Mémoire présenté en vue d’obtenir le diplôme d’Ingénieur CNAM Spécialité : Mesure-Analyse
Option : Sciences et techniques analytiques appliquées à la chimie et au vivant
par
Hélène Malandain
Etude physico-chimique et analytique de l’absorption de
molécules extractantes par des polymères
(méthode SBSE) et expertise par technique LC-MS
Soutenu le : 22 novembre 2012
Jury : Christine Pernelle
Christophe Moulin
François Giraud
David Benanou
Jean-Sébastien Garrigue
Je tiens par ces quelques lignes à exprimer toute ma reconnaissance à Monsieur Christophe Moulin, chef du Service Radioanalyse Chimie Environnement à la Direction des Applications Militaires du Commissariat à l’’Energie Atomique de Bruyère-le-châtel, et Professeur de la chaire de Génie Analytique au Conservatoire National des Arts et Métiers pour ses cours, pour la confiance qu’il m’a accordée en me faisant intégrer son équipe et surtout pour son encadrement et ses conseils. Je remercie très sincèrement Monsieur Xavier Machuron-Mandard, responsable du Laboratoire de Chimie Analytique pour m’avoir guidée dans mes travaux, pour son enthousiasme, ses encouragements, sa confiance au cours du stage et pour son aide précieuse lors de la rédaction de ce mémoire.
Un grand merci également à toute l’équipe du Laboratoire de Chimie Analytique, Françoise Leprince pour ses conseils et sa bonne humeur inaltérable, Fréderic pour nos grands débats chimiques et gastronomiques, Cécile collègue de bureau et de chaine HPLC mais aussi Olivier, Gaël, Françoise Zielinski et les autres. Ce fut un véritable plaisir de partager ces neufs mois à vos côtés, puis mettre au monde ce mémoire.
Je souhaite également dire un grand merci à l’équipe enseignante de la chaire de Génie Analytique, Madame Pernelle, Monsieur Boutin, Madame Agbo, Monsieur Evers, Monsieur Remita et les autres pour leurs enseignements très enrichissants.
Je remercie Novagli Pharma pour m’avoir encouragée dans ma démarche et pour l’organisation de ce stage, ainsi que le Fongecif pour le financement de ces 9 mois.
Mais surtout, que serait le CNAM sans les « Cnameuses », Sylvie, Pascale, Stéphanie. Avec vous l’adage de « l’union fait la force » a pris tout son relief lors de nos journées de révision, et nos soirées de décompression.Jje vous remercie pour ces excellents moments partagés et sais qu’il y en aura bien d’autres.
Merci à ma famille pour son soutien dès le moment où l’idée de me lancer dans l’aventure du Cnam a germé.
Et surtout merci à celui qui m’a soutenue, encouragée et supportée au quotidien avec énormément de patience pendant tout ce parcours. Ce n’était pas facile, tu as été formidable.
PREMIER E PARTIE: INTRODUCTION GENERALE... 1 DEUXIEME PARTIE : PRINCIPES THEORIQUES ET INSTRUMENTATION ... 6 I. PRINCIPES GENERAUX ET INSTRUMENTATIONS SPECIFIQUES POUR LA PREPARATION D’ECHANTILLONS AQUEUX... 7 I.1 Principes généraux de l’extraction liquide/liquide ...8 I.2 Principe de l’extraction sur phase solide ... 10 I.2.1 Principe instrumental de la micro‐extraction sur phase solide ... 11 I.2.2 Principe instrumental de la micro‐extraction sur barreau aimanté... 11
II. THEORIE DE L’EXTRACTION APPLIQUEE AU D2EHPA ... 13
II.1 Analyse thermodynamique de l’extraction du D2EHPA par SBSE ... 13 II.1.1 Constantes d’équilibres thermodynamiques... 14 II.1.2 Influence de la salinité des milieux sur les équilibres thermodynamiques : relation entre activés, concentrations et force ionique ... 17 II.1.3 Modélisation de l’absorption du D2EHPA par du PDMS... 20 II.1.4 Modélisation de la désorption du D2EHPA en milieu aqueux, à partir du PDMS ... 27 II.1.5 Désorption du D2EHPA en milieu organique ... 32 II.2 Considération cinétique ... 35 II.2.1 Schématisation du phénomène cinétique ... 36 II.2.2 Modélisation mathématique ... 37 II.2.3 Simplification de la modélisation cinétique... 38 TROISIEME PARTIE : ETUDE EXPERIMENTALE. ... 42 I. DEVELOPPEMENT ANALYTIQUE... 43 I.1 Détermination des paramètres instrumentaux... 45 I.1.1 Etude des conditions préférentielles d’acquisition spectrométrique du D2EHPA... 45 I.1.2 Etude des conditions de séparation chromatographique... 55 I.1.3 Courbe d’étalonnage de la méthode de dosage du D2EHPA... 61 I.2 Détermination des limites de détection et de quantification... 66 I.2.1 Détermination mathématique des limites de détection et de quantification ... 66 I.2.2 Détermination expérimentale de la limite de quantification ... 67
II. VALIDATION EXPERIMENTALE DU MODELE D’EXTRACTION... 69
II.1 Etude expérimentale de la cinétique... 70 II.1.1 Considérations préliminaires et cinétiques générales ... 70 II.1.2 Etude cinétique préliminaire de l’extraction du TBP par twister... 74 II.1.3 Etude de la cinétique d’extraction du D2EHPA par twister ... 82 II.2 Etude thermodynamique ... 84 III. INTERPRETATION... 86
IV.1.1 Conditions de pH optimales d’absorption en fonction de la concentration ... 91 IV.1.2 Concentration minimum extractible à 95% ... 93 IV.2 Conditions et méthodes de désorption pour l’analyse chromatographique ou directe du D2EHPA prélevé par du PDMS ... 95 IV.2.1 Désorption en phase aqueuse ... 95 IV.2.2 Désorption en phase organique ... 97 IV.2.3 Désorption par source d’ionisation ambiante directement couplée à un analyseur... 98 QUATRIEME PARTIE :...101 CONCLUSION 101 CINQUIEME PARTIE : ANNEXES ...104
ANNEXE I : DONNEES PARTICULIERES ET PROPRIETES PHYSICOCHIMIQUES DU D2EHPA... 105
ANNEXE II : ANALYSE MATHEMATIQUE DES PROCESSUS D’EXTRACTION... 106
ANNEXE III : CHROMATOGRAPHIE LIQUIDE HAUTE PERFORMANCE COUPLEE A LA SPECTROMETRIE DE MASSE ... 126
ANNEXE IV : PRINCIPE DE LA SPECTROMETRIE DE MASSE HAUTE RESOLUTION... 127
ANNEXE V : METHODE D’ANALYSE DU D2EHPA PAR LC/MS... 128
ANNEXE VI : DONNEES BRUTES DE LA COURBE D’ETALONNAGE DE L’ANALYSEUR (LC/MS) ... 129
ANNEXE VII : LIMITES DE DETECTION ET DE QUANTIFICATION POUR LE DOSAGE DU D2EHPA PAR LC/MS... 130
Limite de quantification... 132
TABLE DES ILLUSTRATIONS
Equations :
Équation 1 : Coefficient de partage ...9
Équation 2 : Rapport de distribution ...9
Équation 3 : Coefficient de partage : Equation générale ...14
Équation 4 : Coefficient de partage du D2EHPA entre PDMS et Eau ...15
Équation 5 : Constante de dissociation...16
Équation 6 : Constante de dimérisation...17
Équation 7 : Relation entre activité et concentration ...18
Équation 8 : Expression du logarithme du coefficient d’activité ...18
Équation 10 : Constante de partage en fonction des concentrations...19
Équation 11 : Constante de dissociation en fonction des concentrations et des activités...19
Équation 12 : Constante de dimérisation en fonction des concentrations ...19
Équation 13 : Expression du logarithme du coefficient d’activité à 25°C d’A-...20
Équation 14 : Expression du coefficient de distribution d’absorption...21
Équation 15 : Expression du coefficient de distribution pour l’absorption en fonction de [HA]eq...21
Équation 16 : Expression de [HA]eq en fonction de C0...22
Équation 17 : Expression du coefficient de distribution pour l’absorption en fonction de C0...22
Équation 19 : Expression du rendement d’extraction en fonction de C0...23
Équation 20 : pH minimum permettant une extraction massique de 95% en fonction de C0...26
Équation 22 : Coefficient de distribution dans le cas de la désorption en phase aqueuse...28
Équation 23 : Coefficient de distribution pour la désorption en phase aqueuse en fonction de CPDMS0...29
Équation 24 : Rendement de désorption massique en fonction de CPDMS0...29
Équation 25 : pH permettant une désorption massique de 95% en fonction de C0...31
Équation 27 : Rapport de distribution pour la désorption du D2EHPA du PDMS vers l’hexane ...33
Équation 28 : Valeur du rapport de D’Hexane...33
Équation 29 : Conservation de la matière pour la désorption dans l’hexane...34
Équation 30 : n’hexane exprimé en fonction du volume de solvant de désorption (hexane) ...34
Équation 31 : Première loi de Fick...37
Équation 32 : Intégrale de la cinétique d’absorption exprimant la diminution de la quantité de matière dans l’échantillon ...39
Équation 33 : Equation de la cinétique d’absorption exprimant la diminution de la quantité de matière dans l’échantillon ...39
Équation 34 : Equation de la cinétique d’absorption exprimant l’augmentation de la quantité de matière dans le PDMS ...39
Équation 35 : Expression de limite de détection et quantification en fonction de l’écart-type (sa) sur l’ordonnée à l’origine de la régression...67
Équation 38 : Signal détecteur en fonction du temps de contact du PDMS et de la solution ...72
Équation 39 : Expression du rendement d’extraction en fonction de C0...87
Équation 40 : pH minimum permettant une extraction massique de 95% en fonction de C0...91
Équation 41 : Concentration minimum permettant une extraction de 95%...92
Réactions
Réaction 1 : Partage entre PDMS et milieu aqueux ...14Réaction 2: Réaction de dissociation ...16
Réaction 3: Réaction de dimérisation ...16
Figures
Figure 1 : Extraction liquide/liquide d’un composé acido-basique A. ...9
Figure 2 : Structure du polydimétylsiloxane...10
Figure 3 : Dispositif de fibre SPME [4] ...11
Figure 4 : Barreau de microextraction Gerstel® ...12
Figure 5 : Comparaison des rendements d’extraction par barreau SBSE et fibre SPME pour des analytes de polarité variable exprimée par la constante de partage octanol/eau (Ko/w)[14] ...12
Figure 6 : Formule développée de la molécule de bis (2-ethylhexyl) phosphate ...13
Figure 7 : Coefficient d’activité de A- en fonction de la force ionique I ...20
Figure 8 : Rendement d’extraction massique pour différents γA avec C0 =1 nM -et β=83,3...24
Figure 9 : Rendement d’extraction massique pour différentes C0 avec γA=1-et β=83,3 ...24
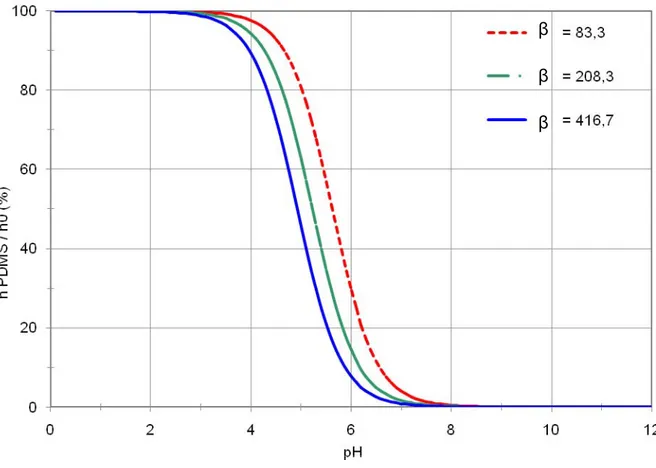
Figure 10 : Rendement d’absorption pour différentes valeurs de β avec C0=1 nM et γA-=1...26
Figure 11 : Partage d’un composé acido-basique lors de sa désorption entre une phase aqueuse et organique ...28
Figure 12 : % Désorption Eau = f (pH) pour D2EHPA Variation de la CPDMS0...30
Figure 13 : % Désorption Eau = f (pH) pour D2EHPA Variation de β (CPDMS 0 = 75 µM) ...31
Figure 14 : Partage d’un composé lors de sa désorption depuis le PDMS vers l’hexane ...33
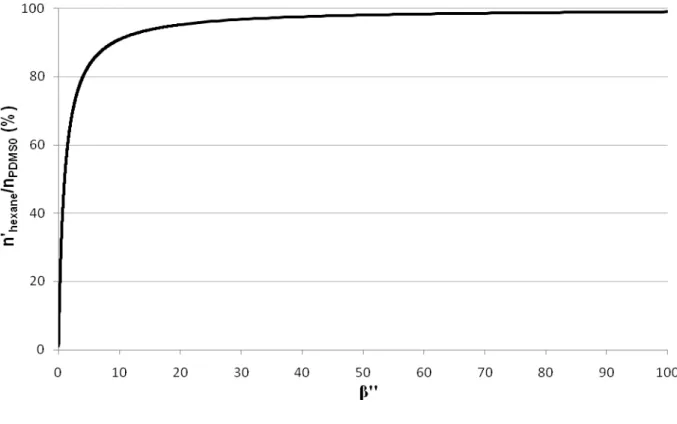
Figure 15 : Rendement de désorption massique dans l’hexane en fonction de β’’ ...35
Figure 16 : Illustration des couches de diffusion (δ) et de la zone de convection à l’interface liquide-solide ...36
Figure 17 : Modélisation de l’évolution théorique de la quantité de matière en fonction du temps...40
Figure 18 : Processus d’ionisation du D2EHPA ...46
Figure 19 : Schéma de principe de l’ionisation par electrospray [27]...47
Figure 20 : Schéma de principe de l’ionisation chimique à pression atmosphérique [28] ...48
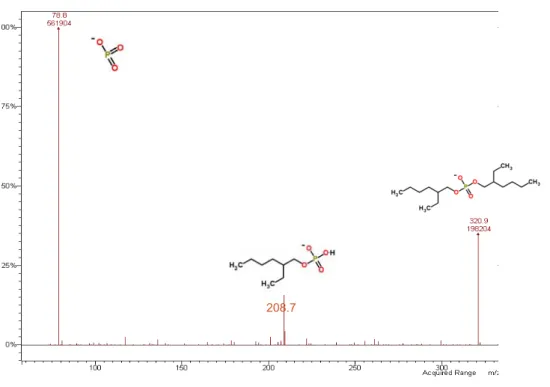
Figure 21 : Spectres de masse (ESI - ) du D2EHPA...49
Figure 22 : Spectre de fragmentation de l’ion m/z=321...50
Figure 23 : Spectre de masse du D2EHPA ionisation ESI en spectrométrie haute résolution ...52
Figure 24 : Spectre de masse (APCI ) du D2EHPA...53
Figure 25 : Comparaison de sensibilité entre MS et MS² ...54
Figure 26 : Chromatogramme du D2EHPA dans le méthanol et la phase mobile ...56
Figure 27 : Volume d’occupation d’une injection de 100 µL dans une colonne (150x2mm) en considérant une porosité de 50% ...56
Figure 28 : Elution du D2EHPA sur colonne C18 (250x4,6 mm) pour différentes conditions chromatographiques...57
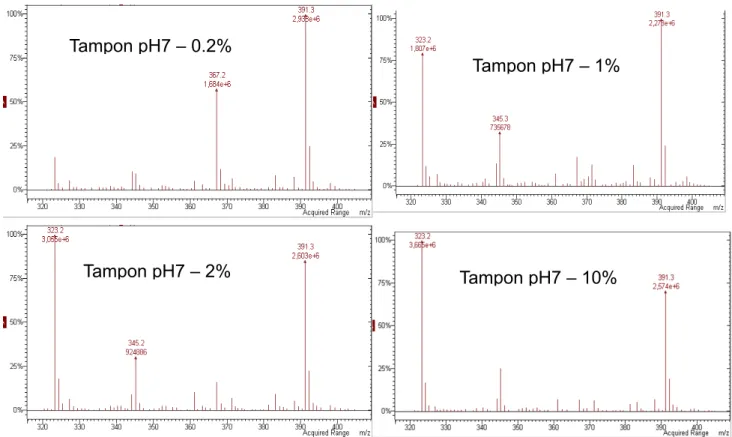
Figure 29 : Spectre de masse du D2EHPA + tampon phosphate après séparation chromatographique...59
Figure 30 : Impact de la concentration en tampon phosphate dans l’échantillon sur l’intensité de la réponse à concentration de D2EHPA constante. ...60
Figure 31 : Courbe d’étalonnage curviligne prouvée par la répartition non aléatoire des résidus...61
Figure 32 : Courbe d’étalonnage linéaire pour une injection dans le flux prouvée par la répartition aléatoire des résidus...62
Figure 33 : Comparaison des calibrations effectuées avec des capillaires en PEEK et en métal ...63
Figure 36 : Calibration du système par une régression linéaire sur le domaine de 7 à 55 µg/L ...66
Figure 37 : Mesure graphique du rapport signal sur bruit d’une solution de D2EHPA à 6,9 µg/L pour confirmation de la valeur de la limite de quantification ...68
Figure 38 : Configuration expérimentale pour l’étude de la cinétique d’extraction ...71
Figure 39 : Influence de la vitesse de rotation du twister sur la cinétique d’absorption du TBP...75
Figure 40 : Quantité de composé absorbé en fonction du temps avec variation du milieu de l’échantillon ...76
Figure 41 : Quantité de composé absorbé en fonction du temps avec variation du volume de l’échantillon...78
Figure 42 : Quantité de composé absorbé en fonction du temps avec variation de la température de l’échantillon ...80
Figure 43 : Cinétique d’absorption du D2EHPA sur le PDMS ...83
Figure 44 : Rendement expérimental d’extraction du D2EHPA en fonction du pH ...85
Figure 45 : Rendement d’extraction en fonction du pH avec Kp = 102,3(trait plein) , avec Kp=10 6,07( pointillés) et points expérimentaux (●) ...88
Figure 46 : pH maximum permettant une extraction de 95% en fonction de la concentration initiale pour différents rapports de volume de phase β...92
Figure 47 : Concentration minimum permettant un rendement d’extraction massique de 95% en fonction de β ...93
Figure 48 : Rendement de désorption massique en fonction du pH pour différentes valeurs de concentration initiale dans le PDMS en tenant compte de la constante de partage apparente du D2EHPA pour β’=83...96
Figure 49 : Rendement de désorption massique en fonction du pH pour différentes valeurs de β’ et en tenant compte de la constante de partage apparente du D2EHPA pour CPDMS0=75 µM ...96
Figure 50 : Vue en coupe d’une source DART (direct analysis in real time) [29] ...98
Figure 51 : Source DART (direct analysis in real time) ...99
Figure 1 : Spectre de masse haute résolution obtenu en plaçant un twister ayant absorbé du D2EHPA dans le flux d’une source DART...98
Figure 53 : Système LC-MS sur lequel les travaux ont été réalisés...126
Figure 54 : Schéma de principe de la spectrométrie de masse haute résolution [32] ...127
Tableaux
Tableau 1 : Recouvrement de la calibration en fin de manipulation et de la calibration initiale de référence...65Tableau 2 : Comparaison des principales caractéristiques entre le TBP et le D2EHPA ...70
Tableau 3 : Paramètres du modèle cinétique pour l’extraction du TBP en fonction de la vitesse de rotation du twister 75 Tableau 4 : Paramètres du modèle cinétique pour l’extraction du TBP en fonction de la nature du milieu...77
Tableau 5 : Paramètres du modèle cinétique pour l’extraction du TBP en fonction du volume de l’échantillon...78
Tableau 6 : Paramètres du modèle cinétique pour l’extraction du TBP en fonction de la température de l’échantillon.80 Tableau 7 : Paramètres physicochimiques et constantes de la littérature appliqués pour le tracé des courbes théoriques d’extraction ...87
LISTE DES NOTATIONS ET ABREVIATIONS
SBSE : Stir bar sorptive extraction
D2EHPA : Bis (2-ethylhexyl) phosphate
TBP : Tributylphosphate
HA: D2EHPA en phase aqueuse
HA : D2EHPA en phase organique
−
A : D2EHPA sous forme basique en phase aqueuse
2
2A
H : D2EHPA sous forme dimère en phase organique
eq
HA ]
[ : Concentration du D2EHPA forme acide à l’équilibre en phase aqueuse
eq
A ]
[ − : Concentration du D
2EHPA forme basique à l’équilibre en phase aqueuse
eq
HA]
[ : Concentration du D2EHPA forme monomère à l’équilibre en phase organique
eq
A
H ]
[ 2 2 : Concentration du D2EHPA forme dimère à l’équilibre en phase organique
P
K : Constante de partage du D2EHPA entre Eau et PDMS
P
K ' : Constante de partage du D2EHPA entre Hexane et PDMS
Di
K : Constante de dimérisation du D2EHPA dans le PDMS
A
K : Constante de dissociation du D2EHPA dans l’eau
A
pK : Inverse du logarithme de la constante de dissociation du D2EHPA dans l’eau
D : Rapport de distribution pour l’absorption
0
n : Quantité de matière initiale dans l’échantillon
PDMS
n : Quantité de matière absorbée par le PDMS après l’étape d’absorption
sol
n : Quantité de matière restant dans la matrice après l’étape d’absorption
0
C : Concentration initiale dans l’échantillon
sol
C : Concentration dans la matrice après l’étape d’absorption
PDMS
C : Concentration dans le PDMS après l’étape d’absorption
sol
V : Volume de l’échantillon
PDMS
V : Volume de PMDS
β : Volume de l’échantillon / Volume de PMDS
0
PDMS
C : Concentration dans le PDMS avant l’étape de désorption
PDMS
n' : Quantité de matière restante dans le PDMS après l’étape de désorption
sol
n' : Quantité de matière présente dans le solvant de désorption aqueux après désorption
sol
V' : Volume du solvant de désorption aqueux
'
β : Volume du solvant de désorption aqueux / Volume de PMDS
Hexane
D'
: Rapport de distribution pour la désorption dans l’hexaneHexane
n' : Quantité de matière présente dans le solvant de désorption organique après
l’étape de désorption
"
β : Volume du solvant de désorption organique / Volume de PMDS
Hexane
V ' : Volume du solvant de désorption organique
s
C : Concentration à saturation en phase aqueuse du D2EHPA
[ ]
HAlim: Solubilité limite de la forme neutre de D2EHPA en milieu aqueuxLC/MS : Liquid Chromatography / Mass Spectrometry
D.E.S.I. : Desorption electrospray ionization
D.A.R.T.: Direct Analysis in real time
psi : Pound per square inch (1 bar = 14,5 psi)
PREMIERE PARTIE:
L’uranium est un élément radioactif naturellement présent dans les eaux et les sols. Hormis pour certains gisements exceptionnels tels que la mine canadienne de « Cigar Lake » dont la teneur en uranium dépasse 20%, dans la lithosphère, la concentration moyenne de l’uranium n’est que de 3 à 4 ppm. L’éventail est toutefois très large selon les matériaux. Ces teneurs (ppm) sont de l’ordre de 1 à 13 pour les schistes, de 1 à 80 dans les schistes carburés, de 3 à 27 dans les bauxites, de 0,1 à 9 dans les roches carbonatées et de 1 à 350 dans les phosphates. Cet actinide est présent dans les systèmes aquatiques continentaux à des concentrations variant de 10-6 à 10-3 ppm. Typiquement, dans l’eau de mer, sa concentration moyenne est de 3,3.10-3ppm.
L’uranium est la matière première indispensable à la fabrication des combustibles nucléaires, après qu’il ait subi un ensemble complexe de transformations chimiques et physiques le conduisant de son état d’élément trace issu du minerai jusqu’à sa forme utilisable dans un réacteur (notamment sous forme d’oxyde UO2, ou d’oxyde mixte (Ux, Pu1-x)O2).
La première étape de ce long processus industriel permettant son emploi comme combustible consiste en son extraction minière et la production d’un concentré uranifère en partie purifié (Yellow cake). Différents procédés d’extraction de l’uranium existent et sont adaptés aux différentes familles chimiques de minerais (roches granitiques, roches calcaires ou mines de phosphates par exemple). De façon générale, ces procédés incluent des étapes d’extraction et de purification de l’uranium par extraction liquide/liquide à l’aide de molécules telles que par exemple le tributylphosphate (TBP) ou l’acide bis-(2-éthylhéxyl) phosphorique (D2EHPA) [1-2]. Ces molécules, dites « agents solvatants » ou « extractants », sont ajoutées au solvant d’extraction, typiquement un solvant hydrophobe de type « kérosène », pour permettre la migration sélective de l’uranium depuis une phase aqueuse initiale et impure (obtenue par dissolution ou lixiviation de minerais) vers la phase organique non miscible.
Les entreprises minières ou de conversion chimique produisant de l’uranium sont, comme toute autre industrie chimique, soumises à une forte règlementation environnementale visant à réduire l’impact de leurs sites sur le milieu naturel. Elles sont notamment soumises à la loi REACH (Registration Evaluation and Autorisation of Chemicals) [3] qui impose la maitrise parfaite du contenu des effluents industriels. Compte tenu de leurs spécificités chimiques et de l’évolution des connaissances toxicologiques et environnementales sur ces produits, les agents extractants font de plus en plus partie des substances jugées comme « sensibles » d’un point de vue environnemental. Certaines pourraient même à terme devoir être remplacées par d’autres, moins toxiques. Cette substitution n’étant pas encore imposée, il est important de pouvoir les détecter et les contrôler dans les effluents, aussi bien pour l’exploitant d’un site (l’industriel lui-même) que pour les organismes de contrôle environnemental.
Dans ce contexte industriel et de surveillance de la qualité de l’environnement, le sujet de ce travail de recherche a donc été le développement et l’étude d’une méthode d’analyse incluant une étape de prélèvement in-situ pour un des extractants de l’uranium : le D2EHPA.
Pour être cohérent avec un objectif de respect environnemental, et compte tenu des nouvelles techniques mises à disposition des chimistes analystes, il a été choisi une technique de prélèvement réduisant l’utilisation de solvants, conforme aux préceptes d’une chimie plus « verte ». Outre ces objectifs environnementaux, la technique de prélèvement ou d’échantillonnage devait permettre une utilisation sur site (in-situ) et être compatible avec une détection de substances à l’état de traces. La technique choisie a été l’extraction par polymère déposé sur barreau agitateur aimanté, plus généralement connue sous le sigle « SBSE » (Stir Bar Sorptive Extraction). Le principe de cette technique réside dans l’absorption du composé d’intérêt par un polymère pouvant être assimilé à un solvant non miscible avec le milieu analysé. Ce processus d’absorption permet le prélèvement de la molécule d’intérêt à partir de sa matrice, sa concentration et très souvent sa pré-purification par rapport à d’autres substances présentes dans l’échantillon et qui peuvent gêner les étapes ultérieures d’une analyse chimique de traces.
Compte tenu de cette nécessité de pouvoir détecter des substances à très bas niveaux de concentration, le choix de la technique instrumentale d’analyse chimique s’est naturellement porté vers les méthodes séparatives (chromatographie) couplées à des analyseurs spectrométriques (spectromètres de masse). Une détection directe par spectrométrie de masse haute résolution a même été mise en œuvre pour illustrer les performances de ces nouveaux analyseurs, notamment par leur couplage avec des sources d’ionisation ambiante en fort développement actuellement.
Outre son intérêt intrinsèque de développement méthodologique, le travail présenté dans ce mémoire repose sur l’étude d’un extractant modèle alliant des propriétés générales d’extraction hydrométallurgique de l’uranium à des propriétés acido-basiques. Ainsi, cette étude a pour objectif d’offrir une démarche générale pouvant être transposée à toute famille de composés similaires au D2EHPA, sur la base de connaissances physicochimiques élémentaires sur ces nouveaux composés. La démarche et les résultats généraux présentés dans ce document ont donc pour ambition de pouvoir être appliqués de manière prédictive et de minimiser les études expérimentales spécifiques qui seraient nécessaires au développement d’une méthode de prélèvement et d’analyse pour un autre extractant analogue au D2EHPA.
mis en place.
Dans la première partie, l’étude théorique vise à étudier l’absorption d’un composé acido-basique qui est régie par plusieurs équilibres physicochimiques qui peuvent être déplacés afin d’améliorer le rendement d’extraction. Celle-ci mène à une modélisation mathématique globale qui permet d’évaluer le rendement d’extraction en fonction de différents paramètres tels que le pH, le rapport des volumes des phases (échantillon/polymère) et la concentration initiale du D2EHPA dans l’échantillon. Comme indiqué précédemment, cette modélisation mathématique s’inscrit dans le cadre d’une démarche globale pouvant être réutilisée dans le cas de molécules semblables, c’est-à-dire de molécules présentant une fonction acido-basique et qui se dimérisent en milieu organique. Il s’agit d’une modélisation thermodynamique qui donne accès à la valeur d’un rendement d’extraction de l’analyte dès lors que l’équilibre de son partage entre phases est atteint (phase aqueuse/phase polymère).
Ces travaux ne pouvaient naturellement ignorer la composante cinétique de tout processus physicochimique, mais la complexité d’une modélisation mathématique de la vitesse d’extraction par barreau SBSE, et l’absence de certaines données diffusionnelles sur la molécule d’intérêt, ont conduit à en limiter et à en simplifier la description cinétique. Une approche expérimentale a ainsi été préférée pour fournir quelques éléments sur le temps d’extraction qui permet d’accéder à un pseudo-équilibre thermodynamique (équilibre apparent, proche de l’équilibre réel).
La seconde partie de ce travail a consisté à réaliser diverses expérimentations pour confronter les résultats de l’étude théorique avec l’expérience et évaluer la crédibilité et la fiabilité du modèle. Ces expérimentations ont nécessité le développement préalable d’une méthodologie analytique spécifique qui n’a pas valeur de méthode parfaitement optimisée ni ultime, mais qui constituait un outil adapté à l’objectif. Les analyses du D2EHPA ont ainsi été réalisées par chromatographie liquide haute performance couplée à de la spectrométrie de masse, après avoir développé préalablement la méthode en vérifiant sa robustesse, sa sensibilité et sa quantitativité.
Compte tenu du temps nécessaire à la réalisation de ces expérimentations et de celui consacré au développement du modèle thermodynamique ou de la méthode d’analyse spécifique, la vérification expérimentale du modèle d’extraction et son ajustement aux données spécifiques du D2EHPA ne se sont faits que par l’étude de l’influence des conditions d’acidité du milieu (pH) sur le rendement d’extraction. Cette acidité peut toutefois être considérée comme le paramètre essentiel régissant le rendement d’extraction, et bien que l’on puisse envisager de compléter cette vérification paramétrique dans le futur, l’étude de l’influence du pH présentée ici permet de relever certaines conclusions essentielles pour la transposition du modèle à d’autres extractants.
constitue naturellement un objectif essentiel. A cet effet, et sur la base des résultats expérimentaux et théoriques obtenus, différents abaques spécifiques permettant de prévoir les conditions d’extraction et de désextraction du D2EHPA ont été calculés et sont présentés dans ce mémoire. Ils permettent d'optimiser les conditions d'extraction sur site, ou de désextraction en laboratoire avant analyse.
Enfin, disposant au laboratoire d’instrumentations hautes performances dédiées au développement de méthodologies d’analyse globales, reposant notamment sur le couplage de la spectrométrie de masse haute résolution (Orbitrap) avec des sources d'ionisation à pression atmosphérique (source d’ionisation ambiante), il était impossible de ne pas chercher à en évaluer et à en présenter tout le potentiel dans le domaine de l’analyse directe d’un extractant prélevé par barreau SBSE. Ces méthodes n’ont pu qu’être effleurées, malheureusement, et le résultat présenté se limite à une illustration qualitative mais somme toute assez démonstrative de l’intérêt de ces nouvelles approches analytiques.
DEUXIÈME PARTIE :
PRINCIPES THEORIQUES
ET INSTRUMENTATION
Cette seconde partie traite des différentes techniques de prélèvement in-situ pouvant être appliquées facilement sur site et présentant un intérêt particulier dans le cadre des travaux que nous avons effectués.
Dans un premier temps, quelques techniques d’extraction adaptées à la préparation in-situ d’échantillon aqueux sont présentées, telles que l’extraction par polymères absorbants.
Dans un second temps, le principe théorique de l’extraction par polymère est appliqué à notre problématique, à savoir l’extraction du bis (2-éthylhexyl) phosphate (D2EHPA) dans des matrices aqueuses complexes. Cette étude théorique consiste en une modélisation mathématique des différents équilibres chimiques qui régissent cette extraction. Ce travail préliminaire a permis de prévoir le rendement d’extraction massique en fonction des conditions de milieu ; ce rendement théorique a alors été confronté à des résultats expérimentaux comme il sera exposé dans la 3ème partie de ce document.
I.
PRINCIPES GENERAUX ET INSTRUMENTATIONS SPECIFIQUES
POUR LA PREPARATION D’ECHANTILLONS AQUEUX
Dans la plupart des processus analytiques globaux, l’analyse chimique est précédée des étapes de prélèvement et de préparation d’échantillons. [4-5]. L’échantillonnage (ou prélèvement) doit être effectué de façon à être le plus représentatif possible de la solution à analyser. La préparation doit quant à elle convertir l’échantillon en une forme convenable pour l’analyse [6].
Dans le cadre d’une extraction, le but recherché peut être l’élimination de l’effet matrice et/ou la concentration si possible quantitative de l’analyte. Cette étape de préparation est souvent l’étape limitante du processus analytique. Malgré l’invention de techniques analytiques de plus en plus sensibles et spécifiques [4], cette étape est en général responsable de 30% des erreurs commises au cours de l’analyse globale et sa réalisation représente 60% du temps consacré à l’analyse.
De plus, les considérations environnementales, et la chimie verte, impliquent une réduction de l’utilisation de solvant organique. La technique adoptée dépend de l’analyte, sa concentration, sa matrice et la précision du résultat recherché. Elle doit être fiable, rapide. Dans ce but, de nombreuses techniques se sont développées ces dernières années afin de remplacer l’extraction liquide/liquide grande consommatrice de solvant.
Parmi les nombreuses techniques récemment développées ces dernières années, on trouve la micro-extraction sur phase solide (Solid Phase Micro Extraction – SPME), la micro-micro-extraction sur barreau aimanté (Stir Bar Sorptive Extraction – SBSE) et l’extraction sur phase solide (Solid Phase Extraction – SPE). Toutes trois se caractérisent par leur simplicité de mise en œuvre, leur efficacité et la réduction des volumes de solvants qu’elles requièrent. Elles sont, à ce titre, plus respectueuses de l’environnement. La SPME et la SBSE seront développées dans cette partie car elles sont les plus adaptées à la préparation d’échantillon sur site dans le cadre d’un emploi spécifique qui nous intéresse particulièrement.
I.1
Principes généraux de l’extraction liquide/liquide
L’extraction liquide/liquide se définit par [7] : la répartition d’un analyte entre deux phases non-miscibles en contact l’une avec l’autre. L’analyte ayant une solubilité différente dans les deux phases, il se répartit de façon inégale dans l’une et l’autre du fait d’interactions différentes qu’il a avec les deux phases en présence. Cette répartition inégale est exprimée par le coefficient de partage KP. Cette
constante thermodynamique représente le rapport des activités de l’analyte dans les deux phases.
Le terme de coefficient de partage a été introduit par Berthelot et Jungfleisch en 1872, d’après leurs études sur la répartition de nombreux composés organiques et inorganiques entre l’eau et le sulfure de carbone ou l’éther. En 1891, Nernst a pris en compte les différentes réactions du soluté dans chaque phase, notamment la dimérisation en phase organique ou la dissociation en phase aqueuse.
L’étude des phénomènes d’extraction liquide/liquide utilise classiquement deux formalismes qui rendent compte du partage de l’analyte entre les deux phases :
- Le rapport de distribution : D
- Le rendement d’extraction massique : next/ n0
Le rapport de distribution exprime le rapport des concentrations des espèces dans la phase aqueuse et dans la phase organique, toutes formes confondues.
Le rendement d’extraction massique exprime le rapport de quantité de matière, là aussi toutes formes confondues. Il introduit donc la notion de rapport de volume de phase.
Les deux formalismes sont équivalents et nécessaires pour exprimer mathématiquement les phénomènes auxquelles nous nous intéressons. Dans la mesure où nous souhaitons dans cette partie
théorique avoir une démarche globale de traitement du problème qui pourrait être réutilisée dans le cas d’extraction de molécules semblables, les deux expressions seront explicitées.
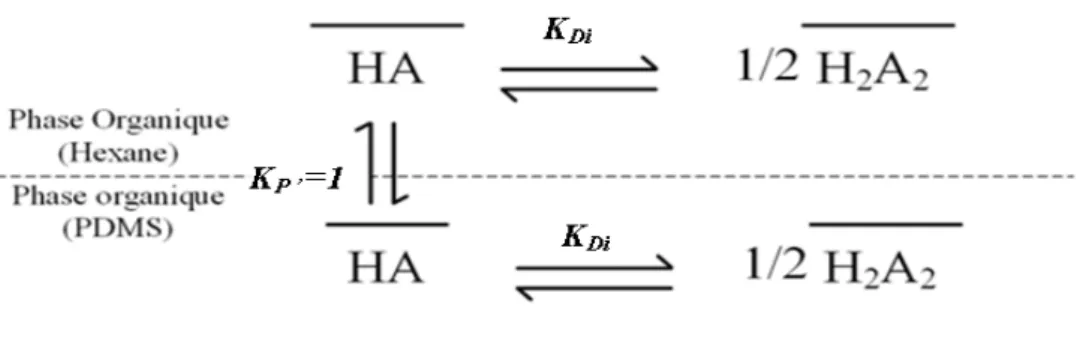
La figure 2 exprime le cas d’une molécule A qui se dimérise en phase organique et se dissocie en phase aqueuse1. C’est le cas typique et général de molécules telles que celle du D2EHPA.
Figure 2 : Extraction liquide/liquide d’un composé acido-basique A.
Dans ce cas, le coefficient de partage et le rapport de distribution s’expriment de deux façons distinctes : eq eq P
HA
HA
K
=
Équation 1 : Coefficient de partage
eq eq eq eq tot eq tot eq
A
HA
A
H
HA
HA
HA
D
]
[
]
[
]
[
2
]
[
]
[
]
[
2 2 , , −+
+
=
=
Équation 2 : Rapport de distribution
Ce principe de partage du composé entre deux phases selon ses affinités est commun aux techniques d’extraction modernes que sont les extractions sur polymère, notamment la SPME et la SBSE.
1 Par respect des conventions habituelles, les espèces chimiques présentes en phase organiques seront symbolisées dans la
I.2
Principe de l’extraction sur phase solide
L’extraction sur phase solide est régie par les principes thermodynamiques de l’extraction liquide/liquide. Une phase solide constituée typiquement d’un polymère aux propriétés extractantes remplace le solvant organique. L’utilisation de solvants d’extraction polluants, de type hexane, dichlorométhane ou chloroforme, n’est alors plus nécessaire. Le développement de ce genre de techniques est donc en accord avec le concept de la « chimie verte » qui favorise un usage restreint de solvants toxiques et la limitation des déchets.
Bien que l’on puisse en principe utiliser divers polymères présentant des propriétés extractantes variables selon leur fonctionnalisation, le polydiméthylsiloxane (PDMS) est l’unique polymère disponible commercialement à ce jour pour la microextraction sur barreau (SBSE). En conséquence, seul ce type de polymère a été étudié lors de ces travaux. Sa structure chimique est illustrée par la figure 3.
Figure 3 : Structure du polydimétylsiloxane
Les groupements méthyles rendent cette structure apolaire et donc adaptée à l’absorption de composés apolaires. L’enchainement d’atome silicium-oxygène induit des propriétés particulières par rapport aux polymères purement organiques [8] :
- Résistance thermique de -70 à +250°C - Elasticité
- Résistance au vieillissement naturel - Perméabilité au gaz
Le PDMS est donc utilisé comme phase extractante dans les dispositifs d’extraction SPME et SBSE qui seront détaillés dans les parties suivantes.
I.2.1 Principe instrumental de la micro-extraction sur phase solide
La micro-extraction sur phase solide (Solid Phase Micro Extraction - SPME) est une technique d’extraction inventée par Janusz Pawliszyn [9]. Le polymère constituant la phase extractante prend la forme d’une fibre cylindrique dont le cœur est constitué de silice, et la gaine est composée de phase absorbante. Cette fibre est placée à l’intérieur d’une aiguille creuse amovible, jouant le rôle de gaine protectrice, comme le présente la figure 4.
Le volume de polymère recouvrant la fibre est faible, de l’ordre 0,5 à 1 µL suivant l’épaisseur de polymère, ce qui définit et limite en partie la capacité d’absorption de la fibre.
Figure 4 : Dispositif de fibre SPME [4]
Ce type de dispositif peut être utilisé pour des prélèvements de natures multiples : prélèvement in-situ d’air ambiant [10], analyses d’échantillons aqueux (biologiques ou environnementaux) [11] ou encore analyses par espace de tête (headspace) [12] dans les domaines environnementaux, agroalimentaires et pharmaceutiques [13].
Avec l’utilisation de cette technique de prélèvement, le processus analytique global s’effectue en deux étapes :
- L’étape de prélèvement par un partage (extraction biphasique) de l’analyte entre l’échantillon et le revêtement de la fibre,
- La désorption de l’analyte vers un solvant ou directement vers l’appareil de mesure.
I.2.2 Principe instrumental de la micro-extraction sur barreau aimanté
La micro-extraction sur barreau (Stir Bar Sorptive Extraction – SBSE) est un dispositif reposant sur les mêmes principes que la micro-extraction sur phase solide (Solid Phase MicroExtraction – SPME). Le film de PDMS, d’un volume de plusieurs dizaines de microlitres, recouvre un barreau magnétique gainé de verre. La figure 5 montre au centre du dispositif le barreau magnétique (a), recouvert de verre ( ). Une couche cylindrique de PDMS (c) gaine le système, sous la forme d’un manchon de polymère.
(c) Film cylindrique de PDMS
(a) Barreau Magnétique
(b) Verre recouvrant le barreau magnétique
Figure 5 : Barreau de microextraction Gerstel®
L’augmentation considérable du volume de PDMS (typiquement 100 fois supérieur par rapport à celui d’une fibre SPME), permet de résoudre la principale limitation de cette précédente technique, à savoir le faible pouvoir extractant du système d’ores et déjà évoqué. Il est ainsi possible d’extraire de plus grandes quantités d’analyte et/ou des analytes ayant un coefficient de partage plus faible. Ces deux aspects sont mis en évidence mathématiquement dans la suite de ce document et sont illustrés par la figure 6 qui représente pour différentes affinités d’analyte (exprimées par leur constante de partage théorique entre l’octanol et l’eau : Ko/w) les rendements d’extraction accessibles sur fibre SPME ou sur
barreau SBSE.
Figure 6 : Comparaison des rendements d’extraction par barreau SBSE et fibre SPME pour des analytes de polarité variable exprimée par la constante de partage octanol/eau (Ko/w) [14]
Cette technique a été décrite par les travaux d’Erik Baltussen et son équipe en 1999 [14] comme une excellente alternative à la SPME pour l’analyse d’échantillons aqueux. Depuis, de très nombreuses applications ont prouvées l’adaptabilité de cette méthode à des problématiques environnementales, en permettant la recherche de traces dans des eaux d’origines variées [15-16-17-18-19].
II.
THEORIE DE L’EXTRACTION APPLIQUEE AU D2EHPA
La théorie de l’extraction d’un composé sur polymère s’étudie à deux niveaux : thermodynamique et cinétique.
Dans un premier temps, l’étude thermodynamique consiste en une exploitation de nos connaissances sur les différents équilibres physicochimiques auxquels participe le D2EHPA, et la modélisation de l’état d’équilibre final du système composé de l’analyte sous ses différentes formes solubilisées dans les deux phases. Cet équilibre s’établit en effet entre les deux phases non miscibles en contact l’une avec l’autre par leur interface.
La seconde partie consiste à étudier l’aspect dynamique, c’est-à-dire le temps d’atteinte de cet état d’équilibre à partir d’un état initial. Il s’agit alors d’une étude cinétique.
Ces deux aspects sont naturellement indissociables et définissent notre capacité à observer l’établissement de l’état d’équilibre, tant dans son principe énergétique (thermodynamique) que dans son accessibilité pratique (cinétique).
II.1
Analyse thermodynamique de l’extraction du D2EHPA par SBSE
L’aspect thermodynamique de l’extraction liquide/liquide, ou de l’extraction sur polymère telle que la SBSE, se traduit par la répartition des espèces chimiques issues du D2EHPA dans les différents milieux en présence.
Cette répartition résulte des principales propriétés physicochimiques du D2EHPA (Figure 7), notamment des propriétés acido-basiques et de polarité dont les constantes sont regroupées en annexe I. Le sens de ces constantes, en termes d’équilibres chimiques est explicité dans les parties suivantes.
II.1.1 Constantes d’équilibres thermodynamiques
Comme indiqué précédemment, le processus global d’extraction résulte de l’établissement d’un équilibre de partage entre phases, lui-même régi par l’ensemble des équilibres physicochimiques établis dans chacune des phases individuellement. Chacun de ces équilibres est un équilibre thermodynamique défini par des constantes qui s’expriment mathématiquement par la loi d’action des masses. Ainsi, les constantes d’équilibre sont les rapports d’activités moléculaires qui traduisent différents phénomènes thermodynamiques.
Pour décrire le partage du D2EHPA entre deux phases, compte tenu de ses propriétés physicochimiques spécifiques, il est ainsi nécessaire de faire appel à :
- Sa constante de partage (KP) entre les deux phases non miscibles,
- Sa constante de dissociation acide (KA) en milieu aqueux,
- Sa constante de dimérisation (KDi) en milieu organique.
II.1.1.1 Constante de partage entre phases (Kp)
Le partage d’un composé entre un milieu organique et un milieu aqueux non miscibles l’un avec l’autre est décrit par un équilibre thermodynamique représenté par la réaction 1.
HA HA↔
Réaction 1 : Partage entre PDMS et milieu aqueux
Cet équilibre thermodynamique est régi par une constante spécifique à l’analyte et aux deux phases en présence. De manière générale, cette constante de partage est égale au rapport des activités de l’analyte dans chacune des phases et s’exprime selon l’équation 3.
Kfs = af / as
Équation 3 : Coefficient de partage : Equation générale
Où f et s se réfèrent aux deux milieux non miscibles en contact.
Dans le cadre de l’extraction sur phase solide, la phase organique (liquide dans les extractions classiques) est remplacée par le polymère (PDMS). Du point de vue du mécanisme d’extraction, le PDMS agit toutefois comme un solvant liquide qui solubiliserait l’analyte. Dans le cas de l’absorption
du D2EHPA en milieu aqueux sur un twister composé de PDMS, le coefficient de partage s’exprimera alors selon l’équation 4.
eq eq P HA HA K =
Équation 4 : Coefficient de partage du D2EHPA entre PDMS et Eau
Le coefficient de partage d’un composé organique entre l’octanol et l’eau (Ko/w) est couramment donné
dans la littérature comme un indicateur de la polarité d’un composé, apparenté à cette constante de partage Kp. Il peut être mesuré expérimentalement ou estimé via différentes méthodes de calcul.
Meylan [20] utilise ainsi un calcul prenant en compte la contribution des fragments de la molécule pour estimer le coefficient de partage KO/W (également appelé P). Ce calcul théorique a été confronté à
l’expérience et une bonne corrélation a été obtenue.
La littérature [21] donne le log Ko/w calculé par la méthode de Meylan.
LOG Ko/w D2EHPA= 6,07
Cette valeur est cohérente avec la valeur que nous avons pu déterminer avec le logiciel Chemsketch disponible au laboratoire : 6,08 ±0,58.
Cette analogie entre la constante de partage KPDMS/Eau et la constante K Octanol/Eau (KO/W) a été mise en
évidence par Baltussen [14]. Selon la théorie de Baltussen, ces valeurs sont équivalentes, en conséquence, en l’absence de données précises sur la valeur du coefficient de partage du D2EHPA entre le PDMS et l’eau, c’est la valeur KO/W qui a été utilisée comme première approximation. Cette
approche est cependant discutée dans ce mémoire, à la fin de la partie expérimentale.
II.1.1.2 Constante de dissociation acido-basique (Ka)
Le D2EHPA possède une fonction acide (O=P-OH) analogue à celles rencontrées dans la molécule d’acide phosphorique. D’un point de vue moléculaire, le D2EHPA est un ester partiel (di-ester) de l’acide phosphorique qui a donc conservé une de ses fonctions acides. Des équilibres acido-basiques dissociatifs s’établissent donc en milieu aqueux.
proton. Pour simplifier l’écriture des équations dans le reste du document, le D2EHPA sera noté HA sous sa forme acide et A- sous sa forme basique. La réaction de dissociation s’écrit alors sous la forme :
HA ↔ H+ + A-
Réaction 2: Réaction de dissociation
La constante de dissociation s’exprime alors selon l’équation suivante :
eq eq eq A HA A H K = − +
Équation 5 : Constante de dissociation
Les données numériques publiées [21] font état d’une valeur du pKA égale à :
pKA D2EHPA = 1,47(soit KA=3,4.10-2)
Cette valeur est cohérente avec la valeur que nous avons pu déterminer avec le logiciel ACD/PkA disponible au laboratoire : 1,47±0,50.
II.1.1.3 Constante de dimérisation (KDi)
La constante de dimérisation d’un composé est la constante de réaction associée à la formation de formes moléculaires doubles (dimères). Elle est d’autant plus importante que le composé s’associe fortement à lui-même pour former ce dimère. Cet équilibre reste en fait réversible car l’association des molécules ne s’effectue pas par des liaisons covalentes, mais plutôt par des interactions hydrophobes ou de type « liaisons hydrogène ». Cet équilibre est illustré par la réaction 3.
2 HA ↔ H2A2
Réaction 3: Réaction de dimérisation
2 2 2 eq eq Di HA A H K =
Équation 6 : Constante de dimérisation
Les constantes de dimérisation du D2EHPA ont été déterminées entre autres pour des solvants tels que le kérosène ou l’hexane, compte tenu de son usage dans des procédés industriels hydro métallurgiques utilisant ces deux solvants de dilution de molécules extractantes. On trouve ainsi publiées les valeurs suivantes :
KDi D2EHPA = 3,39.10-4dans le kérosène [22]
KDi D2EHPA = 3,17.10-4dans le n-hexane [23]
Les polarités de l’hexane ou du kérosène peuvent être considérées comme analogues, les constantes de dimérisation sont alors proches comme en attestent les valeurs publiées. Cette analogie de polarité peut être également appliquée au cas du PDMS d’un point de vue des interactions moléculaires agissant dans le cadre d’une étude d’extraction. La constante de dimérisation sera donc considérée comme identique dans ces trois phases.
Malgré cette analogie et les valeurs comparables de ces constantes, nous n’avons retenu que la constante publiée dans l’hexane pour la suite de nos travaux, car le kérosène est un mélange complexe de plusieurs hydrocarbures, dont certains aromatiques qui peuvent avoir un comportement légèrement différent pour des interactions avec le D2EHPA. Nous avons donc préféré faire référence à un milieu plus simple et mieux défini tel que l’hexane. Dans le cadre de notre étude, cette constante KDi est donc
considérée comme identique à celle publiée pour l’hexane : KDi D2EHPA = 3,17.10-4.
II.1.2 Influence de la salinité des milieux sur les équilibres thermodynamiques : relation
entre activés, concentrations et force ionique
Les précédentes relations d’équilibre ont été exprimées comme il se doit en termes de rapports d’activités. D’un point de vue pratique, compte tenu de la difficulté d’accéder facilement à ces valeurs d’activités, il convient d’établir les relations qui existent entre activités, concentrations et force ionique pour tous les équilibres et les constantes associées mentionnées précédemment. C’est l’objet de ce
II.1.2.1 Principe de la théorie des activités appliquée aux espèces ioniques en solution aqueuse Dans une approche thermodynamique classique, seules les espèces ioniques seront considérées ici. On admettra en effet que les espèces neutres ne présentent pas de différence entre leurs activités et leurs concentrations. En conséquence, on ne rappellera ici que la relation classique entre l’activité d’un composé et sa concentration (Equation 7). Cette relation ne concernera dans le reste de notre étude que les espèces ionisées présentes en solution aqueuse, à savoir les ions H+, OH- ainsi que la forme déprotononée (basique) du D2EHPA.
[ ]
⋅
−=
− − AA
A
γ
Équation 7 : Relation entre activité et concentration
Le coefficient d’activité γ est relié à la charge électrique du composé et à la force ionique du milieu. Ce coefficient d’activité est donc considéré comme égal à 1 pour les molécules non chargées.
L’équation de Davies (1938), s’applique pour le calcul des coefficients d’activité pour une force ionique inférieur à 0,5 [24]. ⎟⎟ ⎠ ⎞ ⎜⎜ ⎝ ⎛ × − + × − = I I I z A i i 0,3 1 log 2 10γ
Équation 8 : Expression du logarithme du coefficient d’activité
A est un coefficient lié à la température. Il vaut 0,5 à 25°C, zi est la charge de la molécule considérée et
I est la force ionique du milieu. Cette force ionique dépend de la charge saline du milieu et s’exprime de la façon suivante :
∑
= Zici I 2 2 1 .Équation 9 : Expression de la force ionique du milieu
Zi et Ci représentent respectivement la charge électrique et la concentration des espèces ioniques
L’expérience montre que pour les composés non chargés, le coefficient d’activité reste très proche de l’unité jusqu’à des concentrations de l’ordre de 1M. Comme indiqué précédemment, on considérera donc ici pour les espèces non chargées que l’activité est égale à la concentration [24].
Compte tenu de ces considérations, toutes les constantes d’équilibre impliquant des espèces ioniques peuvent être exprimées en termes de concentrations, en corrigeant ces valeurs grâce aux coefficients d’activité. On peut notamment exprimer l’activité des ions H+ participant aux équilibres acido-basiques en solution aqueuse, mesurée par le pH de la solution, en fonction de leur concentration [H+]. Cette expression est exprimée ainsi :
[ ]
pHH H
H + = γ + + =10 −
D’autre part, des équations (4), (5) et (6) on déduit aussi l’expression des constantes d’équilibres suivantes : eq eq P HA HA K ] [ ] [ =
Équation 10 : Constante de partage en fonction des concentrations
eq eq A pH a HA A K ] [ ] [ 10− × − − = γ
Équation 11 : Constante de dissociation en fonction des concentrations et des activités
2 2 2 ] [ ] [ eq HA A H K eq Di =
Équation 12 : Constante de dimérisation en fonction des concentrations
Enfin, d’après les équations 10 à 12, pour les formes diverses du D2EHPA, la force ionique du milieu n’impactera que la constante de dissociation via le coefficient d’activité du D2EHPA sous sa forme basique A-.
⎟⎟ ⎠ ⎞ ⎜⎜ ⎝ ⎛ × − + − = − I I I A 0,5 1 0,3 log10γ
Équation 13 : Expression du logarithme du coefficient d’activité à 25°C d’A
-L’expression 13 permet donc de tracer le coefficient d’activité γA− en fonction de la force ionique I.
0,70 0,75 0,80 0,85 0,90 0,95 1,00 1,05 1,10 -15 -14 -13 -12 -11 -10 -9 -8 -7 -6 -5 -4 -3 -2 -1 log10 I C oef ficien t act ivit é
Figure 8 : Coefficient d’activité de A- en fonction de la force ionique I
Lorsque cette force ionique tend vers zéro, le coefficient d’activité de A- tend vers 1. Il s’agit du cas des solutions diluées où les ions n’interagissent pas entre eux. Dans le cas de solution dont la force ionique est comprise entre 10-5 et 0,5 le coefficient d’activité diminue, l’activité se différencie alors de la concentration et ce coefficient est à prendre en compte dans le calcul de la constante de dissociation du D2EHPA.
II.1.3 Modélisation de l’absorption du D2EHPA par du PDMS
Dans cette partie, le phénomène d’absorption du D2EHPA par du PDMS a été étudié de façon théorique, sur la base d’un modèle de mécanisme physicochimique et de l’expression mathématique des lois d’équilibre thermodynamique. Les courbes d’extraction traduisant l’absorption du D2EHPA dans le polymère ont été tracées en fonction des calculs effectués avec les constantes explicitées dans la partie II.1.1. Le modèle d’extraction retenu a priori a été celui classiquement validé dans la littérature pour ce type de molécules, comme présenté dans la figure 1.
II.1.3.1 Expression du coefficient de distribution
Le coefficient de distribution du D2EHPA, dans le cas de son extraction depuis un milieu aqueux vers du PDMS, décrit le rapport de la concentration du composé entre la phase aqueuse et la phase organique (polymère). Ce coefficient de distribution correspond au partage du D2EHPA, toutes espèces confondues, c'est-à-dire en tenant compte de sa dimérisation dans la phase organique et sa dissociation dans la phase aqueuse.
Le coefficient de distribution en fonction de la concentration à l’équilibre s’exprime selon l’équation 14 : eq eq eq eq
A
HA
A
H
HA
D
]
[
]
[
]
[
2
]
[
2 2 −+
+
=
Équation 14 : Expression du coefficient de distribution d’absorption
En combinant les expressions des différentes constantes d’équilibres présentées précédemment, ce coefficient de distribution peut alors s’exprimer en fonction des constantes de partage, de dissociation et de dimérisation comme explicité dans l’équation 15 :
⎟ ⎟ ⎠ ⎞ ⎜ ⎜ ⎝ ⎛ + × × × + = − − A pKa pH eq P Di P HA K K K D
γ
10 1 ] [ 2 1Équation 15 : Expression du coefficient de distribution pour l’absorption en fonction de [HA]eq
Les étapes du calcul mathématique permettant le passage de l’équation 14 à l’équation 15 sont présentées en annexe II.b.1.
Cette équation (15) montre que, le coefficient de distribution varie non seulement en fonction du pH, mais aussi en fonction de la valeur [HA]eq qui correspond à la concentration de la forme acide du
D2EHPA présente à l’équilibre en phase aqueuse.
Afin d’exprimer ce coefficient de distribution en fonction de la concentration initiale C0, il convient d’exprimer [HA]éq en fonction de cette même valeur. Pour cela, la théorie de la conservation de la
matière a été appliquée et s’exprime par les deux relations suivantes :
éq PDMS éq sol sol n n n 0 = +
(
eq eq)
PDMS(
eq eq)
sol sol V HA A V HA H A V C0 = [ ] +[ −] + [ ] +2[ 2 2]La conservation de la matière permet alors d’exprimer [HA]éq par l’équation 16 en fonction des
différentes constantes thermodynamiques et de C0.
2 2 0 2 4 8 10 1 10 1 ] [ P Di P Di P A pKa pH P A pKa pH éq K K K K C K K HA β γ β γ β β + ⎟ ⎟ ⎠ ⎞ ⎜ ⎜ ⎝ ⎛ + + + ⎟ ⎟ ⎠ ⎞ ⎜ ⎜ ⎝ ⎛ + + − = − − − −
Équation 16 : Expression de [HA]eq en fonction de C0
Les étapes du calcul mathématique permettant le passage de l’équation 15 à l’équation 16 sont également présentées en annexe II.b.1.
L’expression finale du coefficient de distribution du D2EHPA par le PDMS peut ainsi se présenter sous la forme de l’équation 17, intégrant les paramètres thermodynamiques (constantes d’équilibre), opératoires (C0 et β) et de milieu (pH et force ionique) :
⎟ ⎟ ⎠ ⎞ ⎜ ⎜ ⎝ ⎛ + ⎟ ⎟ ⎟ ⎠ ⎞ ⎜ ⎜ ⎜ ⎝ ⎛ + ⎟ ⎟ ⎠ ⎞ ⎜ ⎜ ⎝ ⎛ + + − + + − = − − − − − − A pKa pH P Di P A pKa pH P A pKa pH P P K K C K K K K D γ β β γ β γ β 10 1 8 10 1 10 1 2 1 2 0 2
II.1.3.2 Expression du rendement d’extraction massique
Afin de traduire de manière plus explicite la quantitativité de l’extraction, le rendement d’extraction peut être utilisé, plutôt que le coefficient de distribution.
Ce rendement, dans le cas de l’absorption, s’exprime par le rapport de la quantité de matière absorbée par le PDMS sur la quantité de matière initialement présente dans l’échantillon (Equations 18 et 19).
(
)
0 0 0 ] [ 2 1 ] [ 2 2 2 C HA K K HA K n n n n nPDMS HAeq H A eq P eq Di P eq × + = + = βÉquation 18 : Expression du rendement d’extraction en fonction de [HA]eq
⎟⎟ ⎟ ⎟ ⎟ ⎟ ⎟ ⎠ ⎞ ⎜⎜ ⎜ ⎜ ⎜ ⎜ ⎜ ⎝ ⎛ + ⎟ ⎟ ⎠ ⎞ ⎜ ⎜ ⎝ ⎛ + + − ⎟ ⎟ ⎠ ⎞ ⎜ ⎜ ⎝ ⎛ + + − ⎟⎟ ⎟ ⎟ ⎟ ⎟ ⎟ ⎠ ⎞ ⎜⎜ ⎜ ⎜ ⎜ ⎜ ⎜ ⎝ ⎛ + ⎟ ⎟ ⎠ ⎞ ⎜ ⎜ ⎝ ⎛ + + − ⎟ ⎟ ⎠ ⎞ ⎜ ⎜ ⎝ ⎛ + + − = − − − − − − − − P P Di P A pKa pH P A pKa pH P Di P Di P A pKa pH P A pKa pH PDMS K K K C K K K K K K C K K C n n 2 8 10 1 10 1 1 4 8 10 1 10 1 1 2 0 2 2 0 2 0 0 β β γ β γ β β β γ β γ
Équation 19 : Expression du rendement d’extraction en fonction de C0
Les étapes du calcul mathématique permettant le passage de l’équation 18 à l’équation 19 sont présentées en annexe II.B.2.
L’expression du rendement d’extraction en fonction de C0 (équation 19) permet principalement de
tracer la courbe du rendement d’extraction en fonction du pH, et ce pour différentes concentrations initiales de D2EHPA, différentes valeurs de forces ioniques ou différents rapports de volumes de phases (β). Pour se placer dans les conditions des extractions réalisées en laboratoire, pour cette étude, un rapport des volumes de phases a été fixé à 83,33 (soit un volume de PDMS de 24 µL pour un volume de solution de 2 mL).
Figure 9 : Rendement d’extraction massique pour différents γA avec C0 =1 nM -et β=83,3
Cette première étape, exprimant mathématiquement le rendement d’extraction en fonction de différents paramètres, permet de mettre en évidence l’ampleur de leur impact sur la quantitativité de l’extraction.
La figure 9 illustre par exemple l’influence du pH et du coefficient d’activité sur le rendement d’extraction. La figure 10 représente celle de la concentration d’analyte initialement présent dans l’échantillon sur son rendement d’extraction.
On constate aisément, grâce à ces deux graphiques, que pour une concentration et un coefficient d’activité donnés, réduire le pH permet de favoriser l’extraction et d’en accroitre par conséquent le rendement. Ceci s’explique naturellement d’un point de vue chimique par le déplacement de l’équilibre acido-basique en faveur de la forme acide du composé dans la phase aqueuse, lorsque l’on abaisse son pH. Cette forme acide correspondant a priori à la seule pouvant migrer dans le PDMS, favoriser sa formation, donc sa proportion en phase aqueuse permet d’en améliorer l’extraction.
Par ailleurs, à pH constant, augmenter la concentration initiale du D2EHPA en phase aqueuse permet globalement d’en augmenter la quantité extraite par le PDMS, ce qui se traduit également par une plus grande formation d’espèces dimères dans le PDMS. A son tour, cette formation de dimère contribue au déplacement de l’équilibre de partage en faveur de l’absorption du D2EHPA par le polymère.
Enfin, augmenter la force ionique du milieu, en ajoutant un sel à la solution par exemple, permet, à pH fixe, d’augmenter le rendement d’extraction. Dans une gamme de variation réduite pour le coefficient d’activité, c’est-à-dire pour des concentrations salines ne correspondant qu’à des forces ioniques inférieures à 0,1 M, ce phénomène reste toutefois d’une ampleur limitée sur le rendement d’extraction, par rapport aux effets observés pour les autres paramètres (pH notamment).
Parmi les autres paramètres opératoires étudiés figure le paramètre β, qui traduit non pas une des caractéristiques physicochimiques du système, mais une caractéristique géométrique (volumétrique) puisqu’il correspond au rapport du volume des phases en contact. Son influence sur le rendement d’extraction a naturellement été prise en compte et étudié. Les valeurs appliquées dans notre modèle théorique correspondent à un volume de PDMS fixe de 24 µL (Twister Gerstel® 10 mm x 0,5 mm) et à des volumes d’échantillons de 2, 5 et 10 mL. La figure 11 présente le rendement d’extraction en fonction du pH pour différentes valeurs de β. Elle montre qu’à pH constant, la réduction du paramètre β (correspondant à la réduction du volume d’échantillon ou à l’augmentation du volume de PDMS) augmente le rendement d’extraction. En d’autres termes, le rendement d’extraction est favorisé par l’utilisation d’un volume de polymère important par rapport au volume d’échantillon. Plus ce volume sera grand par rapport au volume de la solution analysée, plus le rendement d’extraction sera élevé.
Figure 11 : Rendement d’absorption pour différentes valeurs de β avec C0=1 nM et γA-=1
II.1.3.3 Calcul du pH maximum permettant une extraction de 95% du D2EHPA initial
Au vu des informations précédemment obtenues, il peut être intéressant de calculer le pH maximum de la solution aqueuse permettant d’obtenir un rendement d’extraction massique de 95%. Après différentes étapes mathématiques détaillées dans l’annexe II.B.3, nous pouvons écrire l’équation 20 qui présente l’expression de ce pH particulier :
pKa K C K K C pH Di P Di A ⎟⎟+ ⎠ ⎞ ⎜ ⎜ ⎝ ⎛ ⎟ ⎟ ⎠ ⎞ ⎜ ⎜ ⎝ ⎛ − − + = − 1 1 6 , 7 1 2 , 0 log 0 0 10 β γ
Équation 20 : pH minimum permettant une extraction massique de 95% en fonction de C0
En complément du calcul de ce pH maximum, cette équation permet de montrer également qu’il existe une limitation pour la valeur du rendement d’extraction pour une concentration initiale donnée. En effet, le logarithme décimal d’une valeur négative n’existant pas, il est nécessaire que la relation suivante soit respectée mathématiquement :
β β β
0
1
1
6
,
7
1
2
,
0
0 0−
〉
−
+
Di P DiK
C
K
K
C
β
Cette relation définit donc une valeur limite de concentration en dessous de laquelle une extraction minimale de 95% ne pourra jamais être atteinte. Cette condition de concentration limite est déductible de l’équation précédente et se résume à la relation présentée dans l’équation 21, dont la démonstration mathématique est détaillée en annexe II.B.4.
2 0
10
190
P Di PK
K
K
C
≥
β
−
Équation 21 : Concentration minimum permettant une extraction de 95%
Ceci prouve qu’un rendement d’extraction massique de 95% n’est théoriquement possible qu’à partir d’une certaine concentration initiale de D2EHPA. Lorsque la concentration initiale est inférieure à cette concentration limite, le rendement d’extraction sera plus faible.
Il est par ailleurs, important de noter que cette approche ne fait pas intervenir la cinétique des échanges dans ces calculs. Un rendement d’extraction massique de 95% pour une concentration initiale C0
proche de cette limite sera très long à atteindre.
Les considérations cinétiques sont abordées dans le paragraphe II.2 de cette seconde partie.
II.1.4 Modélisation de la désorption du D2EHPA en milieu aqueux, à partir du PDMS Dans la partie précédente, l’absorption du D2EHPA par le PDMS a été étudiée. Cette partie s’intéresse au phénomène inverse, c'est-à-dire à la désorption du D2EHPA vers une solution aqueuse libre de D2EHPA à l’instant initial. Ce phénomène étant l’inverse de l’extraction, il y aura un équilibre entre le monomère et le dimère en phase organique et un équilibre entre la forme basique et acide en milieu aqueux. La figure 12 illustre ce phénomène de désorption.