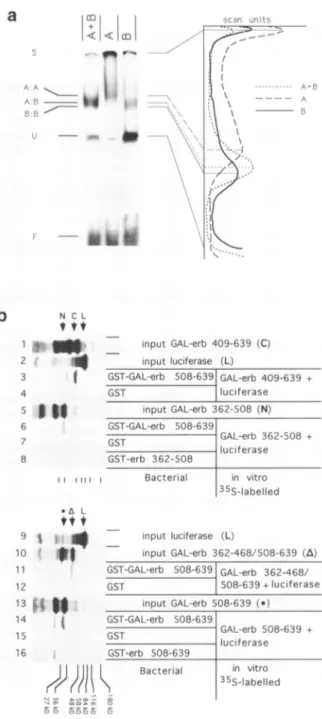
Figure3.(a) Formation ofheterodimersaftercotransfectionofdifferentexpression vectors.Fusionproteins GAL-erb 362-468/508-639orGAL-erb 362-508were
expressedinCOSI cells either alone(lanesAorB,respectively)ortogetherin
oneexperiment(aneA+B). Cellextractswereused inagelretardationexperiment
usinganUASprobeand theautoradiogramwasscanned in ordertodetermine
migration differences of theprotein-DNAcomplexes. The laneswereanalysed
usingtheBio-imagesoftware and thedensity profileisdisplayed.Each band is connectedtothecorrespondingpeakin theprofile.A:A,B:BandA:Bindicate thepositionsof theAhomodimer,theBhomodimer and theA+Bheterodimer,
respectively. Sindicates theslot, Uanunspecific band andF thefreeprobe. (b) GAL-erb fusionproteinsinteractin vitro: GST-Fusionproteins GST-GAL-erb508-639,GST-erb508-639 and GST-erb 362-508 andGSTalonewere
expressed inbacteria,boundtoGlutathione-Sepharosebeads andincubated with theindicatedinvitrotranslated, 35Slabelledproteins.Thebeadswerewashed,
specifically bound proteins wereanalysedonSDS-PAGE and visualisedby
Fluorography. Lanes4,7,8, 9, 10,13(input)showtheinvitrolabelledproteins
utilised,luciferasewasadded in theinteraction reactionasspecificitycontrol.
The arrows indicate thepositions of thecorrectly translated proteins: GAL-erb 362-508 (N), luciferase (L), GAL-erb 409-639 (C), GAL-erb362-468/508-639 (A)andGAL-erb 508-639( ).
a
a
: k.i
A A AFj.k.i.-O
IJ -- P, " C',--1.. T T ..., KD0 L)gel retardation experiments and found to be similar for all the mutants (23 and data not shown).
Transfection of an expression vector coding for the GAL4-DBDhad no effect on UAS driven CAT expression in Ltk- cells (Fig. 1). Incontrast, as we have previously shown, expression of a fusion protein containing GAL4-DBD and v-erbA amino acids 346-639 (GAL-verbA) led to 100-fold repression of the basaltranscription rate. N-terminal deletion in the v-erbA domain of 16 a.a. (GAL-erb 362-639) or of43 a.a. (GAL-erb 389-639) did not reduce the silencing function, in fact a slight but reproducible increase of the repression was detected. Deletion of additional 20 a.a. abolished silencing (GAL-erb409-639). Interestingly,inthis mutant prolines 397 and 399 weredeleted, oneofwhich (Pro 399) is exchanged in a natural mutantwhichis inactive (21). It was previously shown that the C-terminal deletion of 7 a.a. (346-632) retains repression
function, whereas further deletion(346-616) severely reduces silencing (23). These results definetheminimalsilencing domain of v-erbA spanning a.a. 389 to 632.
In order to further define structural features involved in transcriptional silencing,weconstructedinternaldeletionmutants
in thev-erbA silencing domain andcomparedtheir repression activity relativetothecompletedomain(Fig. 2). Deletion ofa.a.
435 to 467 (GAL-erb 362-434/468-639) or a.a. 516 to 614
(GAL-erb 346-515/615-639)abolishesrepression. Deletion of a.a. 390to 433 (GAL-erb346-389/434-639) orofa.a. 469 to507(GAL-erb
362-468/508-639)
reducedsilencing
activity toabout6% oftherepressionconferredbythe wildtypeprotein.This residual activity suggests that some functional features
remain intact, which might be explained by the presence of
subdomains.
Silencing subdomains can
synergize
Forfurthercharacterization we
analyzed
silencing
subdomains forpossible functional complementation. Thereforewemadeuseof thedimerization interface of theGAL4-DBD. Expressionof twodifferent GAL4 fusionproteinsinacell leadstoformation
ofheterodimers between thetwochimericproteins, asvisualized ingel retardationexperiments
(Fig. 3a).
WholecellextractsfromCOSI cellstransfected with GAL-erb 362 -508 (see
Fig. 4)
orGAL-erb 362-468/508 -639 expression vectors formed a
Expressionplasmid A A A+B
corresponding DNA-protein complex withan UAS probe(lane
A or B). Co-expression of both chimeric proteins leads to the
formationof anintermediarycomplexconsistingof a heterodimer
(Fig. 3a, lane A+B).
To further prove the formation of heterodimers via the GAL4-DBD, an independent in vitro assay was performed. Fusion proteins with the Glutathione-S-transferase were
bacterially expressed, boundtoGlutathione-Sepharoseand used to precipitate specifically interacting in vitro translated, 35S-labelled proteins (input Fig. 3b, lanes 4, 8, 10, 13). In each experimentin vitrotranslated, 35S-labelled luciferase (L) (input Fig. 3b, lanes 7 and 9) was added as a specificity control. When
GST-GAL-erb 508-639 was coupledtothe beads, interaction could be detected with labelled GAL-erb 508 -639 (lane 14),
GAL-erb362-468/508-639 (lane 11), GAL-erb 409-639 (lane6) and GAL-erb 362-508 (lane 3). When only GST was
used no radioactive band was visible (lanes 2, 5, 12, 15). In addition,luciferase was not retained inany of these experiments, indicating thattheprotein-proteininteractions are specific. The interaction was mediated by the dimerisation function of the GAL4-DBD, asthefusionproteins GST-erb 508-639or GST-erb 362-508didnotinteract with GAL-erb508-639 (lane 16)
or GAL-erb 362 -508 (lane 1), respectively.
Inorderto measure the effect of heterodimers on UAS-driven transcription, we compared the reporter geneactivity after
co-expression oftwofusionproteinstothereference transcription obtained with a single protein. To avoid differences in the
transfection and expressionconditions, we added equalamounts ofGAL4-DBDexpression plasmidinthe reference transfection
as well. This allows a direct comparison of the effects due to
GAL4-DBD/GAL-fusionprotein heterodimers (in thereference
transfection) to those mediated by GAL-fusion A/GAL-fusion
B heterodimers (in the co-expression experiment).
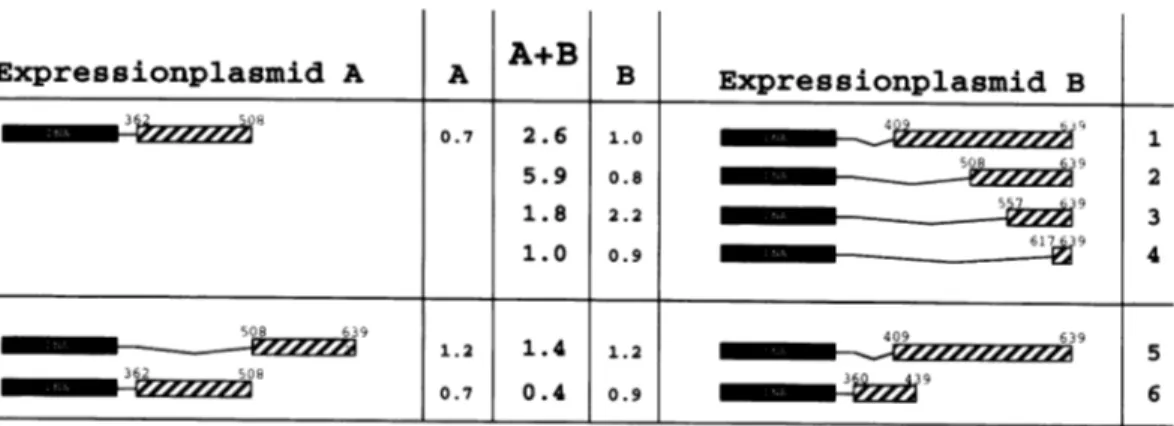
Reference expression of GAL-erb 362-508 or GAL-erb 409-639 is deficient in silencing. Co-expression of both fusionproteinsleadsto a2.6-foldrepressionof CATtranscription (Fig. 4, line 1). Heterodimers between GAL-erb 362-508 and GAL-erb508-639 showedaclear6-foldsynergistic repression
as opposed to the reference transfections (line 2). Further
N-terminal deletion of thesilencing domain in Gal-erb 557 -639 andGal-erb 617-639 resulted in loss of the complementation
B Expressionplasmid B 0.7 2.6 1.o0 1 5.9 0.8
_8
3 2 1.8 2.2 39 1.0 0.9 6739 4 508 639 409 639 1.2 1.4 1.2 5 362 506 ~ mmmmmm*-2zzzzz2z 0.7 0.4 0.9 mm w 6Figure4. Synergism between v-erbAsubdomainsin heterodimers. Expressionvectors coding for the indicated fusion proteins were co-transfected with the UAS-tkCAT reporterplasmid into Ltk- cells either together with a GAL4-DBD expression vector (columns A or B) for reference expression or together with a vector forasecond fusionprotein (column A+B) for synergistic silencing. Numbers indicate fold repression relative to transfection of aGAL4-DBD expression vector withthe UAS-tkCAT reporter (28 i5% CAT conversion).
activity (lines 3 and 4). These data show that thesilencingdomain
ofv-erbA is composed ofat least tworegions which are
non-functional when testedindividually, but which cancomplement eachother evenwhenplaced on twodifferent proteins capable to formheterodimers.
Inorder to ensure that thecomplementation effect is notdue
to ahigher amount of GAL-verbA fusion proteinsincomparison to the reference transfection, we tested combinations of
heterodimeric partners containing identical putative silencing regions. Neither of these combinations led to significant repression (Fig. 4, lines 5+6). In addition, increasing theamount oftransfected GAL-erb 362-508 in the absence of a second expression vector did not result in silencing (data not shown). Furthermore, we were interested to know whether a deletion mutant mediating residual silencing activity could synergize with other subdomains. Therefore we tested GAL-erb 362-468/ 508-639. The repression function of this fusion protein is dramatically increased when co-expressed with different heterodimeric partners (Fig. 5). Both, the N-terminal v-erbA subregion erb 362-508) or the C-terminal region (GAL-erb409-639) fused to theGAL4-DBD synergize efficiently in silencing activity with GAL-erb 362-468/508-639 (Fig. 5). One of these combinations (Fig. 5, line 1) yields near to wild
typeactivity. Another internal deletion mutant with a severe defect insilencing, GAL-erb 362-434/468-639, was unable to com-plement GAL-erb 362-508 (Fig. 5, line 3).
Weconclude that the silencing domain of v-erbA is composed oftwosubdomains(I+11),both of which arenon functional when tested individually, but which can synergize to restore activity
evenwhen placed on two different heterodimeric partners. Sub-domain Iincludes a.a. 389 to 508. Position 389 is determined
Expressionplasmid A A A+B
fromFigure 1, and position 508 from plasmid A in Figure 4,
line 1. Subdomain II containsa.a. 508to 632. Position 508 is suggestedby plasmid B in Figure4, lines 1 and2 andposition 632fromprevious deletions tested in homodimeric GAL fusions (23).
The retinoic acid X receptor (RXR) does not mediate transcriptional silencing
Other members ofthe nuclearreceptorfamilyclosely relatedto
v-erbA, like TR and RAR, show silencing activity in the absence of their specific ligands (6,23). The carboxyterminal domains of TR and RAR are 90 and 30% homologous to v-erbA, respectively. Since thecorrespondingdomain of the retinoidX
receptor(RXR) is27% homologoustothesilencing domain of
RARand sinceRXRisaregular componentof the TR orRAR transcription complex,wewantedtoknow whether RXR confers a silencing function aswell. Since the negativeregulation was morepronounced usingGAL4-DBD fusionproteins, wejoined
theC-terminal part(a.a. 235-462)of RXR to thisheterologous
DNA binding domain. The transcriptional effect on an
UAS-driven tk-CATgeneof this chimericproteinin thepresenceor absence of ligand was tested and compared to the previously
describedregulation mediated byGAL-TR orGAL-verbA (23).
Inordertoavoid interference with endogeneous factors,weused
CVI cells devoid of most of the nuclearreceptors. Expression
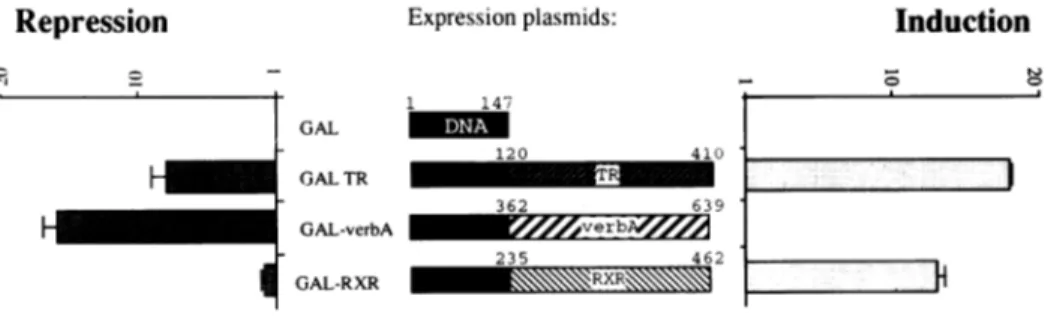
of the GAL4-DBDshowsnoeffect,expressionof GAL-TRleads
to clearrepressioninthe absence ofhormone and clearinduction
in the presence ofhormone (Fig. 6) and GAL-verbA displays
aconstitutive silencing function,asexpected. Incomparisonto
L-cells the CVI cells show less silencing activity. GAL-RXR
mediates induction upon addition of retinoic acid, but no, or a
B Expressionplasmid B
ZozzA
r 6.5 85.5 2.532
0
409 639 29.1 o.9 _ __9M 2 362 434 468 639 362 508 ~M 1.3 2.4 1.7 3Figure 5. Synergismof v-erbA subdomains with internaldeletionmutants. Expressionvectorscodingfor the indicated fusionproteinsweretransfected into Ltk-cellseithertogetherwithaGAL4-DBDexpressionvector(columnsAandB)for referenceexpressionon aUAS-tkCAT reporterplasmidortogetherwitha vector
for another fusionprotein(column A+B)forsynergisticsilencing. ExpressionofUAS-tkCATtogetherwithaGAL4-DBDexpressionvectorwas46 2%CAT conversion.
Repression Expressionplasmids:
1 147 GAL _ 120 GALTR GAL-verbA I GAL-RXR Induction 410 TI 362 639 235 462 RR
Figure6. GAL-RXR mediateshormonalinduction,butnotsilencinginthe absence ofligand.Expressionvectorscodingfor the indicated fusionproteinsweretransfected
into CVIcellstogether withtheindicatorplasmidUAS-tkCAT.FoldinductionofCAT-activityinthe presence of hormone and foldrepressionin the absenceof hormonearecalculated relativetoexpressionofGAL4-DBD.
very low, negative effect inthi induction we used all-trans RA
converted to 9-cis-RA by stereo
These results suggestthat, althi
is functional in hormonedepend
to mediate asignificant repress
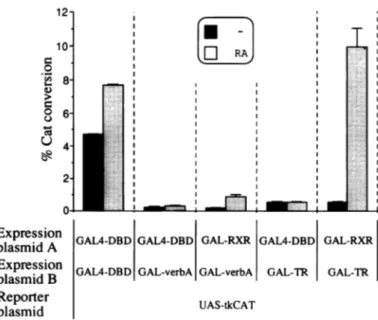
GAL-RXR does not contribul a heterodimer with GAL-TR RXR was shownto form hetero
members of the thyroid hormon we have shown above, it does ni on its own, we wanted to knov to help its heterodimeric partner to exclude effects due to differ
differential dimerization eff heterodimeric complexes, we proteins for this experiment. dimerization is directed by the all the homo- and heterodimers. I
or of GAL-TR along withGALA independent of the addition of GAL4-DBD with thefusion GA silencing mediated by GAL-ver of RA (Fig. 7). Similarly, co-e. to 100-fold relative to GAL-TR did not influence repression int
shown). These results were not since extended assays with lar strongly increased basal level acti RXR effect on repression. Inthe induction expected fromGAL-R, case. The weaker silencing activ relieved. 12-10' 0 ._-4 8' 6' 0 U 4- 2-0* Expression plasmid A Expression plasmid B Reporter plasmid
Figure7. GAL-RXRdoes notsynergize amountsofexpressionplasmids coding
transfectedinto CVI cellstogetherwithtU boxesindicatetheCAT-expressionobta shadedboxes show the CAT-conversior
e absence ofligand. For RXR
(41 which was shown to he
DISCUSSION
Lio
k-rio
n11CVI" cellsY1 I'(>6)
Theresultsin this study describethefunctional silencingdomainough the C-terminusof the RXR ofv-erbA and of the TR/RXR hetero-complex. We utilized the lentgeneinduction, it is unable advantage of fusion proteins consisting of C-terminal parts of
ion. v-erbA joined to the DBD of the heterologous GAL4 yeast
transcription
factor.Expression
ofthedifferent chimericproteins
te to the silencing function in is roughly similar as assessed by gel retardation analysis.
orGAL-verbA Furthermore the GAL4-DBD ensures correct nuclear
dimericcomplexeswithseveral
translocation,
dimerization and DNAbinding
tothespecific
UAS iereceptorfamily. Although, as sequence.otcontaina
repression
function Functionalanalysis
of N-terminal deletion mutants in the v whether RXR would be able v-erbAnegative
domain
revealed the mnalrequirement
of a.a.in its
silencing
activity.
Inorder 389to632 forsilencing
function. Mutants GAL-erb 362-639ential DNA binding
cency
between
affmnity or and GAL-erb 389-639 led to a weak, butreproducible
increase
homo- or in
repression
ascompared
tothecomplete
silencing
domain.Thisused again the GAL4 fusion may suggest the presence of a trans-activation domain around
Teeforgainthe,D AL4bidion
a position 362. Further deletionto a.a. 409 abolished theability
GAL4-DBD
xpreDBDondof
and is identical forei
GLer
bAr
of thefusionprotein to represstranscription. Interestingly this deletion eliminatesPro399,
which had been shownpreviously
EDBDpleadi tofaeatr rLepressio
to beinvolved inrepression,
asits
mutation
toArg
abolished
RA (Fig. 7). Replacement of biological oncogenic and silencing function (21,22). In this context the internal deletion mutant GAL-erb 346-389/
*L-RXR
shows
noeffect onthe
434-639, which has Pro 399 deleted,isimportant. It isstronglyxbA
or GAL-TR in the absence impairedinsilencing function, but, incontrast to deletionmutantxpor
GAL-verbA) of GAL-RXR GAL-erb 409-639, it still retains some negative activity.the
dueabsenetofCT lganss
absenceof ligands (data not Possibly a functionally importantinterruption
of a-helicallimitations structure in the protein isbroughtabout by the two very close
ge amounts of protein extract Pro397 and 399 in the wildtype
protein.
Itcould bepartially
ivitywithoutshowinganyGAL- restored inmutantGAL-erb 346-389/434-639
by
thereplace-presenceofspecific ligandthe ment
with
alinker
sequence,which
wepredict by
Chou-Fasman*R
reduces thesilencingin each andRobson-Garnier analysis
tobecompletely
unstructured andrity ofGAL-TR wascompletely thus
flexible.
Similar
secondary structurepredictions
may allow the following assumptions. Region 360 to 420 is predictedto adopt an a-helical structureinterrupted bythe twoprolines397 and 399 in the wild type v-erbA. The functional mutantGAL-erb389-639retains thisstructurewithashortened N-terminal
helix. Similarly in the hRARa the prolines 170 and 172 are
* ~ T
predicted
toseparate
twohelices,
the N-terminalone ofabout[
i
RA >10
amino acids. Theseobservationssuggest
that
thesilencing
subdomainImight be defined bya structural featureconsisting
oftwo helices
separated
by
a kink.The fact that two of the internal deletion mutants (GAL
: ~~~erb 346-389/434-639 and GAL-erb
362-468/508-639)
retain someoftherepressing functionsuggeststhatthesilencing domain is composed ofatleasttwofunctional domains. Afinal
' proof is given by the co-transfection experiments, generating
heterodimers containing the putative subdomains on different > molecules, presumably in a spatial arrangement different from _2
-_--:
_s s that in theoriginalv-erbA protein. Several results argue for theformation of functional heterodimers from non functional
GAL-RXR GAL4-DBD GAL-RXR components. In vitro interaction assays showed that all tested
GAL4-fusion proteins specifically interact, most importantly GAL-verbA GAL-TR GAL-TR GST-GAL-erb 508-639 and labelled GAL-erb 409-639 which do not restore silencing function when cotransfected (see Fig.
UAS-tkCAT 4, lane 5). The dimerization interface is contributed by the
common GAL4-DBD, as its deletion abolished the interaction
(Fig. 3b, lanes 1 and 16). Gel retardation experiments using withv-erbA or TR insilencing. Equal extracts from COS-1 cells transfected with two different
for theindicated fusionproteinswere
heindicatorplasmidUAS-tkCAT. Dark expresson vectors revealed the presence of heterodimeric linedinthe absence ofhormone, light
complexes.
Simply increasing
the amount of transfected nafter addition ofRA. expression plasmid coding for a single non-functional mutant doesnot lead to repression. Heterodimers formed between
GAL-erb 362/508 and GAL-erb 508-639, which contain perfectly complementary sequences of the silencing domain, showed a
strong synergistic silencing effect. Similarly the repression function of GAL-erb 362-468/508-639, aweakly functional
mutant canbe restoredtonearly wildtypeby co-expressionof
several deletion mutants.
The results obtained using the internal deletion mutants and
theheterodimericcomplexes pointtoregion434-468 asbeing absolutely required for function. It hastobe present inatleast oneof theheterodimericpartners. Deletion ofonlythese amino
acidsinGAL-erb 346-434/468-639 leadstoanon-functional
protein (Fig. 2, line 3), which cannot be complemented by a
fusion protein (GAL-erb 362-508, Fig. 5, line 3) shown to
synergize in other cases (Fig. 4, lines 1 and 2).
Recently, synergismin silencingbetweentwonon-functional
subdomainsof the hTR(3 C-terminalregionwasdescribedusing
GAL4fusionproteinsandareportergenecontainingfour UAS
binding sites (42). In our system, using a single UAS,
co-expression of the corresponding v-erbA fusion proteins
GAL-erb 362-439 and GAL-erb 409-639 led to a weak, but
reproducible synergism as well.
Synergism between transcriptional activating domains is a
general phenomenon. Steroid receptors containtwoormore
trans-activationdomainscooperating forfull activity (43 -45). Incase of the humanestrogen receptor twoactivationfunctions AF 1 and AF2were shownto synergizewhenpresent ondifferent DNA
binding entities (45). RecentlythetranscriptionfactorTEF1was shown to require at least two out of three subdomains for activation function, each of which is inactive when tested
individually in a GALA fusion protein (46). Synergismwithin aheterodimerwasdescribedfor the hormonedependent activation by RXR/RAR complexes (29,30,31,33,39). It was shownthat
heterodimers formed ofonepartner containing the N-terminal AF1 domain and the otherpartnercontaining the C-terminalAF2 domain displayed synergistic activation function (47). This synergism was also seen if one of the partners lacks a
corresponding binding site in the regulatedpromoter. Specific synergistic silencing was described forv-erbA or TR with the
protein Nepl binding close to a thyroid hormone responsive
element in the chickenlysozymesilencer -2.4 kb(11,48). Here we describe a synergism between two silencer subdomains.
The mechanism for this synergism is still unclear. Direct interactionofhTRa 1(49)and the C-terminus of hTR(3 (42)with TFIIBhas been demonstrated. This interaction is relieved in the
presence of T3 (42), suggesting that this interaction may be involved in thesilencingactivity. TheregionofhTR(3 interacting
with TFIIB corresponds roughly tothe synergising subdomain A.A. 508 -639 of v-erbA. Whether thecomplementarydomain 389-508 interacts withsomeother componentofthe
transcrip-tion pre-initiation complex remains to be shown.
V-erbA and ligand-free TR and RAR are able to mediate
silencing. RAR and TR have been shown to function in a
heterodimericcomplex with RXR. In addition RXR is closely
relatedtoTRand RAR. Thereforewewantedtoknow whether
RXRexhibitsasilencingfunctionof itsownand/or whetherRXR
synergizesinrepressionwithone orboth of thesilencingdomains of v-erbA inahetero-complex. TheC-terminalfragmentofRXR, corresponding tothe minimal v-erbAsilencing domain, is able tomediate the RAinducibilityinthechimericprotein GAL-RXR,
but does not show any silencing activity. Usually we observe
thenthe natural proteinsontheirspecific binding site (23). We therefore concludethat the hRXRa doesnotcontainatransferable silencingfunction. Inaddition, we could show that GAL-RXR
inheterodimers with GAL-TR orwith GAL-verbA, respectively,
does notinfluence theirrepression activityintheabsenceofany ligand. Only addition of RA leads to an increase of CAT transcription, due to theligand-induced GAL-RXR.
MATERIALS AND METHODS
Plasmids
The reporterplasmid usedinthisstudycontainsaGAL4 binding site (17mer)(50) inserted in front of the tkCAT gene (23).
The expression vectors pABGAL, pAB-GAL, pGAL-erbA A346, pGAL-erb 362-639, pGAL-erb 409-639, pGAL-erb
508-639, pGAL-erb 557-639, erb 617 -639, pGAL-erb 362-508, pGAL-erb 360-439, pGAL-erb 362-468/ 508-639, pGAL-erb 362-434/468-639, and pGAL-TRwere
described elsewhere (23). Plasmid pGAL-erb 389-639 was
obtainedby cutting pGAL-verbA withSmiaI andBclI, filling in usingthe Klenowfragment of DNA-polymerase I and religating,
thereby deleting a fragment coding for a.a. 346 to 388. The
internal deletion mutant pGAL-erb346-389/434-639 was
constructedby cutting pGAL-verbA withEcoRV andBcII,filling
in using Klenow enzyme and religating, thereby deleting a fragment coding for a.a. 390 to 433. Plasmid pGAL-erb
346-515/615-639 wasobtainedbydigestingpGAL-verbAwith EspIandFspIfollowedbyreligation,thereby deleting a fragment
coding for a.a. 516to 614. pSG-hRXR containing the coding sequence for the human RXRa was kindly provided by H.Stunnenberg (33). The RXR coding region was first cut out
usingEcoRI, filled in usingKlenow enzyme andcloned into the
Eco47III site of pABGAL. Then a
Hincll-HindJI
fragmentcoding for a.a. 235 to 462 was obtained and inserted into the
SmaI-HindI
cut pABGAL to obtain plasmid pGAL-RXR235-462.
The expression plasmids GST-erb 362-508 and GST-erb
508-639 were constructed by insertion of the
SmaIlHindIl
fragment of pGAL-erb 362-508 or pGAL-erb 508 -639,respectively, into the vector pGEX KG (51) digested with
SmaIlHindllI.
The expressionplasmid GST-GAL-erb 508-639 was obtained
by insertion of the
BglHIBamHI
fragment of pGAL-erb508-639, wichwasfilledinusing Klenow,intothevector pGEX
2TK (Pharmacia), digested withEcoRI and filled in.
Vectors for in vitro translation were obtained by insertion of theKpnIlBamHIfragments of pGAL-erb 409-639,pGAL-erb
508 -639 and pGAL-erb 362-468/508-639 into the vector
BluescriptSK+, digested with KpnIlBamHI, and insertion of theKpnIlHindIH fragment of pGAL-erb 362-508 into Bluescript SK+, digested with KpnIlHindIH.
Cellculture and transfections
Ltk- cells, CVI and COSI cells were grown in DME-medium
(Gibco) supplemented with 10% fetal calf serum, 100 U/ml penicillin and 100
pjg/ml
streptomycin.DNAtransfer into CVI cellswasperformed usingthecalcium
phosphateprecipitation method. Ltk- cells weretransfectedas
described(52). 2x106cellswere suspendedinDNA -DEAE-dextran solution (1 pmol reporter and 0.5 pmol expression plasmids) and incubatedfor 30min. Afteraddingdirectly 7ml
medium,cellswereseededon a6cmdish and grown for 36-48 that GAIA fusion
proteins
showhigher silencing
effectson aUAShbefore harvesting. Transfections were done in triplicate and
performed inatleasttwoindependent experiments.Transfections
into COSI cellsweredone by asimilarDEAE -dextran
suspen-sion method using 6 pmole of DNA on 107 cells. After 1 h
incubation inthe DNAsolution, a DMSO shock was performed
for 3 min, the cells were taken up in 30 ml PBS and 10 ml
medium, spundown, seeded on a 15 cm dish and grown for 48
h before harvesting.
Forhormonalinductionexperiments,the serum was depleted
of thyroid hormone and retinoic acid by extensive charcoal
stripping. The cellswerekept foratleast 24hindepleted medium beforetransfection, after transfection l0-7M 3,5,3'-trijodthy-ronine or 10-6M retinoic acid was added. CAT-assays were
performed as described (53). DNA-protein binding assays
Whole cellextracts wereprepared from COSI cells transfected with various expressionvectorsaccordingto(54). Gel retardation experiments were performed using 20000 d.p.m. of kinase-labelled 17-mer DNA probe, 2-4
/g
of whole cell extract inanincubationmixcontaining 1-4,ugofp(dI.dC), 200 mMKCI,
2 mM DTT, 14% glycerol, 5 mM MgCl2 20 mM Hepes pH
7.7. TheDNA-protein complexes formedwereanalysedon a
5% polyacrylamide gel in 0.5xTBE.
Protein-protein interactions in vitro
Protein-proteininteractions wereassayedmainlyasdescribed
(42).
GST-fusion-proteins wereexpressedin E.coliBL21 cells(55).
Cellswereharvestedbycentrifugationandresuspendedin NENT
buffer (100mMNaCl, 1 mM EDTA, 20mMTrispH8.0and 0.5% NP-40). Cellswerelysedbythree freeze-thawcyclesand
the cellular debris was removed by centrifugation.
Glutathione-Sepharose4Bbeadswerewashed withNENTand
10,ulof beads wereincubated with 100 ,ul oflysatecontaining
the GST-fusion protein for 30 min at room temperature. Subsequently,thesupernatantwasremovedand the beadswere
incubatedwith 10% milkpowderin NENTfor 15 minat room
temperature. The beads were washed twice with 1 ml NENT andoncewith 1 mltranscription washingbuffer(20mMHepes pH 7.9, 60 mM NaCl, 1 mM DTT, 6 mM MgCl2, 8%
glycerine and 0.1 mM EDTA). In vitro translated and radiolabeledproteinwereobtainedusingaTNT-kit(Promega).
5
yd
ofcrude lysate were incubated with the beads in 100Al
transcription washingbufferfor 1 hat roomtemperature.Finally
the beads were washed (5x1 mlofNENT) andproteins were
solubilizedinSDS-loadingbuffer andanalyzedonSDS -PAGE. Gelswereamplified with fluorigraphicreagent(Amersham)and bands were visualised by autoradiography.
ACKNOWLEDGEMENTS
Special thanks toH.Stunnenberg for kindly providing us pSG-RXR. Inaddition,wewouldliketothankK.Kriigerforexcellent
technical assistanceand artwork andL.Schafer-Pfeifferfor help in cell culture. We are also grateful to J.Michel for critically
readingthemanuscript.This work was supported by the Deutsche
Forschungsgemeinschaft (SFB249), by the BoehringerIngelheim Fonds (given to B.M.) and by the Fonds der Chemischen
Industrie.
REFERENCES
1. Umesono,K., Murakami,K.K., Thompson,C.C.andEvans,R.M. (1991)Cell,
65, 1255-1266.
2. Naar,A.M., Boutin,J.M., Lipkin,S.M., Victor,C.Y., Holloway,J.M., Glass,C.K. and Rosenfeld,M.G. (1991) Cell, 65, 1267-1279. 3. Nagpal,S., Sanders,M., Kastner,P., Durand,B., Nakshatri,H. and
Chambon,P. (1992) Cell, 70, 1007-1019. 4. Evans,R.M. (1988)Science, 240,889-895.
5. Nunez,E.A. (1989) Current opinion in Cell Biology, 1, 177-185. 6. Damm,K., Thompson,C. and Evans,R.M. (1989) Nature, 339,593-597. 7. Chatterjee,V.K.K., Lee,J.K., Rentoumis,A. and Jameson,J.L. (1989)
Proc. Natl Acad. Sci.USA, 86, 9114-9118.
8. Crone,D.E., Kim,H.S. and Spindler,S.R. (1990) J.Biol.Chem., 265,
10851 -10856.
9. Thompson,K.L., Santon,J.B., Shepard,L.B., Walton,G.M. andGill,G.N.
(1992) Mol.Endocrinol., 6, 627-635.
10. Saatcioglu,F., Deng,T. and Karin,M. (1993)Cell, 75, 1095-1105. 11. Baniahmad,A., Steiner,C., Kohne,A.C.andRenkawitz,R. (1990) Cell,61,
505-514.
12. Sap,J.,Munoz,A., Schmitt,J., Stunnenberg,H. and Vennstrom,B. (1989) Nature, 340, 242 -244.
13. Baniahmad,A., Tsai,S.Y.,O'Malley,B.W. and Tsai,M.J. (1992) Proc.Natl Acad.Sci., 89, 10633-10637.
14. Munoz,K.A., Zenke,M.,Gehring,U., Sap,J., Beug,H. and Vennstrom,B. (1988) EMBO J., 7, 155-159.
15. Sharif,M. andPrivalsky,M.L. (1991) Cell, 66,885-893.
16. Zenke,M., Kahn,P., Disela,C., Vennstrom,B., Leutz,A., Keegan,K., Hayman,M.J.,Choi,H.R., Yew,N., Engel,J.D. andBeug,H.(1988) Cell, 52, 107-119.
17. Gandrillon,O., Jurdic,P., Pain,B.,Desbois,C., Madjar,J.J.,Moscovici,M.G., Moscovici,C. and Samarut,J. (1989) Cell, 58, 115-121.
18. Zenke,M., Munoz,A., Sap,J.,Vennstrom,B.andBeug,H. (1990)Cell,61, 1035-1049.
19. Boucher,P. andPrivalsky,M.L. (1990) Oncogene, 5, 1303-1311. 20. Desbois,C., Aubert,D., Legrand,C., Pain,B.andSamarut,J.A. (1991)Cell,
67, 731-740.
21. Damm,K., Beug,H., Graf,T. and Vennstrom,B. (1987) EMBOJ., 6, 375-382.
22. Damm,K. and Evans,R.M. (1993) Proc.Natl Acad.Sci.USA, 90, 10668-10672.
23. Baniahmad,A., Kohne,A.C. and Renkawitz,R. (1992) EMBO J., 11,
1015-1023.
24. Renkawitz,R. (1990) Trends inGenetics, 6, 192-197. 25. Jackson,M.E. (1991)J.CellSci., 100, 1-7.
26. Schiule,R.,Rangarajan,P., Yang,N., Kliewer,S., Ransone,L.J.,Bolado,J., Vrema,I. andEvans,R.M.(1991)Proc.NatlAcad.Sci.USA,88,6092-6096. 27. Shamol,S.,Brickman,J.M.,Lehming,N. andPtashne,M.(1993) Nature,36,
648-652.
28. Yu,V.C., Deisert,C., Andersen,B., Holloway,J.M., Devary,O.V., Nar,A.M.,Kim,S.Y.,Boutin,J.M., Glass,C.K. and Rosenfeld,M.G. (1991)
Cell, 67, 1251-1266.
29. Zhang,X.K.,Hoffmann,B., Tran,P.B.V., Graupner,G.andPfahl,M.(1992)
Nature, 355,441-446.
30. Leid,M., Kastner,P., Lyons,R., Nakshatri,H., Saunders,M.,
Zachareewski,T., Chen,J.Y., Staub,A., Garnier,J.M., Mader,S. and
Chambon,P. (1992) Cell, 68,377-395.
31. Kliewer,S.A., Umesono,K., Mangelsdorf,D.J. and Evans,R.M. (1992) Nature, 355,446-449.
32. Hallenbeck,P.L., Marks,M.S., Lippoldt,R.E.,Ozato,K. andNikodem,V.M.
(1992)Proc.Natl Acad. Sci.USA, 89, 5572-5576.
33. Bugge,T.H., Pohl,J., Lonnoy,O. and Stunnenberg,H.G. (1992) EMBO J.,
11, 1409-1418.
34. Green,S. (1993)Nature,361, 590-591.
35. Heyman,R.A., Mangelsdorf,D.J., Dyck,R., Stein,R., Eichele,G., Evans,R.M. andThaller,C. (1992) Cell, 68, 397-406.
36. Levin,A.A.,Sturzenbecker,L.J., Kazmer,S., Bosakowski,T.,Huselton,C.,
Allenby,G., Speck,J., Kratzeisen,C.L., Rosenberger,M., Lovey,A. and
Grippo,J.F. (1992)Nature,355,359-361.
37. Lazar,M.A.,Berrodin,T.J. and Harding,H.P. (1991)Mol.CellBiol., 11, 5005-5015.
38. Yen,P.M., Sugawara,A. and Chin,W.W. (1992) J.Biol.Chem., 267, 23248-23252.
39. Durand,B., Saunders,M., Leroy,P., Leid,M. and Chambon,P. (1992) Cell,
40. Lee,I.J.,Driggers,P.H., Medin,J.A., Nikodem,V.M. and Ozato,K. (1994) Pro.NatlAcad.Sci.USA, 91, 1647-165 1.
41. Mangelsdorf,D.J., Ong,E.S., Dyck,J.A. and Evans,R.M. (1990)Nature, 345, 224-229.
42. Baniahmad,A., Ha,I., Reinberg,D., Tsai,S., Tsai,M.-J. and O'Malley,B.W. (1993) Proc.Natl Acad. Sci.USA, 90, 8832-8836.
43. Gronemeyer,H., Turcotte,B., Quirin-Stricker,C., Bocquel,M.T.,
Meyer,M.E.,Krozowski,Z.,Jeltsch,J.M., Lerouge,T., Garmier,J.M. and Chambon,P. (1987) EMBO J., 6, 3985-3994.
44. Hollenberg,S.M. and Evans,R.M. (1988) Cell, 55, 899-906.
45. Tora,L., White,J., Brou,C., Tasset,D., Webster,N., Scheer,E. and Chambon,P. (1989) Cell, 59,477-487.
46. Hwang,J.J.,Chambon,P. and Davidson,I. (1993) EMBO J.,12,2337-2348. 47. Nagpal,S., Friant,S.,Nakshatri,H. and Chambon,P. (1993) EMBO J.,12,
2349-2360.
48. Kohne,A.C., Baniahmad,A. and Renkawitz,R. (1993) J.Mol.Biol., 232,
747-755.
49. Fondell,J.D.,Roy,A.L. andRoeder,R.G.(1993) Genes Dev., 7, 1400-1410. 50. Carey,M., Kakidani,H., Leatherwood,J., Mostashari,F. and Ptashne,M.
(1989) J.Mol.Biol., 209,423 -432.
51. Dixon,J.E. and Guan,L.G. (1991) Anal. Biochem., 192, 262-267. 52. Choi,O-R B. andEngel,J.D. (1988)Cell, 55, 17-26.
53. Gorman,C.M., Moffat,L.F. and Howard,B.H. (1982) Mol. Cell Biol., 2, 1044-1051.
54. Kumar,V. andChambon,P. (1988)Cell, 55, 145-156.
55. Studier,F.W., Rosenberg,A.H., Dunn,J.J. andDubendorff,J.W.(1990)Meth.