Développement de nouvelles méthodes d’identification des sites de SUMOylation par Protéomique
par
Frédéric Lamoliatte
Département de Chimie Faculté des arts et des sciences
Thèse présentée à la Faculté des études supérieures et postdoctorales en vue de l’obtention du grade de Philosophiæ Doctor (Ph.D.)
en Chimie
Aout, 2016
c
La régulation des protéines par les modifications post-traductionnelles (PTMs) est un évé-nement clé dans le maintien des fonctions biologiques de la cellule. Parmi elles, on retrouve les modifications causées par une famille de molécules appelées Ubiquitin Like Modifiers (UBls), in-cluant l’ubiquitination, la neddylation ou encore la SUMOylation. Au contraire des modifications classiques faisant intervenir des petits groupements chimiques, telles que la phosphorylation ou l’acétylation, les UBls sont eux-mêmes des protéines se greffant sur le groupement amine en position ε des lysines des protéines ciblées, générant des protéines ramifiées.
Alors que la principale fonction de l’ubiquitination est la dégradation des protéines par le protéasome, les autres UBls sont encore mal caractérisées. Dans ce contexte, le but de cette thèse était de développer de nouvelles approches protéomiques afin de définir le rôle de la SU-MOylation dans des cellules humaines. En effet, l’identification des sites de SUSU-MOylation par spectrométrie de masse (MS) est un défi. Ceci s’explique par la très faible abondance des pro-téines SUMOylées dans la cellule ainsi que par la longue chaine de 19 à 34 acides aminés laissés sur la protéine ciblée après digestion à la trypsine.
Afin de pallier à ces deux problèmes, un mutant de la protéine SUMO a été généré au sein du laboratoire. La première altération sur ce mutant est l’insertion d’une séquence 6xHis à l’ex-trémité N-terminale de la protéine afin de faciliter l’enrichissement des protéines SUMOylés. La seconde altération de la protéine SUMO est la mutation d’une glutamine en arginine en position 6 à partir du C-terminal. Cette mutation a pour effet de libérer des peptides trypsiques ramifiés contenant seulement 5 acides aminés provenant de SUMO sur le peptide ciblé.
Le premier but de cette thèse était de développer une méthode permettant de cibler spéci-fiquement les peptides SUMOylés lors d’une analyse par LC-MS. Cette méthode repose sur le patron de fragmentation propre de la chaine de 5 acides aminés commune à tous les peptides SU-MOylés et utilise la technologie Sequential Window Acquisition of all THeoretical Mass Spectra (SWATH). Lors d’une telle analyse, l’échantillon est injecté une première fois en fragmentant de larges fenêtres de masses. Ceci permet d’obtenir des spectres MS/MS pour tous les peptides pré-sents dans l’échantillon. Un algorithme est ensuite utilisé afin de détecter les fenêtres de masses contenant des peptides SUMOylés et de recalculer le rapport masse sur charge des peptides can-didats. Les injections subséquentes permettent ensuite de fragmenter uniquement les peptides candidats. Cette méthode s’est avérée être complémentaire aux méthodes conventionnelles et a
permis l’identification d’un total de 54 peptides SUMOylés à partir d’extraits protéiques enrichis sur billes NiNTA.
La seconde approche envisagée était d’ajouter une étape d’enrichissement supplémentaire au niveau peptidique. Pour cela, un anticorps reconnaissant la chaine de 5 acides aminés laissée après digestion tryptique a été produit. Cette étape d’immuno-purification supplémentaire a per-mis l’identification d’un total de 954 sites de SUMOylation dans des cellules humaines lors d’une analyse à grande échelle. Afin de valider les nouvelles cibles identifiées, une étude fonctionnelle de la SUMOylation de la protéine CDC73 a été réalisée. Cette étude a montré que la SUMOyla-tion de CDC73 était requise pour sa rétenSUMOyla-tion nucléaire, confirmant ainsi un rôle important pour la SUMOylation de cette protéine.
Cependant, le principal défaut de la précédente approche était la nécessité de cultiver 500 millions de cellules par condition étudiée. Cette approche a donc été optimisée afin de pouvoir réduire le nombre de cellules utilisées dans une analyse. L’optimisation de chacun des paramètres analytiques nous a permis de réduire ce nombre de 50 fois, permettant ainsi d’identifier plus de 1000 sites de SUMOylation à partir de seulement 10 millions de cellules. De plus, nous avons montré que cette approche permet l’identification concomitante des sites de SUMOylation et d’ubiquitination dans un seul échantillon biologique. Ceci a permis d’identifier un nouveau mé-canisme de régulation des deubiquitinases par les UBls, ainsi que d’élucider les mémé-canismes de translocation du protéasome dans la cellule.
Dans l’ensemble, nous avons développé des méthodes permettant de mieux caractériser la SUMOylation des protéines et avons prouvé que ces méthodes sont applicables à l’étude de plusieurs UBls en parallèle. Nous sommes certains que l’approche par immuno-purification per-mettra à l’avenir d’identifier la SUMOylation à un niveau endogène.
Protein regulation by post-translational modification (PTMs) is a key event in regulating cel-lular function. These modifications include a group termed Ubiquitin-Like modifiers (UBLs) that contain, but is not limited to, ubiquitylation, neddylation and SUMOylation. While conventional modifications, such as phosphorylation or acetylation, involve a small chemical group, UBLs are proteins attached from their C-terminus to the epsilon amine group of a lysine contained in the targeted protein, thus generating branched proteins.
While the main function of ubiquitylation is protein degradation by the proteasome, other UBLs remain mostly unexplored. In this context, the aim of this thesis was to develop new proteomics strategies to characterize SUMOylation in human cells. Indeed, identification of SUMOylation sites by mass spectrometry (MS) is a challenge. This is due to the low abundance of SUMOylated proteins in the cells as well as the long 19 to 34 amino acid SUMO remnant left of the target after trypsin digestion.
In this context, our research group has developed a mutant of SUMO containing two muta-tions. The first mutation consists of a 6xHis tag at the N-terminus of SUMO in order to facilitate SUMOylated substrates enrichment at the protein level. A second mutation was also introduced at the 6th position from the C-terminus and consists in a glutamine to arginine substitution in order to release shorter SUMOylated peptides after trypsin digestion.
The first goal of this thesis was to develop a targeted approach to specifically fragment SUMOylated peptides during an LC-MS run. This was enabled by the common fragmentation pattern of all SUMOylated peptides arising from the five amino acid SUMO remnant. Digested peptides were first analyzed using Sequential Window Acquisition of all THeoretical Mass Spec-tra (SWATH). In this experiment, large mass windows are fragmented. A custom algorithm is then used that detects mass windows in which candidates are located and determine their intact mass. In subsequent injections these peptides were then specifically targeted. This method was complementary to data dependent acquisition and enabled the identification of 54 SUMOylated peptides.
In a second approach, we wanted to enrich for SUMOylated substrates at the peptide level. An antibody was raised against the five amino acid SUMO remnant and used for immunopurifi-cation of SUMOylated peptides. In total, we identified 954 SUMOylation sites in human cells. Moreover, functional analysis of the newly identified substrate CDC73 revealed that
SUMOyla-tion on K136 is required for its nuclear retenSUMOyla-tion, thus showing a new role for the SUMOylaSUMOyla-tion of this protein.
Although this approach gave new insights into the characterization of SUMOylated sub-strates, high amounts of material were still required to obtain such results. The last goal of this thesis was to optimize the previously developed immunopurification. Systematic optimization of every analytical parameter was done and enabled the reduction of the number of cells required by a factor of 50, without affecting the number of SUMOylation sites identified. Moreover, we used this approach to profile for SUMOylation and ubiquitylation dynamics in human cells upon proteasomal inhibition with MG132. This revealed an unexpected regulation mechanism of deu-biquitinating enzymes by UBLs and unraveled translocation mechanisms of the proteasome in the cell.
Our SUMO proteomic approach demonstrates capability for the concomitant analysis of SUMOylation and ubiquitylation. In the future, we hope to extend this approach to endogenous SUMOylation.
RÉSUMÉ . . . iii
ABSTRACT . . . v
TABLE DES MATIÈRES . . . vii
LISTE DES TABLEAUX . . . xi
LISTE DES TABLEAUX SUPPLÉMENTAIRES . . . xiii
LISTE DES FIGURES . . . xv
LISTE DES FIGURES SUPPLÉMENTAIRES . . . xvii
LISTE DES ABRÉVIATIONS . . . xix
CITATION . . . xxiii
REMERCIEMENTS . . . xxv
CHAPITRE 1: INTRODUCTION . . . 1
1.1 Maturation des protéines . . . 1
1.2 Modifications ubiquitin-like . . . 2
1.2.1 Historique . . . 2
1.2.2 Propriété des UBLs . . . 2
1.2.3 Mécanismes de régulation des UBLs . . . 3
1.2.3.1 Maturation et Déconjugaison . . . 4 1.2.3.2 Conjugaison . . . 4 1.2.3.2.1 Enzymes d’activation E1 . . . 5 1.2.3.2.2 Enzymes de conjugaison E2 . . . 6 1.2.3.2.3 Enzymes de ligation E3 . . . 7 1.2.4 Ubiquitination . . . 9 1.2.4.1 Spécificité d’ubiquitination . . . 10
1.2.4.2 Aperçu des fonctions de l’ubiquitine . . . 10
1.2.4.3 Motif d’interaction avec l’ubiquitine . . . 11
1.2.5 SUMOylation . . . 12
1.2.5.1 Nomenclature de SUMO chez l’humain . . . 13
1.2.5.2 Spécificité de SUMOylation . . . 13
1.2.5.3 Aperçu des fonctions de la SUMOylation . . . 14
1.2.5.4 Motif d’interaction avec SUMO . . . 14
1.2.6 Interaction entre ubiquitination et SUMOylation . . . 15
1.2.6.1 PCNA: compétition entre SUMO et ubiquitine . . . 15
1.2.6.3 Corps nucléaires PML . . . 16
1.2.7 Intérêt clinique de l’étude des UBLs . . . 17
1.2.7.1 Leucémie Promyélocytaire Aiguë : une guérison induite par la SUMOylation . . . 17
1.2.7.2 Pathologies causée par des troubles des UBLs . . . 18
1.3 Analyse protéomique par spectrométrie de masse . . . 19
1.3.1 Stratégie d’identification des protéines . . . 19
1.3.2 Extraction des protéines . . . 20
1.3.3 Immunopurification . . . 21
1.3.4 Digestion des protéines . . . 22
1.3.5 Méthode Chromatographiques . . . 22
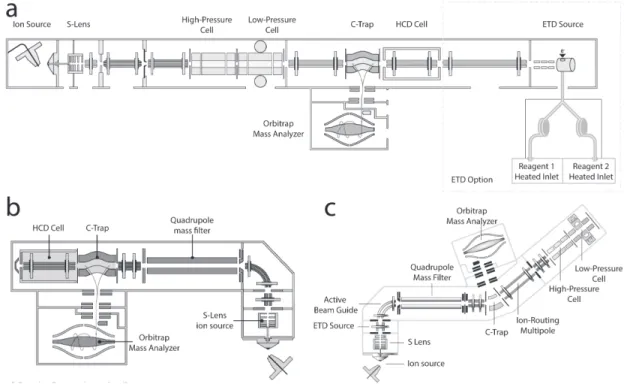
1.3.6 Spectromètre de masse . . . 24
1.3.7 Source d’ionisation . . . 24
1.3.8 Analyseur de masse . . . 25
1.3.8.1 Quadripôle . . . 27
1.3.8.2 Piège ionique Quadripolaire . . . 27
1.3.8.3 Temps de Vol . . . 28
1.3.8.4 Orbitrap . . . 29
1.3.9 Stratégies d’identification des protéines . . . 30
1.3.10 Identification des protéines . . . 33
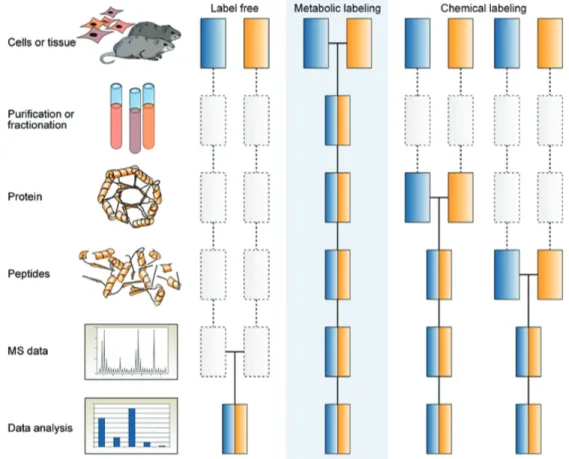
1.3.11 Quantification . . . 34
1.4 Stratégies d’identification des UBLs . . . 37
1.4.1 Approches biochimiques . . . 38 1.4.1.1 Mutagénèse dirigée . . . 38 1.4.1.2 Essai in vitro . . . 40 1.4.2 SUMOylation endogène . . . 40 1.4.3 Utilisation de mutants . . . 41 1.4.3.1 Maturation de SUMO . . . 41 1.4.3.2 Mutations N-terminal . . . 41 1.4.3.3 Mutations C-terminal . . . 41 1.4.3.4 Mutations internes . . . 42 1.4.4 Approches bio-informatiques . . . 42 1.5 Plan de recherche . . . 44 1.6 Bibliographie . . . 46
CHAPITRE 2: TARGETED IDENTIFICATION OF SUMOYLATION SITES IN HUMAN PROTEINS USING AFFINITY ENRICHMENT AND PA-RALOG-SPECIFIC REPORTER IONS . . . 67
2.1 Contribution des auteurs . . . 68
2.2 Abstract . . . 68
2.3 Introduction . . . 69
2.4 Experimental Procedures . . . 71
2.4.1 Peptide Synthesis . . . 71
2.4.3 Protein Digestion . . . 72
2.4.4 Mass Spectrometry . . . 73
2.4.5 Data Processing and Peptide Identification . . . 74
2.5 Results . . . 75
2.5.1 Product Ion Spectra of SUMO Mutant Peptides Revealed Paralog-speci-fic Fragment Ions . . . 75
2.5.2 Increased Identification of SUMO Mutant Peptides Using Conventional Database Search Engines . . . 80
2.5.3 Time and Mass Correlation of Precursor and Fragment Ions Enabled the Identification SUMOylated Peptides from Data Independent Acquisition Experiments . . . 82
2.6 Discussion . . . 88
2.7 Acknowledgments . . . 90
2.8 Bibliographie . . . 91
2.9 Supplementary Data . . . 94
CHAPITRE 3: LARGE-SCALE ANALYSIS OF LYSINE SUMOYLATION BY SUMO REMNANT IMMUNOAFFINITY PROFILING . . . 103
3.1 Contribution des auteurs . . . 104
3.2 Abstract . . . 104
3.3 Introduction . . . 104
3.4 Results . . . 106
3.4.1 Immunoaffinity enrichment of SUMO remnant peptides . . . 106
3.4.2 SUMO proteome dynamics following proteasome inhibition . . . 111
3.4.3 CDC73 SUMOylation affects its nucleocytoplasmic localization . . . 115
3.5 Discussion . . . 117
3.6 Methods . . . 118
3.6.1 Materials . . . 118
3.6.2 Generation of hybridoma cells . . . 119
3.6.3 Cell culture . . . 120
3.6.4 Cell fractionation and enrichment of SUMOylated proteins . . . 120
3.6.5 Immunoblot analysis . . . 121
3.6.6 Plasmid constructs and site-directed mutagenesis . . . 121
3.6.7 Cell transfection . . . 122
3.6.8 Immunofluorescence analysis . . . 122
3.6.9 Mass spectrometry . . . 123
3.6.10 Peptide and protein identification . . . 123
3.6.11 Bioinformatic analyses . . . 124
3.7 Bibliographie . . . 126
3.8 Supplementary Data . . . 130
CHAPITRE 4: UNCOVERING THE SUMOYLATION AND UBIQUITINATION CROSSTALK IN HUMAN CELLS USING SEQUENTIAL PEPTIDE IMMUNOPURIFICATION . . . 143
4.2 Abstract . . . 144
4.3 Introduction . . . 144
4.4 Results . . . 146
4.4.1 Optimization of SUMO peptide immunoaffinity purification . . . 146
4.4.2 Comprehensive SUMO proteome analysis . . . 149
4.4.3 Concomitant profiling of SUMOylation and ubiquitylation . . . 150
4.4.4 SUMOylation and ubiquitylation of ribosome and proteasome . . . 153
4.5 Discussion . . . 159
4.6 Methods . . . 160
4.6.1 Materials . . . 160
4.6.2 Cell culture . . . 161
4.6.3 Protein purification and digestion . . . 161
4.6.4 Antibody cross-linking . . . 162
4.6.5 Dual peptide IP . . . 162
4.6.6 SCX fractionation . . . 163
4.6.7 Mass spectrometry analysis . . . 163
4.6.8 Data processing . . . 163
4.6.9 Bioinformatics analysis . . . 164
4.6.10 Fluorescence imaging and co-localization analysis . . . 164
4.6.11 Western blot . . . 164
4.6.12 Data availability . . . 165
4.7 Bibliographie . . . 166
4.8 Supplementary Data . . . 171
CHAPITRE 5: CONCLUSIONS ET PERSPECTIVES . . . 185
5.1 Conclusions . . . 185
5.2 Perspectives . . . 188
5.2.1 Identification des mécanismes de guérison de l’APL . . . 188
5.2.2 Interaction entre les différentes UBls . . . 188
5.2.3 Interaction entre les UBLs et d’autres modifications . . . 189
5.2.4 SUMOylation endogène . . . 190
1.I Exemples d’enzymes issues de la machinerie d’ubiquitination et de SUMOy-lation humaine . . . 4 1.II Récapitulatif des études à grande échelle visant à identifier les protéines
et/ou sites SUMOylés . . . 39 2.I Examples of identified SUMOylated peptides from LC-MS/MS analyses
S 2.I List of synthetic SUMO peptides identified using different activation modes
(ETD, CID, HCD) . . . 100
S 2.II Characteristic fragment ions observed during HCD fragmentation and their respective distribution of intensities . . . 100
S 2.III Distribution of scores for Mascot searches performed on HCD MS/MS spectra of synthetic peptides with and without removal of fragment ions associated with the SUMO remnant . . . 100
S 2.IV List of identified SUMO peptides from the LC-MS/MS analyses of tryptic digests from HEK293-SUMO3 cells . . . 100
S 2.V Distribution of scores for Mascot searches performed on HCD MS/MS spectra of SUMO3 peptides from HEK293-SUMO3 cells with and without removal of fragment ions associated with the SUMO remnant . . . 100
S 3.I List of identified SUMO substrates and modification sites from large-scale label-free quantitative proteomics analysis . . . 140
S 4.I List of identified SUMO peptides . . . 183
S 4.II List of identified SUMO sites . . . 183
S 4.III List of identified multiply modified peptides . . . 183
1.1 Structures tridimensionnelles de différentes UBL chez Arabidopsis thaliana
. . . 2
1.2 Régulation des UBLs par une cascade enzymatique . . . 3
1.3 Mécanisme de formation de chaine poly-modifiées . . . 5
1.4 Mécanisme de transfert d’une UBL depuis une enzyme E1 vers une en-zyme E2 . . . 6
1.5 Représentation schématique du domaine UBC . . . 7
1.6 Représentation schématique du fonctionnement des ligases de type HECT et RING . . . 8
1.7 Alignement des séquences SUMO1, 2 et 3 humain, Smt3 de levure et ubiq-uitine humaine . . . 12
1.8 Représentation de l’interaction entre un SIM et SUMO . . . 15
1.9 Mécanisme de formation des corps PML . . . 17
1.10 Processus d’identification des protéines par Shotgun Proteomics . . . 19
1.11 Production d’un anticorps monoclonal à partir d’hybridomes . . . 21
1.12 Sources d’ionisation communément utilisées en protéomiques . . . 25
1.13 Schéma des analyseurs de masse communément utilisées en protéomiques . 26 1.14 Schéma des différents Spectromètres de masse utilisant l’Orbitrap . . . 30
1.15 Nomenclature Roepstorff–Fohlmann–Biemann des ions fragments de pep-tides . . . 31
1.16 Méthodes de quantification relatives couramment utilisées en protéomique . 35 2.1 Schematic overview of the work flow used for the analysis of protein SUMOy-lation using DDA and DIA methods . . . 76
2.2 LC-MS/MS analyses of synthetic peptides with SUMO1 and SUMO3 rem-nant chains using ETD, CID, and HCD activation modes . . . 77
2.3 MS/MS spectra of INEILSNALKR with a Lys residue modified with SUMO1 or SUMO3 remnant chains . . . 78
2.4 Distribution of intensity for SUMO-specific fragment ions observed using HCD activation . . . 79
2.5 Enhanced identification and improvement of Mascot scores following the removal of SUMO-specific ions in the HCD MS/MS spectra . . . 81
2.6 LC-MS/MS analysis (DIA) of SUMOylated peptides spiked in a tryptic digest of HEK293 cells . . . 84
2.7 Identification of SUMO3peptides from 2D-LC-MS/MS analyses of a tryp-tic digest of HEK293-SUMO3 cells using DDA and DIA methods . . . 85
2.8 2D-LC-MS/MS analysis of tryptic digest of HEK293-SUMO3 cells (2 µg inj.) using DIA and targeted identification with an inclusion list . . . 86
3.1 SUMO remnant immunoaffinity purification strategy . . . 108
3.2 Enrichment of SUMOylated peptides using SUMO remnant immunoaffin-ity purification . . . 109
3.3 Large-scale LC-MS/MS analysis of SUMOylated peptides following pro-teasome inhibition . . . 112
3.4 Bioinformatic analysis of SUMOylated proteins and SUMO-modified lysines114 3.5 SUMOylation of CDC73 at K136 affects its nucleocytoplasmic localization 116 4.1 Optimization of a SUMO remnant immunoaffinity purification strategy . . 147 4.2 Motif analysis of SUMO-modified lysine residues . . . 151 4.3 Temporal profiling of the SUMOylome and ubiquitylome in response to
MG132 treatment . . . 154 4.4 Crosstalk between SUMOylation and ubiquitylation levels on the histone
deubiquitinase USP22 in response to MG132 . . . 155 4.5 SUMOylated and ubiquitylated proteins show a high degree of
intercon-nectivity . . . 156 4.6 Immunofluorescence of 20S Proteasome and PML or PML-SIM under
S 2.1 Schematic work flow of the algorithm used for data-independent
acquisi-tion (DIA) . . . 94
S 2.2 Relative proportion of fragment ions assigned to backbone sequence ions for different activation modes . . . 94
S 2.3 Impact of the position of the modified lysine residue on the fragmentation of synthetic peptides with SUMO remnant chain . . . 95
S 2.4 Evaluation of optimal ion storage conditions for data-independent acquisi-tion (DIA) . . . 96
S 2.5 Optimization of collision energy for the formation of neutral loss fragment ions from SUMO remnant peptides . . . 97
S 2.6 MS/MS spectra of identified SUMO peptides using MS/MS edition tools (separate pdf file) . . . 97
S 2.7 Sequence motif surrounding modified Lys residues . . . 98
S 2.8 Validation of SUMOylated Ubiquitin identification with synthetic peptide. . 99
S 2.9 Bioinformatics analyses for enriched GO terms representing SUMOylated proteins identified in this study . . . 99
S 3.1 Immunoprecipitation of synthetic SUMOm-remnant peptides using UMO 1-7-7 monoclonal antibody . . . 130
S 3.2 SUMO-remnant immunoaffinity purification with UMO 1-7-7 monoclonal antibody provides high specificity for a wide range of peptide sequences . . 131
S 3.3 Schematic workflow illustrating the selection and identification of SU-MOylated peptides . . . 132
S 3.4 Comparison of Mascot scores from identification of SUMOylated peptides with and without MS/MS editing . . . 133
S 3.5 Dynamic changes in protein SUMOylation of HEK293 SUMO3 mutant cells upon proteasome inhibition . . . 134
S 3.6 Distribution of SUMOylated peptide intensities from replicate injections . . 134
S 3.7 Distribution of all 20 amino acids for their propensity to be found proximal to SUMOylated lysines . . . 135
S 3.8 Confirmation of SUMOylation sites on CDC73 using an Ubc9 in vitro assay 135 S 3.9 CDC73 colocalizes with SUMO3 and PML within the NBs upon MG132 treatment . . . 136
S 3.10 SUMOylation of CDC73 affects its nucleocytoplasmic localization . . . 137
S 3.11 Full images of Western blots . . . 138
S 4.1 MS Optimization . . . 171
S 4.2 Preparation of the Anti K-(NQTGG) Bound Protein A/G Beads . . . 172
S 4.3 Anti K-(NQTGG) IP Optimization . . . 173
S 4.4 Physiochemical Properties of SUMO and Non SUMO Peptides . . . 174
S 4.5 SUMO Peptide Purification Workflow Reproducibility . . . 175
S 4.6 Comparison of the SUMO Sites Identified in this Study to those Reported to Date . . . 176 S 4.7 Temporal Profiling of SUMO and Ubiquitin Changes in Response to MG132177
S 4.8 Specificity of Ni-NTA Purification Step . . . 178 S 4.9 Mapped Residues Involved in PolySUMO and PolyUbiquitin Chain
For-mation . . . 179 S 4.10 List of Proteins Displaying Bidirectional Kinetic Profiles of SUMO and/or
Ubiquitin Levels in Response to MG132 Treatment . . . 180 S 4.11 Role of USP37 SUMOylation on the Levels of Myc Ubiquitination and
SUMOylation . . . 181 S 4.12 3D Mapping of the Modified Lysine Residue Identified on the Ribosome
and the Proteasome . . . 182 S 4.13 Immunofluorescence of 20S and 11S Proteasome and PML under MG132
ADN Acide Désoxyribonucléique AMP Adenosine Monophosphate
APC Anaphase Promoting Complex ARN Acide Ribonucléique
ATG Autophagy-Related Gene ATO Arsenic Trioxide
ATP Adenosine Triphosphate ATRA All Trans Retinoic Acid
BARD1 BRCA1-Associated RING Domain Protein 1 BIRC6 Baculoviral IAP Repeat Containing 6
BPC Base Peak Chromatogram
BRCA1 Breast Cancer Type 1 Susceptibility Protein CDC Cell Division Cycle
CHIP Carbocyl Terminus of Hsc70-Interacting Protein CID Collision Induced Dissociation
DAXX Death Domain-Associated Protein 6 DC Direct Current
DDA Data Dependent Acquisition DESI Desumoylating Isopeptidase DIA Data Independent Acquisition DUB Deubiquitinating Enzyme
E2–25K E2 Ubiquitin Ligase – 25 KiloDalton E6-AP El6-Associated Protein
EI Electron Impact
ELISA Enzyme-Linked Immunosorbent Assay ESI Electrospray Ionization
ETD Electron Transfer Dissociation FA Formic Acid
FDR False Discovery Rate GC Gas Chromatography H2B Histone H2B
HCD Higher Energy Collisional Dissociation HECT Homologous to E6-AP C Terminus
HEK Human Embryonary Kidney
HNRNP Heterogeneous Nuclear Ribonucleoprotein HPV Human Papillomavirus
ISG15 Interferon Stimulated Gene 15 LC Liquid Chromatography LIT Linear Ion Trap
LTQ Linear Trap Quadrupole
MALDI Matrix Assisted Laser Desorption Ionization MDM2 E3 ubiquitin-protein ligase MDM2
MDMX Protein MDM4 MS Mass Spectrometry
MS/MS Tandem Mass Spectrometry
NAE1 NEDD8-activating enzyme E1 regulatory Subunit NEM N-Ethylmaleimide
NF-κB Nuclear factor NF-kappa-B p105 Subunit NLS Nuclear Localization Signal
NTA Nickel Nitriloacetic Acid OTU Ubiquitin Thioesterase
PAGE Polyacrylamide Gel Electrophoresis PBS Phosphate Buffered Saline
PCNA Proliferating Cell Nuclear Antigen PDSM Phospho Dependent SUMO motif
PIAS Protein Inhibitor of Activated STAT PML Promyelocytic Leukemia Protein
PSMD 26S Proteasome non-ATPase Regulatory Subunit PTM Post-Translational Modification
Q-ToF Quadrupole-Time of Flight RanBP Ran Binding Protein
RanGAP Ran GTPase-Activating Protein RAP80 BRCA1-A Complex Subunit RAP80
RARA Retinoic acid receptor alpha RF Radio Frequency
RING Really Interesting New Gene RMN Résonance Magnétique Nucléaire
RNF RING Finger Protein RP Reverse Phase
RPS27A Ubiquitin-40S Ribosomal Protein S27a SAE SUMO Activating Enzyme
SAFB2 Scaffold Assembly Factor B2 SAX Strong Anion Exchange SCX Strong Cation Exchange
SDS Sodium Dodecyl Sulfate
SILAC Stable Isotope Labelling using Amino Acids in Culture SIM SUMO Interacting Motif
SP100 Nuclear Autoantigen Sp-100 SPE Solid Phase Extraction
SP-RING Siz/Protein Inhibitor of Activated STAT-RING STAT Signal Transducer and Activator of Transcription STUBL SUMO-Targeted Ubiquitin Ligase
SUMO Small Ubiquitin-related Modifier
SWATH Sequential Window Acquisition of all Theoretical Mass Spectra TBS Tris Buffered Saline
TFA Trifluoroacetic Acid
TIF1β Transcription Intermediary Factor 1-Beta TMT Tandem Mass Tag
ToF Time of Flight
TRIM Tripartite Motif-Containing Protein
UBA Ubiquitin-Like Modifier-Activating Enzyme UBB Polyubiquitin-B
UBC (domain) Ubiquitin Conjugating Domain UBC (gene) Polyubiquitin-C
Ubc9 E2 SUMO Conjugating Enzyme UBE Ubiquitin-Conjugating Enzyme UBL Ubiquitin-Like Protein
U-Box Ubiquitin Fusion Degradation Protein-Box UCH Ubiquitin Carboxyl-Terminal Hydrolase
UFD Ubiquitin Fold Domain UFM1 Ubiquitin-Fold Modifier 1
UIM Ubiquitin Interacting Motif URM1 Ubiquitin-Related Modifier 1
USP Ubiquitin Specific Peptidase USPL Ubiquitin-Specific Peptidase-Like
WAX Weak Anion Exchange WCX Weak Cation Exchange
∑
n=1
n
= −
12
Tout d’abord, je tiens à remercier le Dr Pierre Thibault pour son accueil. Il m’a accordé sa confiance dès mon arrivée dans son laboratoire malgré mon absence d’expérience avec la pro-téomique et a su m’enseigner les rudiments de la spectrométrie de masse, de la SUMOylation et surtout m’a appris à avoir confiance en moi.
Je souhaite aussi remercier tous les membres actuels et passés du groupe Thibault, et plus particulièrement les membres de la team SUMO avec lesquels j’ai eu le plaisir de collaborer : Louiza Mahrouche pour m’avoir formé lors de mon arrivée, Danielle Caron pour ton apport à la biologie du projet et Francis McManus pour avoir réussi à reproduire des résultats que l’on espé-rait plus. Mention spéciale pour Sibylle Pfammatter et Nebiyu Abshiru pour votre bonne humeur constante, ainsi qu’à Evgeny pour nos longues discussions au sujet de la biologie, de la chimie et du reste.
Merci également aux membres de la plateforme de protéomique: Olivier Caron-Lizotte et Mathieu Courcelles pour les différents outils que je vous ai forcé à développer, Christelle Pomiès pour ces nombreux "équivalents voyages à Cuba" que je t’ai fait commander, et Eric Bonneil pour la formation qu’il m’a offert et pour tous ces bons moments à développer l’approche par SWATH.
Un grand merci à nos collaborateurs à Paris, particulièrement Mounira K. Chelbi Alix, sans qui le projet n’aurait jamais vu le jour, ainsi que Mohamed Ali Maroui pour les cellules SUMO et Ghizlane Maarifi pour tes IF magnifiques.
Enfin, merci à mes collègues du département de Chimie, particulièrement Martin Dufresne pour ces grands moments à se rassurer lors des démonstrations.
INTRODUCTION
1.1 Maturation des protéines
Qu’elles soient procaryotes ou eucaryotes, la plupart des cellules subissent les mêmes trans-formations chimiques leur permettant de transcrire puis traduire l’information génétique conte-nue dans leur ADN en protéines fonctionnelles. Cependant, la plupart des protéines sont initiale-ment exprimées par la cellule sous forme inactive et doivent subir une série de modifications afin de les rendre fonctionelles.
On peut distinguer ces modifications en deux grandes catégories: les clivages protéolytiques et les modifications chimiques. Les clivages sont irréversibles et peuvent avoir différentes fonc-tions telles que le retrait de la méthionine initiatrice [1], l’exportation de la protéine vers le milieu extérieur [2, 3], l’activation de la protéine [4, 5], ou encore la synthèse de différentes protéines à partir d’un seul gène, notamment dans le cas de l’ubiquitine humaine qui peut être produite par quatre gènes distincs (UBB, UBC, UBA52 et RPS27A [6]).
Les modifications chimiques, quant à elles, vont permettre de réguler l’activité de facteurs de transcription [7], la localisation [8] ou encore la stabilité des protéines [9]. Contrairement aux cli-vages protéolytiques, ces modifications sont généralement réversibles et hautement dynamiques. A l’heure actuelle, la banque de données Uniprot [10] comprend 474 modifications post-traductionnelles pouvant affecter les 20 acides aminés naturels, les plus modifiés étant la cystéine (106 modifications), la sérine (56 modifications) et la lysine (55 modifications).
Dans le cadre de cette thèse, nous nous intéresserons à une famille de modifications retrou-vées sur les lysines, à savoir les modifications Ubiquitin-like (UBLs), et plus particulièrement la SUMOylation et l’ubiquitination.
1.2 Modifications ubiquitin-like 1.2.1 Historique
La première protéine membre de la famille des UBL est l’ubiquitine et a été découverte en 1975 par Goldstein et al. [11]. Initialement trouvée dans les cellules de thymus bovin, l’ubiqui-tine et la machinerie d’ubiquitination ont été depuis rapportées dans de nombreuses espèces eu-caryotes allant de la levure jusqu’à l’homme [12–17]. Il faudra cependant attendre 1987, avec la découverte de la protéine ISG15, pour que l’appellation UBL naisse [18]. Les années qui suivent ont permis la découverte de nombreuses autres protéines appartenant à cette famille, incluant FUBI en 1992 [19], ATG8 en 1993 [20], SUMO en 1996 [21], NEDD8 en 1997 [22], ATG12 en 1998 [23], FAT10 en 1999 [24], URM1 en 2000 [25] et UFM1 en 2004 [26]. Cependant, des évi-dences indiquent qu’il pourrait exister d’autres UBLs qui seraient conservées chez les eucaryotes [27].
1.2.2 Propriété des UBLs
Les UBL sont de courts polypeptides allant, chez l’humain, de 74 jusqu’à 165 acides aminés pour FUBI [19] et FAT10 [24] respectivement. Bien que toutes les UBLs aient des séquences très différentes, une des caractéristiques communes à toutes ces protéines est leur structure tridimen-sionnelle de type β β αβ β αβ [28] (Figure 1.1).
Figure 1.1 – Structures tridimensionnelles de différentes UBL chez Arabidopsis thaliana. Figure adaptée de [29].
De plus, la majorité des UBLs présente un motif di-glycine à leur extrémité C-terminale. Lors de la modification d’un substrat par une UBL, le groupement acide carboxylique de ce motif est lié de façon covalente au groupement amine en ε d’une lysine présente sur leur substrat. De plus, les analyses cristallographiques de différents complexes des UBLs liées à leurs enzymes respectives montrent que le motif di-glycine est un des seuls à pouvoir s’insérer dans la poche catalytique, la présence d’acides aminés volumineux rendant impossible la conjugaison et/ou déconjugaison de l’UBL à son substrat [30]. Il existe cependant quelques exceptions. En effet, chez l’humain, les protéines ATG8, ATG12 et UFM1 se terminent respectivement par YG [31] et FG [32], WG [23] et VG [26], ceux-ci n’empêchant pas la conjugaison des UBLs sur leurs substrats respectifs.
1.2.3 Mécanismes de régulation des UBLs
En plus de posséder des structures tridimensionnelles semblables, toutes les UBLs sont régu-lées par une cascade enzymatique similaire [34–37] (Figure 1.2a). Bien que ces réactions soient extrêmement semblables, chaque UBL est régulée par une machinerie enzymatique hautement spécifique [34–37] (Tableau 1.I).
Figure 1.2 – Régulation des UBLs par une cascade enzymatique. a) Cascade enzymatique. b) Liens peptidiques et isopeptidiques. Figure adaptée de [33].
Tableau 1.I – Exemples d’enzymes issues de la machinerie d’ubiquitination et de SUMOyla-tion. Les nombres indiqués entre parenthèses représentent le nombre d’enzymes connues. Adapté de [34–37]
E1 Enzyme E2 Enzyme E3 Enzyme Protéase Ubiquitin UBA1, UBA6 (2) UBE2, BIRC6 ...
(≈30)
RING, HECT ... (>600)
USP, UCH, OTU ... (≈80) SUMO SAE1/UBA2 (1) UBE2I (1) PIAS, RanBP2 ...
(≈10)
SENP, DESI, USPL1 (≈10)
1.2.3.1 Maturation et Déconjugaison
Qu’il s’agisse de l’ubiquitine qui est traduite sous la forme de polyubiquitine, ou SUMO pour qui l’extrémité C-terminale est bloquée par une chaine de 2 à 11 acides aminés, la plupart des UBL sont exprimées sous forme inactives [33]. La première étape consiste donc à cliver, à l’aide d’une protéase, un lien peptidique afin de libérer le motif di-glycine et le rendre accessible aux enzymes de conjugaison.
De plus, les enzymes permettant la maturation des UBLs permettent également leur décon-jugaison [33]. Dans le cas de la maturation, on parle d’activité endopeptidique (clivage d’un lien amide entre l’amine en α d’un acide aminé et le groupement acide en position 1 d’un second acide aminé), alors que l’on parle d’activité isopeptidique dans le cas de la déconjugaison (cli-vage d’un lien amide impliquant une fonction amine ou acide carboxylique sur la chaine latérale d’un acide aminé, Figure 1.2b).
1.2.3.2 Conjugaison
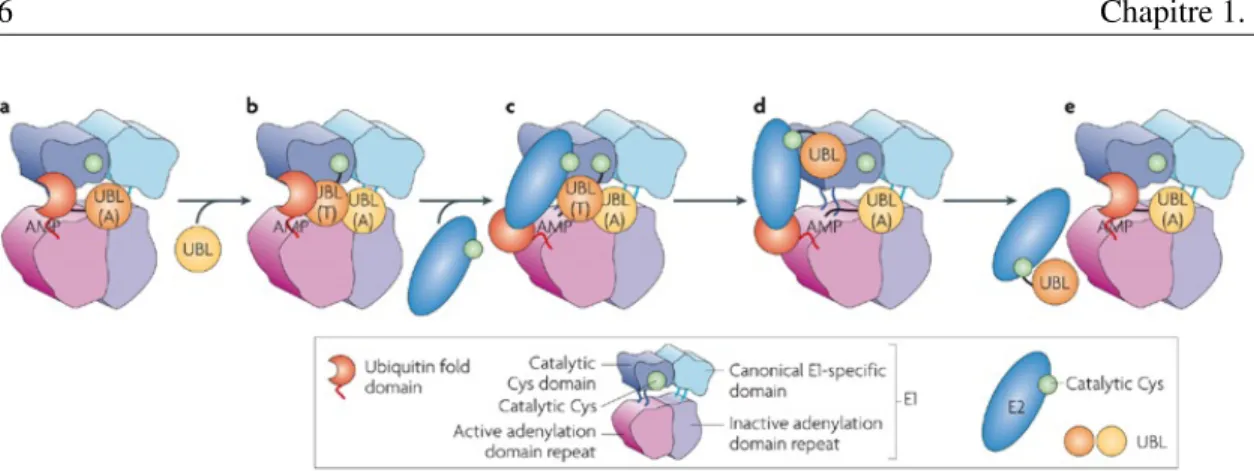
La conjugaison des UBLs se fait en trois étapes. Dans un premier temps, une enzyme E1 permet d’activer l’UBL via consommation d’ATP. Lors de ce processus, le groupement acide carboxylique du motif di-glycine vient se lier à la cystéine présente dans le domaine catalytique de l’enzyme E1 en formant un groupement thioester.
Dans un second temps, l’UBL est transférée de la cystéine de l’enzyme activatrice E1 à la cystéine du domaine catalytique d’une enzyme de conjugaison E2. Enfin, une E3 ligase permet de faire un pont entre le complexe UBL-E2 et le substrat, et ainsi de transférer l’UBL sur le groupement ε d’une lysine portée par la protéine ciblée. Cette dernière étape n’est cependant pas toujours nécessaire. Dans le cas de la SUMOylation notamment, il a été montré qu’UBC9,
l’unique SUMO-E2 connue chez l’humain, peut SUMOyler certain de ses substrats in vitro [38] et in vivo [39] sans l’emploi de E3 ligases.
Une fois une protéine modifiée par une UBL, l’UBL peut elle-même devenir le substrat d’une nouvelle conjugaison, permettant ainsi la formation de chaine poly-UBL homogènes (si la chaine ne contient qu’un type d’UBL) ou mixtes (si plusieurs UBLs interviennent) (Figure 1.3).
1.2.3.2.1 Enzymes d’activation E1
Chez l’humain, on connait 8 protéines et complexes protéiques différents pouvant jouer le rôle de E1 [23, 26, 41–45] . Bien que la machinerie de modification soit hautement spécifique, certaines E1 permettent la reconnaissance de différentes UBLs. C’est notamment le cas de UBA6, cette E1 permettant de modifier les protéines avec FAT10 ou l’ubiquitine in vitro [44].
Chez l’humain, on connait deux E1 pour l’ubiquitine (UBA1 et UBA6) [45], une seule pour les différents paralogues de SUMO (le complexe SAE1/UBA2) [42], une seule pour la NEDD8 (le complexe NAE1/UBA3) [41], ou encore une unique E1 pour ATG8 et ATG12 (l’homodimère ATG7) [23].
Toutes les E1 comportent des domaines similaires. D’abord, le domaine d’adénylation per-met de catalyser la réaction d’adénylation de l’ATP sur le groupement C-terminal de l’UBLs. Ensuite, le domaine catalytique portant la cystéine active favorise le recrutement de l’UBL via la formation d’un lien thioester. Enfin, le Ubiquitin Fold Domain (UFD) permet l’interaction entre les enzymes E1 et E2 [46–48].
Figure 1.4 – Mécanisme de transfert de l’ubiquitine depuis UBA1 vers une enzyme E2. Figure reproduite de [49].
Le mécanisme d’activation le mieux caractérisé à ce jour est celui de UBA1 (Figure 1.4). Dans un premier temps, l’ubiquitine mature est adenylée par une molécule d’ATP, permettant ainsi de former l’intermédiaire ubiquitine-AMP (Ub-AMP) [50]. Ensuite, Ub-AMP est attaquée par la cystéine du domaine catalytique de UBA1, formant ainsi un lien thioester entre le C-terminal de l’ubiquitine et la cystéine de UBA1 [51, 52]. UBA1 va ensuite catalyser l’adénylation d’une seconde molécule d’ubiquitine libre, permettant à UBA1 de charger une autre molécule Ub-AMP, cette fois ci par une interaction non covalente [53]. Une fois chargée avec deux molé-cules d’ubiquitine, l’affinité entre la E1 et une E2 augmente significativement, avec une constante de dissociation passant de l’ordre du milli molaire au nano molaire [54, 55]. UBA1 va ensuite s’associer via son domaine UFD au domaine Ubiquitin Conjugating (UBC) d’une enzyme E2 afin de transférer une molécule d’ubiquitine par trans-thiolation, ceci permettant de diminuer de nouveau l’affinité entre la E1 et la E2 et de dissocier le complexe précédemment formé [53, 56].
1.2.3.2.2 Enzymes de conjugaison E2
Quel que soit les UBLs impliquées, toutes les conjugases E2 portent un même domaine ca-talytique d’environ 150 à 200 acides aminés, le domaine UBC (Figure 1.5) [57]. Celui-ci est composé d’une première hélice α, d’un ensemble de 4 feuillets β associés de façon antiparallèle, suivi par le domaine catalytique replié en épingle à cheveux, pour se terminer avec une seconde hélice α [58] (Figure 1.5).
Chez l’humain, on connait environ 40 E2 ligases dont 30 sont spécifiques à l’ubiquitination [59]. En revanche, on ne connait à l’heure actuelle que 3 enzymes E2 pour ISG15 (UBE2L6, UBE2E1 et UBE2E2) [60], que 2 pour NEDD8 (UBE2M et UBE2F) [61] et une unique enzyme
Figure 1.5 – Représentation schématique du domaine UBC. Figure adaptée de [58].
E2 pour la SUMOylation (UBE2I) [62].
Bien qu’elles présentent toutes un domaine UBC similaire, les séquences en acides aminés des différentes E2 sont extrêmement variables [58]. En effet, le seul acide aminé commun à toutes les E2 est la cystéine permettant le recrutement de l’UBL. Certaines E2 présentent également un ou plusieurs acides aminés polaires (essentiellement Asparagine et Histine) proche de la cystéine catalytique. Ces acides aminés ont comme principale fonction de stabiliser l’oxyanion formé lors de la condensation du groupement di-glycine de l’ubiquitine et de la cystéine de l’enzyme E2 [58, 63].
Les enzymes E2 sont regroupées en fonction des différents groupements se trouvant de part et d’autre du domaine UBC. Ainsi, les enzymes ne comportant que le domaine catalytique sont des membres de la classe I, les enzymes présentant des excroissances N ou C-terminales font parties des classes II et III respectivement et les enzymes présentant les deux font parties de la classe IV [59]. Ces additions permettent de modifier différentes fonctions des E2, incluant la stabilisation des différentes enzymes, leur localisation dans la cellule ou encore la régulation des interactions avec certaines E1 et E3.
1.2.3.2.3 Enzymes de ligation E3
Alors que les E1 et E2 ne présentaient que peu de variété, on estime qu’il existe plus de 600 différentes E3 ligases chez l’humain. Ce grand nombre de E3 permet d’apporter une grande
spécificité aux modifications [64], et ainsi un meilleur contrôle des événements de conjugaison. Les E3 ligases sont classées en fonction du domaine interagissant avec l’enzyme E2. Parmi elles, on retrouve essentiellement des enzymes appartenant au groupe des protéines à domaine RING (Really Interesting New Gene) et HECT (Homologous to E6-AP-Carboxy-Terminus) (Figure 1.6). Il existe également d’autres classes de E3, ces dernières sont cependant faiblement représentées et ne seront pas décrites dans les paragraphes suivants.
1.2.3.2.3.1 E3 ligases à domaine RING
La très large majorité des E3 ligases appartient à cette famille. En effet, on estime que cette famille d’enzyme est potentiellement peuplée par 616 membres distincts chez l’humain [66]. Leur principale fonction est de former un pont entre E2-UBL et leurs substrats en se liant à la E2 par son domaine RING et aux substrats via d’autres domaines présents sur les différentes enzymes.
Initialement décrit en 1991 par Freemont et al. [67], le domaine RING est une séquence riche en cystéines et histidines permettant de lier plusieurs cations bivalents, notamment le Zinc. Contrairement au domaine ZNF (Zinc Finger) qui forme une structure linéaire, le domaine RING permet de former une structure globulaire rigide, permettant des interactions protéines-protéines fortes [68, 69] .
La grande majorité des protéines portant un domaine RING est impliquée dans les méca-nismes de ligation d’une UBL, que ce soit directement ou indirectement. En effet, les protéines telles que PIAS (Protein Inhibitor of Activated STAT) ou encore la plupart des RNF (RING Fin-ger Protein), présentent une activité E3 ligase de façon constitutive et permettent de catalyser la ligation de SUMO et ubiquitine respectivement sur son substrat. En revanche, certaines pro-téines, telles que BARD1 [70] ou MDMX [71], possèdent un domaine RING mais ne possèdent
Figure 1.6 – Représentation schématique du fonctionnement des ligases de type HECT et RING. Figure reproduite de [65].
pas d’activité de ligation. Ces protéines doivent dans un premier temps s’associer sous forme d’hétérodimère (avec BRCA1 pour BARD1 ou avec MDM2 pour MDMX) afin de permettre la ligation d’une UBL sur leur substrat.
Cependant, le rôle des E3 à domaine RING ne consiste pas uniquement de rapprocher E2-UBL et son substrat. En effet, certains complexes E2-E3, comme UbcH7-ubiquitine et Brca1-Bard1 chez la levure [72], sont capable de se former avec une très grande stabilité, mais ne pré-sentent aucune activité catalytique. D’autres complexes sont au contraire extrêmement instables mais présentent une forte activité catalytique. Ceci suggère que la E3 permet également d’activer la E2 et que la formation du complexe seule n’est pas suffisante à la ligation d’une UBL sur son substrat [73].
1.2.3.2.3.2 E3 ligases à domaine HECT
Bien qu’étant le deuxième groupe le plus représenté par les E3 ligases, celui-ci ne contient qu’environ 30 protéines différentes chez l’humain. Originellement découvert sur la protéine E6-AP, le domaine HECT est une séquence d’environ 350 acides aminés présente à l’extrémité C-terminale de certaines E3 ligases.
Contrairement au domaine RING, le domaine HECT agit selon un mécanisme similaire aux enzymes E1 et E2 et permet de former une liaison thioester avec le motif di-glycine. Le domaine HECT est composé de deux lobes [74–76]. Le grand lobe N-terminal contient le domaine de liaison avec la E2 alors que le plus petit lobe C-terminal contient le domaine catalytique.
Une particularité des ligases portant un domaine HECT est leur rôle dans les événements de polyubiquitination. En effet, et bien qu’elles puissent former des chaines polyubiquitinée de façon séquentielle, ces enzymes ont la possibilité d’accumuler des chaines polyubiquitines sur leur site catalytique, avant de transférer cette chaine sur un substrat non modifié [77, 78]. Ainsi, E6AP assemble des chaines polyubiquitinées en K48 sur sa cystéine avant de transférer le tout sur un substrat, alors que UBE3C permet de polyubiquitiner en K48 et K29 une ubiquitine libre, puis de transférer la chaine complète sur un substrat.
1.2.4 Ubiquitination
L’ubiquitine est une petite protéine d’environ 7 kDa extrêmement conservée chez les euca-ryotes. Bien qu’il n’existe aucun homologue de l’ubiquitine chez les procaryotes,
l’ubiquitina-tion trouverait son origine dans les protéines bactériennes ThiS (Thiamine biosynthesis protein) et MoaD (Molybdopterin synthase sulfur carrier subunit) [79, 80]. Ces protéines ne présentent qu’une très faible identité de séquence avec l’ubiquitine, cependant leur structure tridimensio-nelle est similaire aux UBLs. De plus, les protéines ThiS et MoaD sont généralement associées à ThiF (Sulfur carrier protein ThiS adenylyltransferase) et MoeB (Molybdopterin-synthase ade-nylyltransferase) ayant un rôle similaire aux enzymes d’activation E1 des UBLs. Cependant, ces deux protéines ont une fonction très différente de l’ubiquitine, celles-ci étant associées au transfert de sulfures sur leurs substrats.
Chez l’humain, quatre gènes distincts codent pour l’ubiquitine (UBB, UBC, UBA52 et RPS27A [6]). Parmi eux, UBA52 et RSP27A sont des protéines de fusion entre un monomère d’ubiquitine et une protéine ribosomale alors que UBB et UBC sont des polymères d’ubiquitine, codant pour 4 et 9 molécules d’ubiquitine respectivement.
1.2.4.1 Spécificité d’ubiquitination
A ce jour, on ne connait pas de motif consensus d’ubiquitination [81]. Il est cependant admis que la spécificité d’ubiquitination est essentiellement apportée par le grand nombre d’enzyme E3 répertoriées pour l’ubiquitine [64].
1.2.4.2 Aperçu des fonctions de l’ubiquitine
L’ubiquitination est la première des UBLs à avoir été découverte [11]. Originellement, cette protéine était associée à la différenciation des pro-thymocytes ainsi que des cellules B de la moelle osseuse. Cependant, le rôle majeur qui lui est attribué de nos jours est de permettre la dégradation des protéines par le protéasome [82–84].
Dans des conditions basales, l’ubiquitination permet notamment de contrôler le cycle cel-lulaire. Un des exemples les plus étudiés à ce jour concerne la E3 ligase Anaphase-promoting complex (APC). En effet, cette E3 est régulièrement activée et désactivée lors des différentes transitions du cycle cellulaire, permettant la dégradation successive de la Cycline A pour le pas-sage de la prophase à la métaphase, puis de la Sécurine et la Cycline B pour le paspas-sage et la métaphase à l’anaphase et enfin de différents facteurs de transcription, kinases et ligases afin de permettre le retour en phase G1 [85].
de stress. En effet, l’ubiquitination permet notamment d’envoyer les protéines dénaturées lors d’un choc thermique vers le protéasome [86, 87], ou encore de permettre la régulation des en-zymes de réparation de l’ADN en cas de détection d’une mutation [37, 88, 89]. Cependant, si les dommages causés par le stress sont trop important, l’ubiquitination peut également permettre d’activer l’apoptose [90–92].
Ces mécanismes de dégradations font généralement intervenir la polyubiquitination des sub-strats sur la lysine K48 [93]. Tel que mentionné précédemment, la polyubiquitination du substrat peut avoir lieu de façon séquentielle, l’ubiquitine devenant le nouveau substrat d’une E3 ligase, ou encore avoir lieu en une seule étape pour certaines E3 ligases de la famille des HECT (voir la section 1.2.3.2.3.2 pour plus de détails à ce sujet). Cependant, il a également été démontré que la monoubiquitination de certains substrats était une condition suffisante pour leur dégradation par le protéasome [94].
La polyubiquitination peut également avoir lieu en K6, K11, K27, K29, K33, K63 et, plus rarement, en M1, formant de ce fait des chaines linéaires de polyubiquitination. A ce jour, les différentes chaines de polyubiquitination sont encore mal caractérisées. Cependant, il a été mon-tré que ces chaines pourraient être directement liées à la fonction de l’ubiquitination sur les différentes protéines [95]. Ainsi, la polyubiquitination en K6 a été associée à la réponse aux dommages à l’ADN [96, 97], en K11 pour le contrôle du cycle cellulaire [98, 99], en K63 pour l’endocytose [100] ou encore en M1 la réponse immunitaire innée par la régulation de la signali-sation de NF-κB [101, 102].
1.2.4.3 Motif d’interaction avec l’ubiquitine
L’ubiquitine peut également interagir de façon non covalente avec certaines molécules via une courte chaine d’environ 20 acides aminés appelée motif d’interaction avec l’ubiquitine (UIM), ou encore séquence LALAL en référence avec l’UIM identifié dans la sous-unité PSMD4 du pro-téasome [103, 104]. L’analyse de différents UIM a permis de déterminer la séquence consensus xAcAcAcAcΦxxAxxxSxxAcxxxx où Ac représente un acide aminé acidique (D ou E), Φ un acide aminé hydrophobe volumineux (généralement L) et x n’importe quel acide aminé. De plus, des analyses réalisées par RMN montrent que cette séquence forme une hélice α interagissant avec les feuillets β de l’ubiquitine [105].
Cette séquence permet de recruter des protéines ubiquitinée à leur destination. C’est notam-ment le cas de PSMD4 dont le rôle est de recruter les protéines polyubiquitinées en K48 afin de
les dégrader [103, 104].
1.2.5 SUMOylation
Tout comme l’ubiquitine, SUMO est une protéine exprimée chez tous les eucaryotes. Cepen-dant le nombre de paralogue de SUMO peut varier d’un organisme à l’autre, allant d’un seul chez S. cerevisae (Smt3) [106] jusqu’à 8 chez Arabidopsis thaliana (SUMO1 à 8) [107]. Chez l’humain, on retrouve trois paralogues exprimés de façon ubiquitaire dans toutes les cellules (SUMO1, 2 et 3) et un quatrième exprimé uniquement dans les reins, les ganglions lympha-tiques et la rate (SUMO4) [108]. Des évidences montrent également qu’un cinquième paralogue, SUMO5, pourrait exister dans certains tissus [109]. Ce cinquième paralogue n’a cependant pas encore été observé au niveau protéique, les évidences de ce dernier se limitant à une observation de l’ARNm codant pour SUMO5.
Les formes matures de SUMO2 et 3 ne diffèrent que par 3 résidus, alors que SUMO1 et SUMO2/3 partagent environ 50% de leur séquence (Figure 1.7). A ce jour, la fonction des diffé-rents paralogues de SUMO ne sont pas claires. Cependant, la délétion du gène SUMO2 est létale au stade embryonnaire chez la souris [110], alors que SUMO1 ou SUMO3 sont dispensables et leurs délétions n’empêchent pas le développement des organismes [110, 111].
Les récentes analyses à grande échelle réalisées par spectrométrie de masse révèlent qu’un grand nombre de substrats sont communs aux différents paralogues de SUMO [113], suggérant ainsi une redondance de fonction des différentes formes. En revanche, SUMO4 se comporte très différemment des autres paralogues. En effet, la proline située à proximité du motif di-glycine de SUMO4 limite sa conjugaison à ses substrats [114]. Malheureusement, peu d’étude à ce jour se sont portées sur l’étude de SUMO4, limitant ainsi les connaissances à son sujet.
Figure 1.7 – Alignement des séquences SUMO1, 2 et 3 humain, Smt3 de levure et ubiquitine humaine. Figure reproduite de [112].
1.2.5.1 Nomenclature de SUMO chez l’humain
La nomenclature des différents paralogues de SUMO chez l’humain n’est pas consistante. La plus utilisée à ce jour a été initialement introduite par Saitoh et Hinchey [115]. Celle-ci pro-pose que la forme immature de SUMO1 comprenne 101 acides aminés, alors que les formes matures de SUMO2 et SUMO3 sont respectivement de 92 et 93 acides aminés. Cependant, cette nomenclature porte à confusion. En effet, la nomenclature officielle utilisée par Uniprot repose sur les homologies de séquences entre les différentes espèces. Ainsi, SUMO2_human est l’ho-mologue de Sumo2_mouse, les deux comportant en réalité 93 acides aminés. Pour des raisons de consistance, cette thèse utilisera exclusivement la nomenclature Uniprot. De ce fait, lorsque l’on référencera les ouvrages et publications de différents groupes, il se peut que les noms des paralogues mentionnés dans cette thèse soient différents de ceux utilisés originellement.
1.2.5.2 Spécificité de SUMOylation
Rapidement après la découverte des premiers substrats SUMOylés, Rodriguez et al. ont ob-servés que la SUMOylation survenait dans des séquences ψ-K-x-E/D [116, 117] (ou ψ est un acide aminé aliphatique et x n’importe quel acide aminé). Ce motif consensus est de loin do-minant et représente la majorité des substrats les plus abondants [118]. Ceci s’explique par le fait que ce motif peut être reconnu par UBC9 seul et ne nécessite aucune E3 ligases pour être SUMOylé [38, 39].
En 2006, Hietakangas et al. ont étendu le motif consensus en identifiant un sous-groupe de substrat présentant un motif de SUMOylation phospho-dépendent (PDSM) ψ-K-x-E/D-x-x-pS-P [119]. C’est ensuite avec le développement de nouvelles méthodes protéomiques que d’autres groupes vont caractériser de nouveaux motifs consensus tels que le motif dit inversé E/D-x-K-ψ [120].
Plusieurs autres motifs ont également été observés, tel que le motif de SUMOylation à do-maine négatif (NDSM) [119], le motif de SUMOylation à cluster hydrophobique (HCSM) [120] ou encore le motif de SUMOylation Zinc-Finger [121]. Cependant, ces motifs consensus ne semblent être observés que dans des études isolées.
1.2.5.3 Aperçu des fonctions de la SUMOylation
Initialement identifiée sur la protéine RanGAP [122], la principale fonction associée à la SUMOylation a longtemps été le transport de protéines du cytoplasme vers le noyau [123–125]. En effet, les premières études portées sur SUMO1 semblait révéler que, pour être modifiée, une protéine devait porter à la fois un motif consensus et un NLS [116]. De plus, il a également été montré chez la levure que la délétion d’Ubc9 perturbait l’importation des protéines vers le noyau [126].
On sait depuis que la SUMOylation peut être impliquée dans un grand nombre de fonctions tels que la réponse aux dommages à l’ADN [127], la régulation du cycle cellulaire [128], ou encore la biogénèse des ribosomes [129].
Tout comme l’ubiquitination, SUMO peut s’auto-SUMOyler pour former des chaines poly-SUMO sur différentes lysines [130]. De plus, de nombreuses évidences montrent qu’il existe des chaines mixtes poly-SUMO entre les différents paralogues, y compris avec SUMO1 qui a long-temps était reporté comme terminant des chaines poly-SUMO [121, 131–133]. Il est cependant nécessaire de préciser que ces chaines poly-SUMO impliquant SUMO1 ont été identifiées dans le cadre d’études à grande-échelles utilisant des mutants surexprimés de SUMO et pourraient en réalité correspondre à des artéfacts.
Bien qu’observées depuis longtemps, on ne connait pas encore la fonction de ces chaines. Une étude récente réalisée chez la levure montre cependant que la polySUMOylation de la co-hésine est nécessaire pour son ubiquitination subséquente, entrainant ainsi sa dégradation [134]. Cette dernière étude suggère donc un fort lien entre les différentes UBLs et montre que la po-lySUMOylation pourrait être un signal de dégradation (ce point sera détaillé dans la section 1.2.6.2).
1.2.5.4 Motif d’interaction avec SUMO
Tout comme pour l’ubiquitination, un grand nombre de protéines peuvent interagir de façon non-covalente avec SUMO par l’intermédiaire d’un motif d’interaction avec SUMO (SIM). Il s’agit d’une courte chaine composée d’acides aminés hydrophobes ou acidique [135–137] for-mant un feuillet β pouvant se lier à SUMO de façon parallèle ou antiparallèle [136] (Figure 1.8). Cette interaction peut avoir de multiples rôles, notamment de permettre le recrutement de E2-SUMO vers une E3 ligase [138], ou encore de former des complexes protéiques [139].
Figure 1.8 – Représentation de l’interaction entre un SIM et SUMO. Figure adaptée de [140].
1.2.6 Interaction entre ubiquitination et SUMOylation
La SUMOylation et l’ubiquitination présentent énormément de similarité, que ce soit dans leur structure ou dans leurs mécanismes de régulations. De plus, ces deux modifications affectent un même acide aminé, à savoir la lysine. De ce fait, il est assez fréquent d’observer un même substrat pouvant présenter l’une ou l’autre de ces modifications en fonction des conditions cel-lulaires. Il y a donc une interaction permanente entre SUMO et l’ubiquitine, que celle-ci soit coopérative ou compétitive.
1.2.6.1 PCNA: compétition entre SUMO et ubiquitine
On retrouve notamment cet effet de compétition avec la protéine Proliferating Cell Nuclear Antigen(PCNA). Lors de la réplication de l’ADN, PCNA forme un homo-trimère entourant la molécule d’ADN, ceci permettant de détecter les dommages présents sur l’ADN et permet leur correction en recrutant les enzymes de réparation de l’ADN [141]. Chez la levure, PCNA peut être modifiée sur les lysines 127 et 164 par SUMO et ubiquitine, contrôlant ainsi les protéines qui seront recrutées à l’ADN.
Dans des conditions normales, PCNA est SUMOylée, permettant ainsi le recrutement de l’hélicase Srs2 et assurant la réplication normale de l’ADN par la polymérase δ [142]. En re-vanche, lorsqu’un dommage à l’ADN est rencontré, PCNA est déSUMOylée pour être ubiquiti-née [143, 144]. Dans le cas d’une monoubiquitination, PCNA recrute la polymérase η, permettant de forcer la réplication malgré cette erreur, laissant ainsi une mutation dans le génome qui pourra par la suite être corrigée par le système de réparation par excision de nucléotides [145, 146]. En
revanche, en cas de polyubiquitination en K63, PCNA recrute la machinerie de réparation par template switching, assurant une réplication sans erreur [147, 148].
1.2.6.2 SUMO-Targeted Ubiquitin Ligase
Une classe particulière de E3 ligase de l’ubiquitine a pour rôle de modifier spécifiquement les lysines portées par des protéines polySUMOylées. Cette classe de E3 ligase est dénommée SUMO-Targeted Ubiquitin Ligase (STUBL) et est caractérisée par la présence d’un domaine RING permettant d’exprimer leur activité de ligation, ainsi qu’un ou plusieurs domaines SIM assurant la reconnaissance des substrats SUMOylés [149, 150].
Une fois SUMOylées et ubiquitinées, ces protéines peuvent suivre deux voies différentes. Certaines seront transférées vers le protéasome afin d’y être dégradées. D’autres vont pouvoir se lier spécifiquement à des protéines possédant un domaine SIM et un domaine UIM, comme c’est le cas pour RAP80, ce qui permettra de former de nouveaux complexes protéiques avec les protéines nouvellement ubiquitinées [89].
1.2.6.3 Corps nucléaires PML
PML est un suppresseur de tumeur exprimé dans toutes les cellules humaines [151–153]. Ini-tialement présente dans les cellules sous la forme de monomères ou de dimères, PML forme des corps nucléaires dans différentes conditions de stress, incluant le choc thermique [154], les dom-mages à l’ADN [155–157], la réponse antivirale [158], l’inhibition du protéasome [159–161] ou encore le stress oxydatif [162, 163], notamment en réponse au trioxyde d’arsenic [139, 164, 165]. Ces corps sont ensuite SUMOylés, provoquant le recrutement de protéines contenant un SIM (en interagissant avec les sites de SUMOylation de PML) ou étant SUMOylés (en interagissant avec le SIM de PML) [166] (Figure 1.9).
Parmi les protéines recrutées dans les corps PML matures, on retrouve la STUBL RNF4 et le protéasome [167, 168]. RNF4 va permettre l’ubiquitination des substrats SUMOylés, incluant le suppresseur de tumeur SP100 [169], le répresseur de facteur de transcription DAXX [170], ou même PML elle-même [150]), entraînant ainsi leur dégradation par le protéasome [171, 172].
Figure 1.9 – Mécanisme de formation des corps PML. Figure reproduite de [166].
1.2.7 Intérêt clinique de l’étude des UBLs
Alors que l’ubiquitination est une modification très abondante dans les cellules, les autres UBLs ne sont visibles qu’à de très faibles stœchiométries. Cependant, les troubles de la régu-lation de ces différentes modifications ont étés observées dans différentes pathologies, incluant différents types de cancer. Dans d’autres cas, ce sont les traitements utilisés contre différentes maladies qui peuvent avoir entraîné des changements des niveaux de SUMOylation ou d’ubiqui-tination.
1.2.7.1 Leucémie Promyélocytaire Aiguë : une guérison induite par la SUMOylation La leucémie promyélocytaire aiguë (AML) est une maladie induite par la translocation du gène codant pour le récepteur de l’acide rétinoïque (RARA). Sous sa forme la plus répandue, tou-chant près de 95% des patients, le gène RARA est transloqué sur le gène codant pour la protéine promyelocytic leukemia(PML), altérant de ce fait les fonctions des deux protéines. En l’occur-rence, les corps PML/RARA sont plus petits et plus nombreux et la cinétique de SUMOylation de PML/RARA est réduite en comparaison avec PML sauvage [173]. De plus, PML/RARA va se lier à l’ADN avec une affinité supérieure à RARA sauvage, bloquant la transcription des gènes ciblés [174].
A ce jour, la leucémie provoquée par la translocation PML/RARA est l’une des mieux trai-tées, notamment grâce à l’utilisation combinée de l’acide rétinoïque all-trans (ATRA) et du tri-oxyde d’arsenic (ATO). L’ATRA induit de façon passive la formation des corps PML, mais per-met également d’induire la sénescence des cellules cancéreuses [175]. De son côté, l’ATO va induire un stress oxydatif, augmentant encore la formation des corps PML, tout en entrainant l’apoptose des cellules via un mécanisme indépendant de PML [139, 176]. De ce fait, ce trai-tement combiné permet de rétablir une structure similaire aux corps PML sauvages, menant à la dégradation de PML/RARA ainsi que des partenaires interagissant avec PML. Cependant, les mécanismes de reconnaissance et la nature d’un grand nombre de ces partenaires restent encore à déterminer [177].
1.2.7.2 Pathologies causée par des troubles des UBLs
Un grand nombre de troubles de la régulation des UBLs entraine la formation de tumeurs [79]. Par exemple, la stabilité de nombreux gènes suppresseurs de tumeurs et d’oncogène est régulée par leur ubiquitination et/ou SUMOylation. Par exemple, on peut noter que la protéine virale E6-AP codée par le virus HPV (Human papilloma viruses) entraine l’ubiquitination puis la dégradation de p53, augmentant ainsi les risques de cancer chez les patients infectés par ce virus [178–180]. De plus, les perturbations du cycle cellulaire peuvent également mener à la formation de tumeurs. L’ubiquitine et SUMO ayant un rôle essentiel dans la régulation de ces processus, des perturbations de la cascade enzymatique de l’ubiquitination [181] et de la SUMOylation [182] peuvent être impliquées dans différents cancers.
D’autres pathologies sont également en lien avec ces deux modifications, notamment les troubles neurologiques tels que la maladie de Parkinson [183, 184] ou la maladie d’Alzheimer [185, 186], ainsi que différentes maladie auto-immune [187] ou même l’insuffisance cardiaque [188].
Bien que la découverte de ces modifications soit encore récente, les preuves montrant qu’elles sont impliquées dans un grand nombre de pathologie sont déjà connues. Une meilleure explora-tion de la SUMOylaexplora-tion et l’ubiquitinaexplora-tion permettrait donc d’avoir une meilleure compréhension de ces différentes maladies, ainsi que de nouvelles pistes pour des voies thérapeutiques.
1.3 Analyse protéomique par spectrométrie de masse
Avant l’avènement de la spectrométrie de masse (MS), la majorité des analyses d’échantillons biologiques étaient réalisées par chromatographie liquide (LC) ou gazeuse (GC) couplée à des détecteurs spectroscopiques ou électrochimiques. Bien que ces méthodes permettaient de faire des analyses d’un très grand nombre de composés simultanément, celles-ci étaient promptes à générer un grand nombre de faux positifs [189–191].
D’autre part, les méthodes immunochimiques, telles que le test Enzyme-Linked Immuno-sorbent Assay(ELISA), permettent d’identifier des composés biologiques sans ambiguïté. Ce-pendant ces approches ne permettent l’identification que d’un très faible nombre de molécules simultanément. De plus, ces méthodes reposent sur l’utilisation d’enzyme ou d’anticorps et peuvent donc produire des résultats variables selon les lots d’anticorps [192].
La chromatographie couplée à la MS combine les avantages des deux approches. En ajou-tant une dimension supplémentaire à la séparation chromatographiques avec l’identification des masses des composés analysés, ces méthodes permettent d’identifier un grand nombre de com-posés sans ambiguïté.
1.3.1 Stratégie d’identification des protéines
L’approche la plus communément utilisée pour l’identification des protéines par spectromé-trie de masse consiste en un processus multi-étapes appelé Shotgun proteomics (Figure 1.10).
Dans un premier temps, l’échantillon (cellules, tissus, organes ...) doit être lysé afin d’en
extraire les protéines. Ces dernières sont ensuite digérées en peptides afin de faciliter leur analyse et identification par les outils de MS. Enfin les peptides sont injectés sur un système LC-MS/MS. Ces étapes génériques peuvent être additionnées de plusieurs méthodes de purification ou de séparation afin d’augmenter la couverture de séquence de différentes familles de protéines.
1.3.2 Extraction des protéines
Après avoir récolté des cellules ou extrait un organe d’intérêt d’un animal, la première étape consiste à briser les membranes des cellules afin d’en extraire les protéines qui les composent. Cette extraction peut se faire de façon mécanique (dounce homogenizer [194], bead beating [195] ...) ou par l’utilisation de tampons adéquats fragilisant la membrane cellulaire (tampon hypertoniques [196], digestion enzymatique [197] ...). Les membranes des organites présentant des résistances différentes à certaines conditions de stress, il est également possible de réaliser des lyses successives afin de fractionner les protéines en fonction de leur compartiment cellu-laire d’origine. Un fractionnement souvent utilisé pour les cellules humaines consiste à lyser les membranes cytoplasmiques avec un tampon hypotonique puis des membranes nucléaires avec un tampon hypertonique [121].
Une fois les protéines extraites, il est commun de fractionner l’échantillon afin d’identifier les protéines d’intérêt. La méthode la plus communément utilisée pour séparer les protéines est l’électrophorèse sur gel de polyacrylamide en présence de dodécylsulfate de sodium (SDS-PAGE). Alors que les méthodes conventionnelles d’électrophorèse permettent d’obtenir une sé-paration des molécules en fonction de leur structure tridimensionnelle et de leur charge, l’ajout SDS dénature les protéines, permettant ainsi de les linéariser et de leur apporter une charge glo-bale négative. Ainsi, la séparation sera exclusivement basée sur la longueur des protéines et donc sur leur poids moléculaire.
Des méthodes de séparation par gel à deux dimensions (2D) étaient autrefois couramment uti-lisées pour des applications en spectrométrie de masse, notamment avec l’utilisation de MALDI comme source d’ionisation (voir 1.3.7) [198]. Ces approches par gel 2D offrent la possibilité de séparer les protéines selon leur point isoélectrique lors d’une première séparation, puis en fonction de leur poids moléculaire lors d’une seconde. Les protéines ainsi séparées sont relative-ment pures, permettant une identification à l’aide de méthodes d’empreinte de masse (voir 1.3.9). Cependant, cette approche laborieuse est de moins en moins utilisée de nos jours au profit des méthodes beaucoup plus simples et rapide utilisant la 2DLC (voir 1.3.5).
On peut également décider de purifier certaines protéines à l’aide de maqueurs artificielle-ment ajoutés sur les protéines, ou encore à l’aide anticorps reconnaissant les protéines endogènes.
1.3.3 Immunopurification
L’immunopurification est une technique qui consiste à enrichir une substance, l’antigène, à l’aide d’un anticorps. Alors que l’anticorps est lui même une protéine, l’antigène peut être une protéine, un peptide, une chaine d’acides nucléiques, un métabolite ou même une substance exogène. En laboratoire, il est possible de générer des lignées cellulaires produisant des anticorps: les hybridomes (Figure 1.11).
La première étape de production d’un hybridome est d’exposer un animal de laboratoire (gé-néralement souris ou lapin) par injection de l’antigène en intra-veineuse. Dans le cas où l’antigène seul n’est pas immunogène, il est possible de coupler l’antigène à une large protéine étrangère à l’animal, telle que l’Ovalbumine. Le système immunitaire de l’animal commence ainsi à produire des cellules B présentant différents anticorps dirigés contre l’antigène.
Quelques jours après l’injection, l’animal est sacrifié pour récupérer ses cellules B splénique qui sont alors fusionnées avec des cellules de myélome immortalisée.
Après cette étape d’hybridation, les hyridomes en cultures forment une population hétérogène produisant un large panel d’anticorps. Afin d’obtenir un anticorps monoclonal, il est possible d’isoler les cellules et de les sélectionner afin de ne conserver qu’un seul clone d’hybridome en culture.
Figure 1.11 – Production d’un anticorps monoclonal à partir d’hybridomes. Figure adaptée de [199].

![Figure 1.3 – Mécanisme de formation de chaine poly-modifiées. Figure adaptée de [40].](https://thumb-eu.123doks.com/thumbv2/123doknet/7703094.245972/31.918.206.778.763.999/figure-mécanisme-formation-chaine-poly-modifiées-figure-adaptée.webp)
![Figure 1.9 – Mécanisme de formation des corps PML. Figure reproduite de [166].](https://thumb-eu.123doks.com/thumbv2/123doknet/7703094.245972/43.918.180.808.138.498/figure-mécanisme-formation-corps-pml-figure-reproduite.webp)
![Figure 1.10 – Identification des protéines par Shotgun Proteomics. Figure adaptée de [193].](https://thumb-eu.123doks.com/thumbv2/123doknet/7703094.245972/45.918.182.811.770.1005/figure-identification-protéines-shotgun-proteomics-figure-adaptée.webp)
![Figure 1.11 – Production d’un anticorps monoclonal à partir d’hybridomes. Figure adaptée de [199].](https://thumb-eu.123doks.com/thumbv2/123doknet/7703094.245972/47.918.189.809.760.919/figure-production-anticorps-monoclonal-partir-hybridomes-figure-adaptée.webp)
![Figure 1.12 – Sources d’ionisation communément utilisées en protéomiques. Figure adaptée de [208].](https://thumb-eu.123doks.com/thumbv2/123doknet/7703094.245972/51.918.186.802.821.1003/figure-sources-ionisation-communément-utilisées-protéomiques-figure-adaptée.webp)