Université de Montréal
Synthèse stéréosélective de pipéridines
par
Alexandre Larivée
Département de Chimie, Université de Montréal Faculté des Arts et des Sciences
Thèse présentée à la Faculté des études supérieures en vue de l’obtention du grade de Phulosophia Doctor (Ph.D.)
en Chimie
avril, 2007
7œ+
v.ezi
e ‘s,t:
s h,Université
de Montré al
Direction des bibliothèques
AVIS
L’auteur a autorisé l’Université de Montréal à reproduire et diffuser, en totalité ou en partie, par quelque moyen que ce soit et sur quelque support que ce soit, et exclusivement à des fins non lucratives d’enseignement et de recherche, des copies de ce mémoire ou de cette thèse.
L’auteur et les coauteurs le cas échéant conservent la propriété du droit d’auteur et des droits moraux qui protègent ce document. Ni la thèse ou le mémoire, ni des extraits substantiels de ce document, ne doivent être imprimés ou autrement reproduits sans l’autorisation de l’auteur.
Afin de se conformer à la Loi canadienne sur la protection des renseignements personnels, quelques formulaires secondaires, coordonnées ou signatures intégrées au texte ont pu être enlevés de ce document. Bien que cela ait pu affecter la pagination, il n’y a aucun contenu manquant.
NOTICE
The author of this thesis or dissertation has granted a nonexclusive license allowing Université de Montréal to reproduce and publish the document, in part or in whole, and in any format, solely for noncommercial educational and research purposes.
The author and co-authors if applicable retain copyright ownership and moral rights in this document. Neither the whole thesis or dissertation, nor substantial extracts from it, may be printed or otherwise reproduced without the author’s permission.
In compliance with the Canadian Privacy Act some supporting forms, contact information or signatures may have been removed from the document. While this may affect the document page count, it does not represent any loss of content from the document.
Cette thèse intitulée:
Synthèse stéréosélective de pipéridines
présentée par: Alexandre Larivée
a été évaluée par un jury composé des personnes suivantes:
Hélène LebeL président-rapporteur André B. Charette, directeur de recherche
Shawn Collins, membre du jury Éric Fillion, examinateur externe Hélène Lebel, représentant du doyen de la FES
111
Résumé
Cette thèse traite principalement de la synthèse stéréosélective de pipéridines. Le motif pipéridine est présent dans plusieurs composés naturels et synthétiques d’intérêts pharmaceutiques. Notre stratégie consiste à réduire des dérivés de type pyridinium de façon contrôlée afin d’obtenir des pipéridines hautement fonctionnalisées. Cette thèse est divisée en cinq chapitres.
Dans le premier chapitre, nous présentons un éventail de produits naturels et synthétiques possédant une pipéridine 2,5-substituée. De plus, nous résumons diverses approches modernes employées afin de préparer des pipéridines 2,5-substituées.
Dans le second chapitre, nous présentons une méthodologie permettant de préparer des pipéridines 2,5-cis-disubstituées non racémiques dans une séquence de cinq étapes à partir de la pyridine. Les étapes clés sont un couplage de Suzuki et une hydrogénation diastéréosélective. L’application de la méthodologie à la synthèse d’un agoniste non naturel des récepteurs Œ-adrénergiques est également présentée.
Dans le troisième chapitre, nous présentons une nouvelle réaction permettant de préparer des 1,2,3 ,4-tétrahydropyridine-N-imidate-2,6-di substituées non racémiques en trois étapes. Ce chapitre traite également de l’aspect mécanistique de la réaction et l’isolation d’un palladacycle tend à démontrer l’implication d’un mécanisme d’insertion C H.
Dans le quatrième chapitre, nous révisons brièvement les réactions d’activation de liaisons C-H des noyaux pyridines ainsi que les mécanismes impliqués lors de ces réactions.
Dans le cinquième chapitre, nous présentons le développement d’une nouvelle réaction d’aryÎation directe sur des ylures de N-iminopyridinium. Nous exposons également
les implications mécanistiques de cette réaction et proposons un mécanisme réactionnel.
L’application de la méthodologie à la synthèse de l’anabasine en trois étapes est également présentée. Mots-clés: -Insertion C-H -Couplage de Heck -Halogénation -Couplage de Suzuki -Hydrogénation diastéréosélective -Pipéridine -Époxydation -Ylure de pyridinium -Anabasine -Palladium
V
Abstract
This thesis addresses the stereoselective synthesis of piperidines. The piperidine ring system is present among many natural and synthetic compounds of pharmaceutical interest. Our strategy consists of the controlled reduction of pyridinium derivatives to obtain highly functionalized piperidines. This thesis is divided ïnto five chapters.
In the first chapter, we present a large number of natural and synthetic products possessing the 2,5-substituted piperidine motif. Also, we review numerous modem synthetic approaches to access 2,5-di substituted piperidines.
In the second chapter, we present a methodology developed to synthesize non
racemic 2,5-cis-disubstituted piperidines in five steps from pyridine. The key steps are a Suzuki cross-coupling and a diastereoselective hydrogenation. The application of this methodology to the synthesis of a non natural Œ-adrenergic feceptor agonist is also presented.
In the third chapter, we present a new reaction allowing for the preparation of 2,6-substituted-l ,2,3,4-tetrahydropyridine-N-imidates in three steps from commercially available pyridine. This chapter also examines the mechanism of the reaction and the isolation of a palladacycle suggesting a C-H insertion mechanism.
In the fourth chapter, we briefly review pyridine C-H bond activation reactions and their mechanisms.
In the fifth chapter, we present a new reaction consisting of the direct arylation of N-iminopyridinium ylides. We also discuss the mechanistic implications of that reaction and propose a reaction mechanism. The application of this methodology to the synthesis of racemic anabasine in three steps is also presented.
-Heck coupling -Halogenation -Suzuki coupling -Diastereoselective hydrogenation -Piperidine -Epoxidation -Pyridinium Ylide -Anabasine -Palladium
vii
Table des matières
Liste des tableaux x
Liste des schémas xiii
Liste des figures xviii
Liste des abbreviations xx
C’hapitre J Pipéridines 2,5-substituées importantes et leur préparation 1
1.1 Introduction 1
1.2 Les pipéridines 2,5-substituées 2
1.3 Préparation des pipéridines 2,5-subtituées 5 1.3.1 Synthèse de pipéridines par réactions péricycliques ou par cycloadditions .. 6
1.3.1 .1 La réaction d’imino Diels-Alder 6 1.3.1.1 .1 La méthodologie du professeur Barluenga 7 1.3.1.1.2 La méthodologie du professeur Kobayashi 9 1.3.1.2 Les azadiènes dans la réaction de Diels-Alder 12 1.3. 1.2.1 Les réactions d’hétéro Diels-Alder des I -azadiènes 13 1.3.1.2.2 Les réactions d’hétéro Diels-Alder des 2-azadiènes 15 1.3.1.3 Les réactions d’électrocyclisation 17 1.3.2 Synthèse de pipéridines par réaction de fermeture de cycle 20 1.3.2.1 Lactamisations, aminations réductrices et formation d’hémiaminaÏs 20 1.3.2.1 .1 Synthèse de pipéridines par lactamisation d’acides s-aminés 20 1.3.2.1.2 Synthèse de pipéridines par amination réductrice et formation
d’hémiaminals 21
1.3.2.2 Additions nucléophiles et addition conjuguées 27 1.3.2.2.1 Cyclisation par réaction de SN2 27
1 .3.2.2.2 Cyclisation intramoléculaire par addition de Michael 29 1.3.2.2.3 Cyclisation par addition de Mannich 32
1 .3.2.3 Cyclisation radicalaire 35
1.3.2.4.1 Cyclisation par réaction de métathèse 38 1.3.2.4.2 Cyclisation par hydroamination 46 1.3.2.4.3 Cyclisation catalysée au palladium 49 1.3.3 Synthèse de pipéridines par réactions d’expansion de cycle et de
réarrangements 53
1.3.4 Synthèse de pipéridines par réduction de pyridines 57 1.3.4.1 Réduction de pyridines par hydrogénation 58 1.3.4.1.1 Hydrogénations diastéréosélectives de pyridines 58 1.3.4.1.2 Hydrogénations énantiosélectives de pyridines 60 1.3.4.2 Réduction de sels de pyridiniums par l’addition de nucléophiles 65 1.3.4.2.1 Réactions d’additions de nucléophiles dirigées du professeur
Marazano 65
1.3.4.2.2 Réactions d’additions de nucléophiles diastéréosélectives par le
professeur Comins 68
1.3.4.2.3 Additions nucléophiles sur des ylures de N-iminopyridinium 72 1.3.4.2.4 Additions nucléophiles sur des sels de triflate de pyridinium N
imidate 74
1.4 Conclusion 78
Chapitre 2 Synthèse de pipéridines 2,5-substituées 79
2.1 Introduction 79
2.2 Synthèse de pipéridines 2,5-cis-substituées par couplage de Suzuki 79
2.2.1 Halogénation de 1,2,3,4-tétrahydropyridines 2-substituées 80
2.2.1.1 Résultats 80
2.2.1.2 Mécanisme 85
2.2.2 Couplage de Suzuki des 5-iodo-1.2.3.4-tétrahydropyridine-N-imidates
2-substituées 87
2.2.2.1 Résultats 88
ix
2.2.3 Réduction des I ,2,3.4-tétrahydropyridines 2,5-substituées 94
2.2.3.1 Résultats 94
2.2.3.2 Mécanisme 97
2.2.4 Application à la synthèse d’un agoniste des récepteurs a-adrénergiques.. 100 Ozapitre 3 Synthèse de tétrahydropipéridines 2,6-substituées 107
3.1 Introduction 107
3.2 Réactions des 1.2.3,4-tétrahydropyridines dans les conditions de couplage de
Heck 107
3.2.1 Résultats 10$
3.2.2 Mécanisme 114
Chapitre 4 Activation des liaisons C-H du noyau pyridine 124
4.1 Introduction 124
4.2 Arylation directe de pyridines de façon intramoléculaire 125 4.3 Arylation directe de pyridine de façon intermoléculaire 130
4.4 Conclusion 13$
Chapitre 5 Arylation directe des ylures de N-iminopyridinium 139
5.1 Introduction 139 5.2 Résultats 140 5.3 Mécanisme 166 5.4 Applications 171 Conclusion et perspective 176 Partie expérimentale 17$ Annexe I ccxxxii
Analyse cristallographique complète pour la structure de 24 ccxxxii
Annexe 2 xxx
Analyse cristallographique complète pour la structure de 30 xxx
Annexe 3 xliv
Liste des tableaux
Tableau 1. Synthèse en phase solide de pipéridines 2,5-substituées par cycloaddition de
type imino Diels-Alder $
Tableau 2. Synthèse énantiosélective de 4-pipéridones-2.5-substituées par la réaction
d’imino Diels-Alder 11
Tableau 3. Synthèse énantiosélective de 2-pipéridones polysubstituées 17 Tableau 4. Synthèse diastéréosélective d‘hexahydroquinolines par une condensation de
Knoevenagel et une électrocyclisation en tandem 19
Tableau 5. Synthèse de dihydropyridines à partir d’époxydes vinyliques 23 Tableau 6. Conversion de cétones propargyliques 3-aminés en tétrahydropyridines 32 Tableau 7. Synthèse énantiosélective de lactames bicycliques par métathèse 41 Tableau 8. Synthèse énantiosélective de pipéridines par métathèse 42 Tableau 9. Application de la métathèse à la synthèse de pipéridines par Grigg 45 Tableau 10. Préparation de pipéridines 2,5-substituées par réaction d’hydroamination 48 Tableau 11. Utilisation de complexes 3t-allyles pour la préparation de pipéridines
2.5-substituées 52
Tableau 12. Hydrogénation diastéréosélective de Glorius 59 Tableau 13. Hydrogénation catalytique énantiosélective de quinolines 2-substituées 61 Tableau 14. Réactivité de la pyridine et espèces dérivées en hydrogénation catalysée à
l’iridium 62
Tableau 15. Amination de pyridines par la O-(2,4-dinotrophényl)hydroxylainine 63 Tableau 16. Hydrogénation énantiosélective des ylures de N-iminopyridinium 64 Tableau 17. Réactions d’addition de réactifs de Grignard à des sels de pyridinium N
benzylés 66
Tableau 1$. Réactions d’addition diastéréosélectives de réactifs de Grignard à des sels de
xi
Tableau 19. Iodation de 2,3-dihydro-4-pipéridones par Comins 70 Tableau 20. Réaction de Nozaki-Hiyama-Kishi pour la synthèse de 4-pipéridones
2,5-disubstituées 72
Tableau 21. Additions de réactifs de Grignard sur des ylures de N-iminopyridinium 73 Tableau 22. Additions de réactifs organométalliques sur un sel de triflate de pyridinium N
imidate 75
Tableau 23. Monohydrogénation chimiosélective de l,2-dihvdropyridines N-imidate 77 Tableau 24. Différents agents d’iodation pour l’halogénation de la tétrahydropyridine 1.. $1 Tableau 25. Optimisation du solvant de réaction pour l’iodation de la tétrahydropyridine 1 $2 Tableau 26. Optimisation de la base pour l’iodation de la tétrahydropyridine I $3 Tableau 27. Optimisation finale pour l’iodation de la tétrahydropyridine I $4 Tableau 2$. Iodation de différentes 1 .2,3,4-tétrahydropyridine N-imidates $5 Tableau 29. Optimisation du catalyseur de palladium pour le couplage de Suzuki $9 Tableau 30. Optimisation du solvant de réaction pour le couplage de Suzuki 90 Tableau 31. Conversions en fonction du temps pour différentes bases 91 Tableau 32. Couplage de Suzuki dans les conditions optimisées 92 Tableau 33. Optimisation des conditions d’hydrogénation de la tétrahydropyridine 19 95 Tableau 34. Couplage au palladium entre le diazonium de phényle et la tétrahydropyridine
1 110
Tableau 35. Optimisation du solvant de réaction pour le couplage au palladium entre l’iodobenzène et la 1,2.3.4-tétrahydropyridine I 111 Tableau 36. Optimisation de la base pour le couplage au palladium entre l’iodobenzène et
la 1,2,3,4-tétrahydropyridine I 112
Tableau 37. Arylation directe intermoléculaire catalysée au palladium de l’oxyde de
pyridine 134
Tableau 39. Utilisation de différents métaux de transition pour catalyser l’arylation d’ylures
de pyridinium 141
Tableau 40. Optimisation de la source d’halogénures d’aryle 142 Tableau 41. Optimisation du solvant de réaction 143
Tableau42. Optimisation de la base 144
Tableau 43. Optimisation de la source de palladium 145 Tableau 44. Utilisation de différents ligands monodentates 146 Tableau 45. Utilisation de différents ligands bidentates 147 Tableau 46. Optimisation de la stoechiometrie de la phosphine 14$ Tableau 47. Effet de la concentration sur les rendements 149 Tableau 4$. Effet de l’utilisation de tamis moléculaire comme additif 151 Tableau 49. Effet de la variation du nombre d’équivalents d’ylure utilisé 152 Tableau 50. Résultats des couplages de bromoarènes riches en électrons 153 Tableau 51. Optimisation de la quantité d’ylure utilisé pour les couplages avec des
bromoarènes déficients en électrons 154
Tableau 52. Résultats des couplages de bromoarènes pauvres en électrons 155 Tableau 53. Optimisation de la quantité d’ylure utilisée pour les couplages avec des
bromures d’hétéroaryle 156
Tableau 54. Résultats des couplages de bromures d’hétéroaryle 157 Tableau 55. Résultats de l’insertion du palladium dans des liaisons C-H aliphatiques 162
XIII
Liste des schémas
Schéma I. Synthèse diastéréosélective de pipéridines 2,3,5-trisubstituées par cycloaddïtion
de type imino Diels-Alder 9
Schéma 2. Diels-Alder intramoléculaire dans la synthèse de la (±)-tylophorine 13 Schéma 3. Hétéro Diels-Alder et addition diastéréosélective d’un allyle boronate en tandem 14 Schéma 4. Préparation des dihydro-1,4-oXazin-2-ones 15 Schéma 5. Préparation de pipéridïn-5-ols 2,5-substitués à partir de
dihydro-1,4-oXazin-2-ones 16
Schéma 6. La méthodologie d’électrocyclisation du prof. Hsung 1$ Schéma 7. Approche utilisant une lactamisation par Davis 21 Schéma 8. Synthèse de tétrahydropyridines à partir de l’allysine éthylène acétal 24 Schéma 9. Application de la méthodologie de Blaauw à la synthèse de produits naturels.. 25 Schéma 10. Synthèse de la coniine par la méthodologie CN(R,S) 26 Schéma 11. Synthèse de la pumiliotoXine C par la méthode CN(R,S) 27 Schéma 12. Synthèse de la (—)-slaframine par substitution nucléophile intramoléculaire.. 2$ Schéma 13. Addition conjuguée d’une amine sur un vinyle sulfoxyde chiral 29 Schéma 14. Addition de Michael intramoléculaire appliquée à la synthèse d’alcaloïdes ... 30
Schéma 15. Addition conjuguée intramoléculaire d’une amine sur une cétone a,3-insaturée 31 Schéma 16. Synthèse de la (—)-indolizidine 209B par une réaction de Mannich
intramoléculaire 34
Schéma 17. Synthèse de l’indolizidine (—)-223A par une réaction de Mannich
intramoléculaire 35
Schéma 18. Cyclisation radicalaire 5-exo-trig sur une 2-méthylèneaziridine 36 Schéma 19. Synthèse de pipéridines 2,5-substituées par cyclisation radicalaire 37 Schéma 20. Résolution cinétique par métathèse de fermeture de cycle 40
Schéma 21. Application de la métathèse à la synthèse de lactames par Beak 44 Schéma 22. Application de la métathèse à la synthèse de lactames par Marco 44 Schéma 23. Hydroamination/cyclisation en tandem 49 Schéma 24. Réaction de Heck intrarnoléculaire dans la synthèse de la strychnine de Rawal 50 Schéma 25. Cyclisation intramoléculaire catalysée par le palladium dans la synthèse de la
strychnine de Padwa 51
Schéma 26. Synthèse de la pumiliotoxine C par réarrangement de Beckmann 53 Schéma 27. Synthèse de pipéridines par réarrangement d’aza-Achmatowicz 54 Schéma 28. Synthèse de 2-pipéridones par déamination réductive d’esters Œ-aminés 55 Schéma 29. Synthèse de 2-piperidinones par réaction de Schmidt intramoléculaire 56 Schéma 30. Synthèse d’une pipéridine 2,5-disubstituée tricyclique par réaction de Schmidt
intramoléculaire 57
Schéma 31. Hydrogénation diastéréosélective de 2-a-hydroxypyridines 58 Schéma 32. Synthèse d’une pipéridine 2,5-trans-substituée par Marazano 67 Schéma 33. Synthèse de 4-pipéridones 2,5-disubstituées par couplage au palladium 71 Schéma 34. Additions diastéréosélectives de réactifs de Grignard sur des ylures de N
iminopyridinium 74
Schéma 35. Synthèse de 5-amino-4-pipéridones 2-substituées 76 Schéma 36. Époxydation de tétrahydropyridines suivi d’une ouverture d’époxyde par
addition nucléophile en tandem 78
Schéma 37. Mécanisme proposé pour la réaction de Suztiki catalysée au palladium 93 Schéma 38. Hydrogénation des tétrahydropyridines 2,5-substituées 13 et 15 96 Schéma 39. Protonation de l’énamine menant à la formation de pipéridines
2,5-cis-substituées 99
Schéma 40. Mécanisme proposé pour l’hydrogénation des 1,2,3,4-tétrahydropyridines
2,5-substituées 100
xv
Schéma 42. Époxydation et ouverture d’époxyde en tandem de la tétrahydropyridine
2.5-substituée 12 103
Schéma 43. Époxydation et réduction en tandem de la tétrahydropyridine 5 105 Schéma 44. Époxydation et réduction en tandem de la tétrahydropyridine 2.5-substituée 20 105
Schéma 45. Couplage de Heck 108
Schéma 46. Couplage de Heck anticipé sur les 1,2,3,4-tétrahydropyridines N-imidates.. 109 Schéma 47. Equilibre entre des espèces de palladium(II) à 14 et 16 électrons 113 Schéma 48. Couplage au palladium entre l’iodobenzène et la tétrahydropyridine 1 113 Schéma 49. Tentative de couplage au palladium entre l’iodobenzène et la dihydropyridine
24 114
Schéma 50. Mécanisme classique d’une réaction de Heck et produit normalement attendu 115 Schéma 51. Mécanisme possible de couplage de Heck entre l’iodobenzène et une tétrahydropyridine suivi par l’isomérisation de l’alcène 116 Schéma 52. Déprotonation intramoléculaire par l’amidine du proton équatorial benzylique 11$ Schéma 53. Mécanisme possible d’insertion C-K pour le couplage entre le iodobenzène et
une tétrahydropyridine 121
Schéma 54. Préparation du carbopalladacycle 30 à partir de la tétrahydropyridine 29 122 Schéma 55. Couplage entre une pyridine 2-halogénée et un dérivé aryle organométallique 124 Schéma 56. Premier exemple d’arylation directe intramoléculaire d’un noyau pyridine.. 126 Schéma 57. Arylation directe intramoléculaire entre une pyridine et une chloropyridine 127 Schéma 5$. Réactions en cascade entre le norbornène et des iodopyridines catalysées au
palladium 12$
Schéma 59. Formation de carbopalladacycle en présence de norbornène et d’un iodoaryle 12$
Schéma 60. Deux mécanismes possibles pour les réactions en cascade entre le norbomène
et 1’ iodopyridine 129
Schéma 61. Homocouplage de type Ullmann catalysé au palladium 131
Schéma 62. Couplage catalysé au palladium entre le chlorobenzène et la pyridine 131
Schéma 63. Mécanisme proposé par Sasson pour le couplage catalysé au palladium entre le
chlorobenzène et la pyridine 133
Schéma 64. Influence des substituants de l’oxyde de pyridine 136
Schéma 65. Influence des substituants de l’oxyde de pyridine sur la vitesse de la réaction
136
Schéma 66. Cycle catalytique proposé par Fagnou pour l’arylation directe d’oxydes de
pyridine 137
Schéma 67. Expérience d’effet cinétique isotopique intermoléculaire 13$
Schéma 6$. Réactivité des acétanilides et de leurs complexes 140 Schéma 69. Résultat pour l’utilisation de 1.2 equiv de Ph-Br 150 Schéma 70. Résultat de l’utilisation d’ylure en excès 150 Schéma 71. Arylation directe de l’yture de N-iminoisoquinolinium 45 157 Schéma 72. Arylation directe de l’ylure de N-iminoquinolinium 47 15$ Schéma 73. Arylation directe de l’ylure de 3-méthyl-N-iminopyridinium 49 159 Schéma 74. Arylation directe de l’ylure de 2-propyl-N-iminopyridinium 52 161 Schéma 75. Compétition entre l’activation de liens C-H aliphatiques et de liens C-H
aromatiques 161
Schéma 76. Effet cinétique isotopique du deutérium observé pour la réaction d’arylation directe des ylure de pyridinium avec le bromobenzène 163 Schéma 77. Arylation directe de l’oxyde de pyridine dans nos conditions réactionnelles 164 Schéma 7$. Expérience de compétition entre l’yltire de pyridinium et l’oxyde de pyridine
164
Schéma 79. Cycle catalytique pour l’arylation directe des ylures de pyridinium basé sur
xvii
Schéma $0. Cycle catalytique proposé pour l’arylation directe des ylures de N
iminopyridinium 167
Schéma 81. État de transition de l’étape de transfert d’aryle entre deux complexes de
palladium(II) 16$
Schéma $2. Le ligand carbène: une forme limite de résonance du carbopalladacycle 169 Schéma $3. Espèces à l’équilibre au cours du cycle catalytique 170 Schéma $4. Hydrogénation chimiosélective du noyau pyridinium 172 Schéma 85. Réduction de la liaison N-N avec Sm12 et protection en un seul pot 173 Schéma $6. Déprotection du t-butylcarbamate par l’acide trifluoroacétique 173 Schéma $7. Préparation de l’anabasine N-BOC en 3 étapes et 61% de rendement global 174 Schéma $8. Arylation directe, addition de Grignard et réaromatisation en un pot 175
Liste des figures
Figure 1. Produits naturels biologiquement actifs possédant l’unité pipéridine 1 Figure 2. Pipéridines possédant le motif de substitution 1,2,5 2 Figure 3. Produits naturels possédant une pipéridine polysubstituée avec des groupements
aux positions 2 et 5 3
Figure 4. Composés synthétiques importants possédant le motif pipéridine 2.5-substituée.. 5 Figure 5. La réaction d’imino Diels-Alder 6 Figure 6. La réaction de Diels-Alder des azadiènes 12
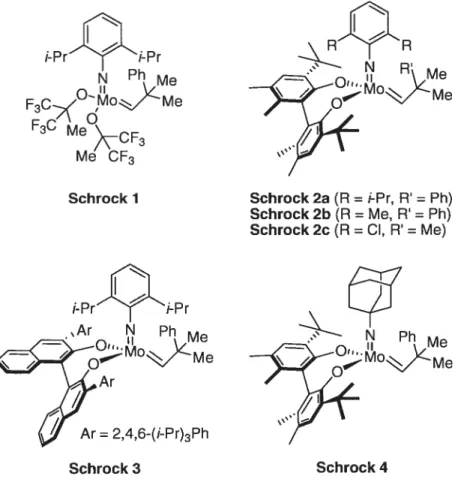
Figure 7. Catalyseurs de Schrock 39
Figure 8. Catalyseurs de Grubbs 43
Figure 9. Exemples de catalyseurs pour la réaction d’hydroamination 47 Figure 10. Synthèse de l’alcaloïde 6-épi-indolizidine 223A par Padwa 55 Figure 11. Expansion de cycle par réaction de Schmidt 56 Figure 12. Régiosélectivité d’addition nucléophile au noyau pyridinium 65
Figure 13. Approche rétrosynthétique aux pipéridines 2,5-cis-disubstituées $0 Figure 14. Formation d’un intermédiaire iminium lors l’iodation des tétrahydropyridines $6 Figure 15. Conformères possibles pour l’intermédiaire iminium formé 87 Figure 16. Effets nOe observés dans le spectre NOESY de 21 96 Figure 17. Représentation POV-Ray du spectre de diffraction des rayons X de la
2-phényl-1 ,2-dihydropyridine-N-imidate 24 98
Figure 18. Agoniste des récepteurs a-adrénergiques 101 Figure 19. La norépinéplirine, un neurotransmetteur naturel des réceptetirs adrénergiques 101 Figure 20. Effets nOe d’importances observés dans le spectre de 25 104 Figure 21. Rendement global de la synthèse de l’agoniste d’un récepteur Œ-adrénergique
jusqu’à ce jour 106
xix
Figure 23. État de transition d’une métathèse de liens sigma tel que proposé par Echavarren 120 Figure 24. Représentation du spectre de diffraction des rayons X du complexe de palladium
30 122
Figure 25. Structures d’un ylure de N-benzoyl-N-iminopyridinium et d’un acétanilide... 139
Figure 26. Représentation du spectre de diffraction aux rayons X de l’ylure de
5-méthyl-2-phényl-N-iminopyridinium 50 160
Figure 27. Conversions en fonction du temps pour l’ylure de pyridinium et l’oxyde de
pyridine 165
Liste
des abbreviations
Ac acétyle
acac acétylacétonate
Acc. Chem. Res. Accounts of ChernicaÏ Research Adv. Heterocyci. Chem. Advances in Heterocvclic Ctzemistiy
AIBN azobis-i-butyronitrile
Angew. Chem. lut. Ed. Angewandte Chemie International Edition
Ar aryle
atm atmosphère
BArF tétrakis(3 ,5-bis(trifluorométhyl)phényl)borate Bfl\TAP 2,2’-Bis(diphenylphosphino- 1,1’ -binaphtalène Bioorg. Med. Chenz. Bioorganic & Medicinal Chemistiy
Bioorg. Med. Chem. Lett. Bioorganic & Medicinal Chemistiy Letters Boc carbamate de t-butyle
BiPy 2,2’-bïpyridine
Can. J. Cheni. Canadian Journal of Oiemistry Cbz carbamate de benzyle
Chem. Ber. Chemisehe Berichte
Chem. Commun. Chetnical Communications Chem. Eur J. Chemistrv—A EuropeanJouruai
Chem. Lett. ChemistiyLetters
Chem. Rev. Chemical Reviews
Chem. Soc. Rev. ChemicaÏ Society Reviews
cod cyclooctadiène
Cp* pentaméthylcyclopentadiènyle
Cy cyclohexyle
xxi
DMA acétamide de N,N-diméthyle DMEDA N,N ‘-diméthyléthylènedi amine DMF formamide de N,N-diméthyle DTBMP 2.6-di-t-butyl-4-méthylpyridine ee excès énantiomère ed excès diastéréomérique equiv équivalent Et éthyle Fur furyle h heures Net hétéroaryle HTIB hydroxy(tosyloxy)iodobenzène i- iso
j
joursJ. Am. C’hem.Soc. Journal ofthe Americcm C7iemicaÏ Socierv
J. C. S., Chem. Commun. Journal ofthe Ozemical Societv, Chentical Communications J. Mol. Cat. A Journal ofMotecutar Catatysis A: Chemical
J. Mot. Cat. Journal oJMotecutar Catalvsis J. Org. Chem. Journal of Organic Chemistrv
J. Organomet. Cizem. bumal of Organometaliic Chemistrp
LC-MS chromatographie liquide—spectroscopie de masse
LDA di-i-propylamidure de lithium MCPBA acide m-chloroperbenzoïque
Me méthyle
Monats. Chem. Monatshefteflir Chemie Ms méthylsulfonyle (mésyle)
MSH O-mésitylènesulfonylhydroxylamine
Napht naplityle
n.d. non déterminé
NIS N-iodosuccinimide
NOE effet nucléaire Overhauser
NOESY effet nucléaire Overhauser et spectroscopie d’échange
Nu nucléophile
Org. Lett. Organic Lette rs Org. React. Organic Reactions Org. Synth. Organic Syntheses Bpin borane de pinacolyle
Ph phényle
pH mesure de l’acidité
Pr propyle
psi pounds I square inch
Pure Appt. Chem. Pure and Apptied Chemistni
Py pyridyle
r.d. rapport diastéréomérique r.r. rapport régioisomérique
SES 2-triméthyl s ilyléthanesulfonyle
t- tertio
t.a. température ambiante
TBAF Fluorure de tétrabutylammonium TBS t-butyldiméthylsilyle
Tetrahedron Lett. Tetrahedron Letters
Tf trifluorométhanes ulfonyle (triflyle) TFA acide trifluoroacétique
THF tétrahydrofurane
T.M. tamis moléculaire
TMEDA N,N,N ‘,N ‘4étraméthyléthylènediamine
TMS triméthylsilyle
Toi 4-méthylphényle (toluyle)
Ts 4-méthylbenzènesulfonyle (tosyle)
“Theonlvsource ofknowÏedge is experience”
xxv
Remerciements
Les travaux de recherche présentés dans cette thèse ont été facilités par la collaboration de mes pairs et le support de mes proches. Ces remerciements ont pour but de reconnaître leurs contributions autant scientifiques que morales.
Tout d’abord, je tiens à exprimer ma gratitude à l’endroit de mon directeur de recherches, le professeur André B. Charette, pour m’avoir accueilli au sein de son groupe de recherches et m’avoir accordé son soutien financier tout au long de mes études graduées. Par ailleurs, la formation académique et l’éthique de travaille que m’ont fournies l’environnement du groupe Charette et l’université de Montréal m’ont très bien préparé, je crois, à relever tous ces défis. Le prof. Charette a eu une grande influence sur mon épanouissement professionnel autant pour sa rigeur scientifique, la liberté du champ de recherches qu’il m’a acordé. que par le partage de son opinion personnelle sur une foule de sujets.
Je tiens également à remercier tous les membres du groupe de recherches passés et actuels pour l’atmosphère familial, grouillant et chaleureux qu’ils ont créé à toutes heures du jour et de la nuit. Un merci particulier aux docteurs Michel Grenon, Jonathan Martel et Alexandre Lemire qui ont démarré le projet d’addition de réactifs organométalliques aux sels de pyridinium et au Dr Claude Legault qui est le père du projet des ylures de pyridinium. Leurs résultats et leurs conseils ont été grandement bénéfiques à mes recherches. Je voudrais également remercier les gens que j’ai cotoyés de plus près dans le laboratoire, Dr. Marie-Christine Lacasse, Sébastien Francoeur, Dr. Jean-Manuel Cloarec, Dr. Dm0 Alberico et Olga Lifchits. Et un merci tout particulier à Dr. Alessandro Boezio, Patrick L. DeRoy, Dr. Alexandre Lemire, Dr. Benoït Moreau. Christian Perreault, Guillaume Barbe et Alexandre Côté pour plusieurs discussions scientifiques enrichissantes. Je voudrais également remercier deux personnes qui ont participé à ces travaux de
recherches en tant qu’étudiants d’été. Justin Vinh Doan et Guillaume Pelletier, ainsi que James Mousseau qui a repris certains de mes travaux en débutant son doctorat.
Je voudrais remercier Barbara Bessis pour son efficacité, son dévouement ainsi que sa persévérance à nous épauler pour mille et une tâches administratives de façon presque quotidienne. Je tiens à souligner la participation du corps professoral à ma formation universitaire, par l’excellence des cours gradués dispensés et la disponibilité des professeurs du département de chimie. Je remercie également Dr. Tan Phan Viet, Sylvie Bilodeau, Robert Mayer et Cédric Malveau du laboratoire de résonance magnétique nucléaire pour le soutien apporté dans les expériences RMN, Francine Bélanger-Gariépy du laboratoire de diffraction des rayons X, les membres du personnel de l’atelier électronique et mécanique, les membres du personnel du Centre de spectrométrie de masse et du laboratoire d’analyse élémentaire et le personnel courtois de l’administration, tout particulièrement Lyne Laurin.
Je voudrais aussi remercier mes collègues qui ont accepté de relire et corriger cette thèse, soit Sébastien Goudreau, Guillaume Barbe, Isabelle Bonnaventure, Dr. Maryon Ginisty et Dr. James Buil.
Cette liste de remerciements serait incomplète sans remercier les membres de ma famille. Tout d’abord, ma conjointe Fanie Lauzon que je remercie pour son amour inconditionnel, les voyages et son soutien psychologique et financier. Je voudrais également remercier mes parents Johanne Larivée et Pascal Tellier, ainsi que leurs conjoints Daniel et Louise, ma tantine Josée Larivée et mes grand-parents Jeannine et Gilles Larivée pour leurs encouragements pendant ces longues années d’études. Finalement, je remercie mes amis de tous horizons qui ne comprennent rien à ce que je fais, mais qui veulent toujours connaître le titre de ma thèse!
Chapitre 1
Pipéridines 2,5-substituées importantes et
leur préparation.
1.1 Introduction
L’unité pipéridine forme une famille de composés hétérocycliques incluant une
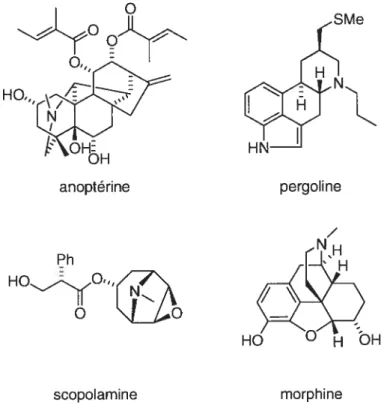
multitude de produits naturels.1’2 L’intérêt de la communauté scientifique vis-à-vis cette classe de composés est décuplé par le fait que plusieurs d’entre eux présentent une activité biologique intéressante, notamment l’anoptérine, la pergoline, la scopolamine et la morphine (Figure 1). HO,,, HO,O’‘ ‘ HO 0HOH scopolamine morphine
Figure 1. Produits naturels biologiquement actifs possédant l’unité pipéridine.
‘J. W. Daly. T. F. Spande, M. Garraffo, J. Nat.Pmd. 2005. 68. 1556-1575. 2
D. R. Dalton, dansTheAÏkaloids. P. G. Gassmani EU.: Marcel Dekker. mc: New York and Basel, 1979. Vol. 7, p. 97-760.
Une statistique intéressante montre que plus de 12000 molécules possédant une unité pipéridine ont étés soumises à des essais cliniques ou précliniques entre juillet 1988 et décembre 199$.
1.2 Les pipéridines 2,5-substituées
Bien qu’ un nombre important de pipéridines parsèment la littérature scientifique, très peu de produits naturels possèdent le motif de substitution carboné 2,5 exclusivement. Néanmoins, la solanidine, un alcaloïde stéroïdien, et la (—)-déoxynupharidine, un alcaloïde diterpénoïde, sont deux molécules naturelles I ,2,5-trisubstituées (Figure 2).
R
R1
Motif 1,2,5
Me
solanidine
Figure 2. Pipéridines possédant le motif de substitution 1,2,5.
(—)-deoxynupharidine
3
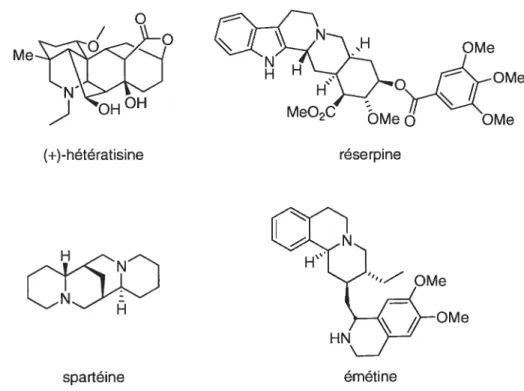
En effet, la majorité des produits naturels possédant des substituants en position 2 et 5 sont des pipéridines polysubstituées. On peut citer en exemple plusieurs alcaloïdes; comme la spartéine dérivée de la lupine, l’émétine dérivé de la tyrosine, la réserpine dérivée du tryptophane ou encore des alcaloïdes générés à partir de l’incorporation d’un atome d’azote dans un squelette terpénoïde comme l’hétératisine (Figure 3)•2
spartéine
OMe
H
éméti ne
Figure 3. Produits naturels possédant une pipéridine polysubstituée avec des groupements aux positions 2 et 5
Compte tenu que les pipéridines dotées d’un motif de substitution 2.5 d’origine naturelle sont peu abondantes. il n’est pas étonnant de remarquer que la plupart des pipéridines 2,5-disubstituées connues sont des produits synthétiqties souvent brevetés. Bien
Me-(+)-hétératisine OMe OMe MeO2CMeQ OMe réserpine
que ces composés ne soient pas issus de milliers d’années d’évolution, certains d’entre eux sont biologïquement actifs ou possèdent des propriétés physiques intéressantes. Par exemple, certains pipéridinols 2,5-substitués ont été synthétisés pour leurs activités vis-à-vis des récepteurs adrénergiques.4’5’6 Pfizer a également rendu public la structure de pipéridines 2,5-disubstituées synthétisés dans leurs centres de recherche.7 Un brevet rapportant la synthèse de composés agonistes des récepteurs lA et 1D de la sérotonine comme étant des agents psychothérapeutiques puissants, utilise une octahydropyrido-1,2-pyrazine disubstituée comme intermédiaire avancé dans la synthèse d’une quarantaine de composés biologiquement actifs.8 Au début des années 70, la 2,5-diméthylpipéridine 1-substituée fut brevetée pour différents usages allant de son utilisation comme herbicide à ses propriétés anti-inflammatoires (Figure4)•9
A. Balsamo, P. L. Barili, M. Gagliardi, A. Lapucci, B. Macchia, F. Macchia, M. Bergarnaschi, CIiii,i. I,id. 1976,58, 222-222.
A. Balsamo, P. L. Barili, M. Gagliardi, A. Lapucci, B. Macchia, F. Macchia, M. Bergamaschi, Eur. J. Med. Chem. 1982, 17, 285-289.
6
B. Macchïa, M. Macchia, A. Martinelli E. Martinotti, E. Orlandini, F. Romagnoli, R. Scatizzi, Fur, J. Med. Chem. 1997, 32, 23 1-240.
M. C. Desai, L. M. S. Stramiello, Tetrahedron Lett. 1993, 34, 7685-7688. $G. M. Briglit, Brevet International, WO 9952907.
a) W. J. Houlihan, brevet américain, US 3,334,104; b) W. J. Houlihan, brevet allemand, DE 1 964 441; c) W. J. Houlihan, brevet américain. US 3,709,677; d) H. Tilles, brevet francais, FR 2 095 399.
5
activité -bIocante des récepteurs adrénergiques
c
LN(BOC) breveté par Pfizer
Me
R=SO2NH2 R=COSAr
N Me R=SH
R
breveté comme herbicide et comme anti-inflammatoire H( 02N agoniste de récepteur a-adrénergïque N Me H Ph Ph synthétisé chez Pfizer-PGRD
Figure 4. Composés synthétiques importants possédant le motif pipéridine 2,5-substituée.
1.3 Préparation des pipéridînes 2,5-subtituées
Plusieurs méthodes de synthèse permettent l’accès à une grande variété de pipéridines.10’’12 Néanmoins, peu d’entres elles peuvent prétendre être générales et
10M. G. P. Buffat. Tetrahedron. 2004. 60. 1701-1729.
‘
P. M. Weintraub. J. S. Sabol. J. M. Kane. D. R. Borchering. Tetrahedivn. 2003, 59. 2953-2989.
12
permettre l’accès à tous les motifs de substitution envisageables. Cette section présentera les différentes méthodes qui ont été utilisées pour les élaborer.
1.3.1 Synthèse
de
pipéridines
par
réactions
péricycliques
ou
par
cycloadditions
Une méthode populaire pour synthétiser les cycles à six membres est évidemment la réaction de Diels-Alder et la synthèse de pipéridines n’échappe pas à cette tendance.’3 D’autres types de réactions péricycliques ont aussi été utilisés à cette fin, comme la réaction ène,14 les électrocyclisations et les cycloadditions dipolaires.’5 Seules la réaction de Diels Alder et les électrocyclisations seront résumées dans cette introduction.
1.3.1.1 La réaction d’imino Diels-Alder
La réaction d’imino Diels-Alder s’effectue normalement entre une imine et un diène riche en électrons. Elle est souvent catalysée par un acide de Lewis et génère normalement des dérivés de type 4-pyridone (Figure 5).
Acide de Lewis
R2NR1 R2 N
Figure 5. La réaction d’imino Diels-Alder.
13
K.-I. Takao, R. Munakata, K.-I Tadano, Chem. Rev. 2005. 105, 4779-4807.
‘
7
Cette approche fut utilisée dans différentes méthodologies de synthèse de pipéridines achirales,16 en plus d’être employée à plusieurs occasions pour la synthèse de composés complexes.17 L’utilisation d’auxiliaires chiraux fut également exploitée avec succès.18 Néanmoins, cet ouvrage s’attardera davantage aux méthodologies développées par les professeurs Barluenga et Kobayashi.
1.3.].].] La méthodologie du professeur Bartuenga
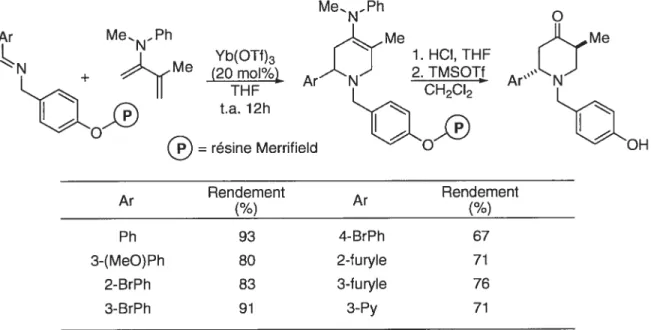
Une méthodologie développée en Espagne dans les laboratoires du professeur Barluenga utilise comme diènophile une imine dérivée d’une benzyle amine supportée sur une résine de type Merrifield.’9 Cette approche semble limitée aux arylimines et les substituants en position 5 sont peu variés. Néanmoins, elle offre une alternative intéressante pour la synthèse de trans-4-pipéridones-2,5-disubstituées en plus d’offrir les avantages de purification inhérents à la synthèse en phase solide (Tableau 1).
‘ R. E. Looper. R. M. Williams, Tetrahedron Lett. 2001. 42. 769-77 1. ‘
a) P. D. Bailey. P. D. Smith. F. Pederson. W. Clegg. G. M. Rosair. S. J. Teat, Tetrahedron Lett. 2002, 43, 1067-1070: b) Y. Huang. V. H. Rawal, Org. Leu. 2000, 2. 3321-3323: c) S. C. Schûrer. S. Blechert. Tetrahedron Leu. 1999, 40, 1877-1880.
‘
a) P. D. Bailey, P. D. Srnith, K. M. Morgan, G. M. Rosair. Tetrahedron Lett. 2002, 43. 1071-1074: b) L. F. Tietze, G. Kettschau, Topies in Carrent Chemistiy; P. Metz, EU.; Springler-Verlag: Berlin. 1997; Vol. 189, pages 1-20; b) G. R. Heintzelman, S. M. Weinreb, J. Org. Chem. 1996, 61, 4596-4599.
18
a) W. Maison, G. Adiwidjaja, Tetraïtedron Leu. 2002, 43, 5957-5960; b) F. A. Davïs. Y. Wu, H. Yan, K. R. Prasad, W. McCoull, Oig. Lett. 2002, 4, 655-658: c) R. Badorrey, C. Cativiela, M. D. Dfaz-de-Villegas, J. A. GIvez, Tetrahedron, 1999,55. 7601-7612.
Tableau 1. Synthèse en phase solide de pipéridines 2,5-substituées par cycloaddition de type imino Diels-Alder.
Me...NPh Me 1.HCI,THF 2. TMSOTt Ar N CH2CI2
o
Me Ar” N OHCette méthodologie est une variante d’une méthodologie d’abord développée en phase liquide pour la synthèse de dérivés de la famille des indolizidines et des quinolizidines.2° Une autre variante de cette méthodologie a permis la synthèse diastéréosélective de pipéridines 2,3,5-trisubtituées à l’aide d’un auxiliaire chiral dérivé de la proline (Schéma 1)21
J. Barluenga, C. Mateos, F. Aziiar, C. Valdés. Org. Lett. 2002, 4. 3667-3670. 20J Barluenga, C. Mateos. F. Aznar. C. VaJds. Org. Lett. 2002,4. 197 1-1974.
21
j• Barluenga. F. Aznar, C. Ribas, C. Valdés, J. Org. Cheni. 1999. 64. 3736-3740.
LN
(20 moI%Yb(OTt)3 THF t.a. 12h =résine Merrifield Ar Rendement(¾) Ar Rendement (¾) Ph 93 4-BrPh 67 3-(MeO)Ph 80 2-furyle 71 2-BrPh 83 3-furyle 76 3-BrPh 91 3-Py 719
o—’
1.ZnCI2,THFÏ
Me -80°Cat.a. 2. NaHCO3 (aq) rJ 3. SiO(j
TMSo
OTMS 51%,>99%ee//
étapes 12% gIoba Me (—)-nupharamineSchéma 1. Synthèse diastéréosélective de pipéridines 2,3,5-trisubstituées par cycloaddition de type imino Diels-Alder
1.3.1.1.2 La méthodotogie du professeur Kobavashi
La méthodologie la plus élégante fut toutefois développée dans les laboratoires du
professeur Kobayashi. Elle consiste en une réaction d’imino Diels-Alder énantiosélective catalysée par un complexe de zirconium auquel est greffé un ligand chiral de type binaphtol. Une première publication exploitant ce système permettait d’obtenir des excès énantiomères modérés (65-89% ce) avec des quantités de catalyseurs importantes (20 mol%).22 Dans une seconde publication, des modifications apportées au ligand du complexe de zirconium ont permis d’améliorer les excès énantiomères (80-89% ce) sans
+
22
toutefois diminuer les quantités du catalyseur utilisées (Zr(OtBu)4: 20 mol%, ligand t 40
mol%).23
Plus récemment, le professeur Kobayashi a publié une nouvelle amélioration à sa méthodologie basée une fois de plus sur des modifications apportées au ligand.24 L’ajout de groupement CF3 au substituant aryle en position 3,3’ du ligand binaphtol et le remplacement des ligands tBuO du zirconium par des ligands cyano (CN) permis d’abaisser drastiquement les quantités du catalyseur utilisés tout en conservant des excès enantiomères élevés. Le diène utilisé dans cette réaction est limité aux diènes de type Danishefsky et seules des 4-pipéridones avec un groupement methyle en position 5 ont été synthétisés. Les imines doivent être dérivées des 2-aminophenols et le groupement R semble limité atix substituants difficilement isomérisables à l’énamine (Tableau 2).
23
S. Kobayashi, K.-I. Kusakabe, S. Komiyama. H. Ishitani,]. Org. Chem. 1999,64, 4220-422 1.
24
11
Tableau 2. Synthèse énantiosélectïve de 4-pipéridones-2,5-substituées par la réaction d’ imino Diels-Alder. OTMS HO-- Me RN + OMe R Me LOH (2-5 mol%) C6H6, MS 3A,t.a. rendement ee R (¾) (%) Ph 81 91 2-(Me)Ph 72 88 1-Napht 71 84 <0 68 90
‘
75 84Le professeur JØrgensen a publié une méthodologie similaire employant un catalyseur de cuivre auquel est lié un ligand de type BINAP, ce qui lui a permis d’obtenir également d’excellents excès énantiomères.2
1.3.1.2 Les azadiènes dans la réaction de Diels-Alder
Les azadiènes ont été fréquemment utilisés pour générer des pipéridines.26 Deux types d’azadiènes sont utilisés, les 1-azadiènes et les 2-azadiènes (Figure 6).
R2
1 -azadiène
R1
N N
2-azadiène
figure 6. La réaction de Diels-Alder des azadiènes
25
• Yao, M. Johannsen, R. G. Hazeli, K. A. JØrgensen. Angew. Chem. bit. Ed. 1998, 37, 3121-3124.
26
13
1.3.1.2.1 Les réactions d’hétéro Diets-Aider des 1-azadiènes
Les 1-azadiènes sont des précurseurs attrayants pour préparer des pipéridines qui ont été utilisés dans la synthèse de plusieurs produits naturels.27 Notamment, les professeur Fukumoto et Kametani au Japon ont développé une méthodologie utilisant un amide
a,f3-insaturé comme précurseur d’un 1-azadiène qu’ils ont utilisé pour effectuer la synthèse racémique de la tylophorine (Schéma 2).2829TBSOTf Et3N CH2Cl2 1500 68% 4 étapes rendement global
Schéma2. Diels-Alder intramoléculaire dans la synthèse de la (±)-tylophorine
27
M. Behforouz, M. Ahmadian, TetraÏiedron2000,56, 5259-528$.
28
M. Ihara, T. Kirihara, A. Kawaguchi, K. Fukumoto, T. Kametani, Tetrahedron Lett. 1984, 25, 4541-4544.
29
M. Ihara, M. Tsuruta, K. Fukumoto, T. Kametani, J. Chem. Soc., Chem. Commun. 1985, 1159-Ï 161.
OMe
/
/7% OMe OMe (±)-tylophorineCependant, très peu de réactions de Diels-Alder des 1-azadiènes offrent la possibilité d’obtenir des produits énantiomériquement purs. Parmi les exemples diastéréosélectifs intéressants, une méthodologie développée par le professeur Ghosez en France utilise un auxiliaire chiral dérivé de la proline.3° Quelques années plus tard, le professeur Hall au Canada a modifié cette méthodologie en utilisant un I -azadiène avec un substituant ester boronique en position 4. Ceci permet d’effectuer en tandem l’hétéro Diels Aider et l’addition stéréosélective de l’allyle borane généré sur un aldéhyde (Schéma 3)•31
Ghosez (7994) Me + Me° MeMe N O’0Me OMe R = H, Me X =O, NMe, NPh 51-84%, 93-98 ed Hall (2000) O
I
NPh H NPh + 8O03j PhLN Me Me PhCHO OHç;_MeQ-(
OMe OMe 55%, >95% edSchéma 3. Hétéro Diels-Alder et addition diastéréoséiective d’un allyle boronate en tandem
30
15
1.3.1.2.2 Les réactions d’hétéro Diels-Alder des 2-azadiènes
Les 2-azadïènes ont été largement utilisés pour la synthèse de pipéridines, notamment pour la synthèse de produits naturels complexes.32’33 Une méthodologie développée par le professeur Hoomaert en Belgique utilise des dihydro-1,4-oxazin-2-ones comme 2-azadiènes pour préparer des composés pipéridinols polysubstitués.34 Les dihydro I ,4-oxazin-2-ones peuvent être préparées en deux étapes à partir de cyanohydrines (Schéma
4)35
(COCD2, Et3NHCI
R1 (R2)4Sn, (Ph3P)4Pd R
R’ Phd, 90 oc, 4h toluène, 1 10 °d
HOCN 12 exemples dl”NC 4 exemples CI’N’R2
30-85% 79-90%
Schéma 4. Préparation des dihydro- 1 ,4-oxazin-2-ones.
Ces hétérocycles réagissent avec l’éthylène pour former des composés de type bicyclo[2,2,2], qui une fois réduits donnent des pipéridin-5-ols 2,5-substitués. Cette méthodologie ne permet pas l’accès à d’autres motifs de substitution en plus d’être limitée à la synthèse de ces composés sous forme racémiques (Schéma 5).
31
j• Tailor, D. G. Hall. Org. Lett. 2000,2, 3715-3718. 32
D. A. Poweil, R. A. Batey, Org. Lett. 2002,4, 291 3-2916.
K. C. Nicolaou, M. Nevalainen. B. S. Satina. M. Zak, S. Bulat. Angew. Chen,. Int. Ed. 2002. 41, 194 1-1945.
X. Wu. K. Dubois. J. Rogiers, S. Toppet, F. Compemolle, G. J. Hoonaert, Tetrahedron 2000, 56. 3043-3051.
ToIuèn11O oc
[ Ï
THF,°C R1pH R1 = Me R2 = Ph 72% R1 = Ph, R2 =Ph, 78% R1 Me, R2 = Me, 56% R =Ph, R2 =H, 60%Schéma 5. Préparation de pipéridin-5-ols 2,5-substitués à partir de dihydro-1,4-oxazin-2-ones.
Une autre méthodologie très efficace impliquant une réaction de Diels Aider des 2-azadiènes a été développée par le professeur Ghosez. Elle consiste en la synthèse énantiosélective de 2-pipéridones polysubstituées à l’aide d’un catalyseur bisoxazoline de cuivre.36 Bien qu’un faible éventail de substituants ait été incorporé pour démontrer l’étendu de cette méthodologie, elle n’en demeure pas moins une façon très élégante d’accéder à ces composés (Tableau 3).
36
17
Tableau 3. Synthèse énantiosélective de 2-pipéridones polysubstituées.
R2 Me3SiO’ + 1. cat.” (8 mol%) T.M.,CH2CI2 2. MeOH N R1
R1 R2 R3 température exo: endo rendement ee
(°C) (%) (%) Ph Me Me —45 >99: 1 80 95 Ph H H —45 6.1 : 1 83 98 Ph Me H —45 >99: 1 96 98 Ph H Me t.a. >99: 1 80 93 Me>’Ph Me Me t.a. >99: 1 98 90 Me Me cat.* = t-Bu
P4
t-Bu TfO OTf1.3.1.3 Les réactions d’électrocyclisation
L’électrocyclisation à six électrons des 1-azatriènes a été utilisée avec succès dans la synthèse de divers produits naturels. Notamment le professeur Katsumura a développé une réaction diastéréosélective utilisant un auxiliaire curaI qui lui a permis d’effectuer la synthèse formelle de la 20-épiuléine.37 Néanmoins, cette méthodologie n’a pas démontré sont efficacité dans la synthèse de pipéridines 2,5-disubstitutées.
exo
Le professeur Hsung, quant à lui, a rapporté la synthèse de quelques produits naturels (4,8-diépipumiliotoxine C, 2-épiperhydrohistrionicotoxine) en utilisant une méthodologie exploitant également une électrocyclisation.38’ Un iminium a,f3-insaturé est mis en présence d’un amide vinylogue pour générerin situ l’azatriène par condensation de Knoevenagel. Celui-ci effectue l’électrocyclisation attendue pour générer en un seul pot l’hexahydroquinoline (Schéma 6). L’isolation par les auteurs d’un intermédiaire azatriène leur a permis d’élucider la voie mécanistique et surtout d’écarter un autre mécanisme possible impliquant l’addition conjuguée de l’amine sur l’iminium a,13-insaturé suivit d’une condensation intramoléculaire de type Knoevenagel.
Q +J2 O NR O
NHR* R1 [Rl R1
Schéma 6. La méthodologie d’électrocyclisation du prof. Hsung
Cette méthodologie donne accès en une seule étape à des hexahydroquinolines
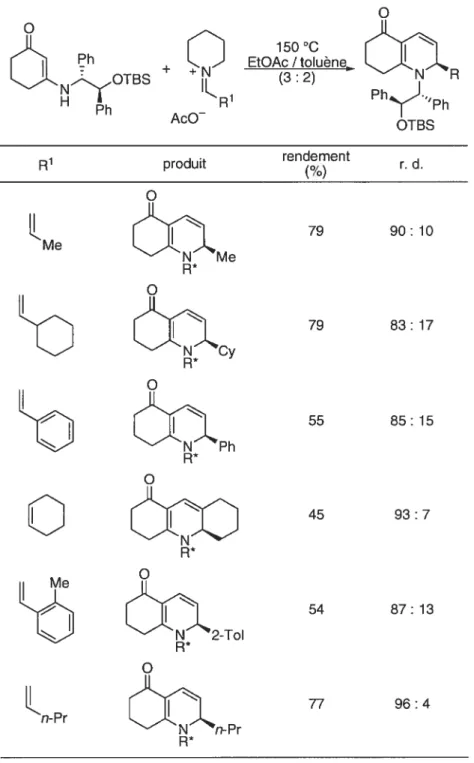
tétrasubstituées qui peuvent facilement être réduites aux octahydroquinolines correspondantes. L’utilisation d’un auxiliaire chiral branché sur l’azote de l’amide vinylogue permet la préparation de façon diastéréosélective de composés possédants différents groupements aryles et alkyles en position 2 (Tableau4)•40
38
M. J. McLaughlin, R. P. Hsung, K. P. Cole, J. M. Halin, J. Wang, Org Lett. 2002, 4, 2017-2020.
H. M. Sidenicka, R. P. Hsung, M. J. McLaughlin, L.-I. Wei, A. I. Gerasyuto, W. B. Brennessel, J. Am. CÏte,,z. Soc. 2002, 124, 10435-10442.
°
H. M. Skelenicka, R. P. Hsung, L-1. Wei, M. J. McLaughlin, A. I. Gerasyuto, S. J. Degen, Org. Lett. 2000,
19
n
Tableau 4. Synthèse diastéréosélective d’hexahydroquinolines par une condensation de Knoevenagel et une électrocyclisation en tandem.
150 oc EtOAcI toIuène (3 : 2) Ph + Ac0 R R’ produit rendment r.
u.
(/o) LM 79 90:10 N Mecy
79 83: 17 Ph 55 85: 15o
45 93 $ 7[i%
2-ToI 54 87: 13 n-Pr n-Pr 77 96: 4(n
1.3.2 Synthèse de pipéridines par réaction de fermeture de cycle
La synthèse de pipéridines par fermeture de cycle est très répandue dans la littérature. Elle consiste généralement à l’installation des chaînes latérales de la pipéridine voulue sur un squelette carboné acyclique pour ensuite former le cycle à six membres par une réaction de cyclisation ïntramoléculaire.
1.3.2.1 Lactamisations, aminations réductrices et formation d’hémiaminals
1.3.2.1.] Synthèse de pipéridines par lactainisation d’acides 3aminés
Il serait possible de citer de nombreux exemples de fermeture de cycle à six membres par une réaction de lactamisation. Déjà en 1954, le professeur Woodward utilisait cette réaction dans la synthèse de la strychnine.1’
Plus récemment, le professeur Davis a utilisé une approche de lactamisation pour la synthèse de pipéridines non racémiques.42 La chiralité du produit acyclique est induite par un auxiliaire de type sulfoxyde lors d’une réaction de Mannich diastéréosélective. Une lactamisation permet ensuite la fermeture du cycle et la réduction subséquente de la lactame donne accès à différents motifs de pipéridines (Schéma 7).
41
R. B. Woodward, M. P. Cava, W. D. Ollis, A. Hunger, H. U. Daeniker, K. Schenker, J. Am. Client. Soc. 1954, 76,4749-4751.
42
21
o
o
1. TFA I MeOH 3 steps
Ar NH Q Q 2. NaHCO3
t
1
llU
90% ,...Ç 52% Ph N e H OH 3 steps Ar’NH OH O %&0Me 67% (—)-SS20846ASchéma 7. Approche utilisant une lactamisation par Davis
Les 2-pipéridones peuvent aussi être transformées en triflates ou phosphonates d’énols et générer des tétrahydropyridines fonctionnalisées en position six par couplage de Suzuki43 ou de Stille.44 Il est à noter que les travaux du professeur Davis n’ont jamais menés à la synthèse de composés 2,5-disubstitués.
1.3.2.1.2 Synthèse de pipéridines par amination réductrice et formation d’hémiaminats La réduction diastéréosélective ou énantiosélective d’imines a de nombreux précédents.
Notamment, l’hydrogénation énantiosélective de 3 ,4-dihydroisoquinolines avec la méthode développée par le professeur Noyori permet d’obtenir des tétrahydroisoquinolines avec des excès énantiomères élevés.46 Cependant, cette thèse
°E. G. Occhiato, A. Trabocchi, A. Guarna, J. Org. Chem. 2001, 66, 2459-2465.
J. Jiang, R. J. De Vita, G. A. Doss, M. T. Goulet, M. T. Wyvratt, J.Am. Chem. Soc. 1999, 12], 593-594. S. Kobayashi, H. Ishitani, Chem. Rev. 1999, 99, 1069-1 094.
46
tentera de s’attarder uniquement aux méthodologies qui semblent plus générales et qui ont démontrées leurs applications en synthèse totale de produits complexes.
Une fois de plus, l’induction asymétrique observée lors de la cyclisation est induite par des centres stéréogènes qui sont présents dans la chaîne acyclique. Il est d’ailleurs bien connu, tel que démontré par Yamamoto, qu’une 3,4,5,6-tetrahydropyridine 2,6-disubstituée peut être réduite sélectivement au produit cis ou trans grâce au contrôle de la tension allylique.47 Le professeur Ellman utilise cette approche pour construire des pipéridines 2,4,6-substituées à partir de chaînes acycliques chirales.48 Cette méthodologie tire avantage de l’accessibilité aux sulfinimines chirales tout comme la méthodologie de Davis décrite à la section 1.3.2.1.1.
1.3.2.1.2.1 La méthodologie du professeur Lautens
Le professeur Lautens a développé une méthodologie pour préparer des 1,2-dihydropyridines impliquant la condensation intramoléculaire d’une amine secondaire avec un aldéhyde Œ,f3-insaturé.49 Cette réaction met en évidence le réarrangement d’époxydes vinyliques en présence d’un catalyseur de scandium (H[1,2] puis énolisation) pour générer une espèce diénolate. Ce diénolate effectue une addition de Mannich sur un imine N
benzylé ce qui produit un aldéhyde s-aminé qui condense in situ sur lui-même (Tableau 5).
K. Maruoka, T. Miyazaki, M. Ando, Y. Matsumura, S. Sakane, K. Hattori, H. Yamamoto, J. Am. Cheni. Soc. 1983, 105, 283 1-2843.
48
H. M. Peltier, I. A. Eliman,J. Org. CÏtem. 2005, 70,7342-7345. B. Brunner, N. Stogaitis, M. Lautens,Org. Lett. 2006,8, 3473-3476.
23
Tableau 5. Synthèse de dihydropyridines à partir d’époxydes vinyliques
o II O EtO’t> + Me N Ph RH Sc(OTf)3 (15 mol%) THF T.M. 5A 2-3h O °C à 50 oc Me PhPh rendement rendement R R fol .fo) /o 4-(F)Ph 61 2,4-(cI)2Ph 53 4-(CI)Ph 60 2-(Bpin)Ph 19 4-(Br)Ph 62 COOEt 63 Ph 37 2-Fur 37 4-(02N)Ph 43
Ce protocole permet la synthèse racémique de dihydropyridines avec des rendements moyens à faibles. Toutefois. cette réaction pourrait éventuellement menée à une version asymétrique.
1.3.2.1.2.2 La méthodologie de Blaauw
Une méthodologie développée au Pays-Bas utilise une approche chiron pour synthétiser des tétrahydropyridines par condensation intramoléculaire d’un dérivé de l’allysine (Schéma 8).°
50P. N. M. Botman, F. J. Dommerholt, R. de Gelder.
Q.B. Broxterman. H. E. Schoemaker. F. P. J. T. Rutjes, R. H. Blaauw, Org. Leu. 2004. 6, 4941-4944.
2 étapes TsoH(1orno/o) toluène, 110 00 2h
bz CO2Me
H2N CO2H CbzHN CO2Me
allysine éthylène acétal
7Z3 étapes HO,,
HCI
Schéma 8. Synthèse de tétrahydropyridines à partir de l’allysine éthylèneacétal
L’allysine est impliquée dans la biosynthèse des protéines et est synthétisée à partir de la lysine et de la lysyle oxydase dans la dernière étape de la biosynthèse du collagène. La seule synthèse chimique de l’allysine éthylène acétal comporte huit étapes pour conduire au produit racémique et le rendement global est de 14%.’ Blaauw rapporte la synthèse de quelques produits naturels en utilisant cette méthodologie, notamment la
(+)-épiquinamide52 et la (—)-dysibetaine PP53 (Schéma 9).
51
A. Rumbero, J. f. Martfn, M. A. Lumbreras, P. Liras, C. Esmahan, Bioorg. Med. Chem. 1995, 3, 1237-1240.
52
M. A. Wijdeven, P. N. M. Botman, R. Wijtmans, H. E. Schoemaker, f. P. J. T. Rutjes, R. H. Blaauw, Org. Lett. 2005, 7, 4005-4007.
25 H2NH 5 étapes 76% CO2Me 9 étapes 20% + H2N CO2Me __ H __
(J 2étapes Cbz—NN CO2Me 4 étapes
66% . 81% HN CO2H Cbz
J
(r.d.,10 1) HfltE:)1;-Ç
(—)-dysibetaine PPSchéma 9. Application de la méthodologie de Blaauw à la synthèse de produits naturels
1.3.2.1.2.3 La méthodologie CN(R,S)
Une méthodologie de cyclisation intermoléculaire entre un aminoalcool curai et un diaidéhyde a été développée en France dans les laboratoires du professeur Husson. Cette méthode permet la préparation en deux étapes de la 2-cyano-6-phényloxazolopipéridine, un composé chiral, stable et équivalent à une 1 ,4-dihydropyridine.54 Ce synthon fut utilisé pour la synthèse de plusieurs alcaloïdes. L’alkylation en a du groupement nitrile, son élimination, l’ouverture de l’oxazoiidine puis une débenzylation permettent la synthèse de pipéridines 2-aikylées enantiopures avec 77% de rendement global (Schéma I
54M. Bonin, D. S.Grierson, J. Royer, H.-P. Husson, Org. Synth.1992, 70, 54-5$.
L. Guerrier, J. Royer, D. S. Grierson, H.-P. Husson, J. Am. Chem. Soc. 1983,105, 7754-7755. (+)-épiquinamide
1. KCN, H20, pH 4 2. ZnBr2, CH2CI2 75-83% 1. LDA —78 oc 2. PrBr 99% cN Ph
Schéma 10. Synthèse de la coniine par la méthodologie CN(R,S)
Cette méthodologie a démontré son efficacité pour la synthèse de plusieurs alcaloïdes comportant des pipéridines 2- et 2,6-dïsubstituées. Son efficacité pour la synthèse de pipéridines 2,5-disubstituées reste cependant mitigée. Effectivement, la synthèse de la pumiliotoxine C publiée par Husson comporte peu d’étapes mais le rendement global du produit final est très faible (Schéma 11).56
‘Ph NH2 + cHo cHo
Q
‘Pr H2, Pd/c HCI 4 MeOH H >98% ee NaBH4 EtOH 80°c 98% ‘Ph27
H
H2, Pd(OH)2/C
rn
+CH2CI2IMeOH Pr”Nl
HH
Schéma 11. Synthèse de la pumiliotoxine C par la méthode CN(R,S)
1.3.2.2 Additions nucléophiles et addition conjuguées
1.3.2.2.] C’yclisation par réaction de SN2
La réaction de substitution nucléophule d’un halogénure d’alkyle par une amine pour former une pipéridine est très bien documentée et c’est une méthode souvent utilisée pour procéder à la cyclisation dans les dernières étapes d’une synthèse. Il est possible de trouver des références de substitutions nucléophiles intramoléculaires dans lesquelles un atome
MeO OMe Li B F4 0”CN_Me5’ O MeCN IH20 60 °c, 95% LDA, -78 oc Ph 65% A1203, cH2cI2 40 °c, 55% PrMgBr Et20 15°c rendement non-spécifié Ph 4 (r.d.=7:3) H MecN Na, NH3 —78 °c, 99% Pr o Ph H Me PrÇD pumiliotoxine c 29% 67%
d’azote (amine, amide, sulfonamide et phosphinoylamine) déplace soit un chlonire,57 un bromure58’59 ou un iodure d’alkyle.6° Toutefois, les groupements tosylates et mésylates sont davantage utilisés comme groupements partants.6’ Dans la plupart des cas, les centres chiraux de la molécule sont élaborés lors de la synthèse du précurseur acyclique. Une illustration intéressante de cette stratégie est la synthèse de la (—)-slaframine par la professeure Cossy (Schéma 12)•62
NBoc . NHBoc OBn
9etapes HOOH 1. MsCI, DMAP, Py 2. H2, Pd/C, Et3N, MeOH 67% H OH H OBn 2 étapes AcHN N 73% BocHN N (—)-slaframine
Schéma 12. Synthèse de la (—)-slaframine par substitution nucléophile intramoléculaire
S. Hanessian, A. M. Griffin, L. D. Cantin, Chircilitv2000. 12, 342-345.
‘ H. Mao, G. J. Joly, K. Peeter, G. J. Hoonaert, f. Compemolle, Tetrahedmn Lett. 1989, 30, 907-910. L. T. Lïu, P.-C. Hong, H-L. Huang, S.-F. Chen, C.-L. J. Wang, Y-S. Wen, Tetrahedron: Asyinmetiy2001,
12, 419-426. 60
S. Suginone, S. Yamada, J. B. Wang, J. Oig. Chem. 1990,55. 2170-2176. 61
Najdi, M. J. Kurth, Tetrahedron Lett. 1990,31. 3279-3282. 62
29
L’ addition nucléophule intramoléculaire d’ amines sur des époxydes a également été publiée.63 Cette approche est extrêmement efficace pour la préparation de 3-pipéridinols, mais elle demeure peu utilisée.
1.3.2.2.2 C’ycÏisation intramolécuÏaire par addition de Michael
La formation de pipéridines par addition de Michael intramoléculaïre a été
largement utilisée. Le problème majeur de cette approche demeure le contrôle de la diastéréosélectivité. Bien que des additions de Michael intramoléculaires soient utilisées pour la formation de liaisons carbone-carbone,64’65 la majorité des exemples impliquent l’addition conjuguée d’une amine.
L’addition d’amines sur des sulfoxydes a,j3-insaturés permet parfois un bon contrôle de la sélectivité. Tel que démontré par le professeur Pyne, l’utilisation de sulfoxydes chiraux a conduit à la préparation de pipéridines 2-substituées avec de bons rapports diastéréomériques (Schéma 13)66
BnNEt3OH
o
MeN> L CH2CI2,-40 °C
F3CO O’Ph r.d. = 91: 9 Me
Schéma 13.Addition conjuguée d’une amine sur un vinyle sulfoxyde chiral
63A. Kilonda, F. Compemolle, K. Peeters, G. J. JoIy, S. Toppet, G. J. Hoomaert, Tetrahedron 2000,56, 1005-1012.
64
F. Hughes, R. B. Grossman, Org. Lett. 2001, 3, 2911-2914.
65
A. Barco, S. Beneti, A. Casolari, G. P. Pollini, G. Spalluto. Tetrahedron Lett. 1990,3], 3039-3042. 66
Le professeur Ma en Chine a utilisé, quant à lui, l’addition conjuguée d’une amine sur une sulfone a,f3-insaturée dans la synthèse convergente des clavepictines A et B ainsi que de la pictamine.67 Le contrôle de la diastéréosélectivité du nouveau centre créé est excellent grâce au substituants équatoriaux en position 2 et 3 de la pipéridine. La pipéridine trisubstituée de départ a été préalablement synthétisée par une amination réductrice intramoléculaire (Schéma 14). (—)-clavipectine A R =Ac, R =C6H13 RO” N (+)-clavipectine B Me ‘-... R =H, R =C6H13 (—)-pictamine -R=Ac, R =C4H9 R
Schéma 14. Addition de Michael intramoléculaire appliquée à la synthèse d’alcaloïdes
L’addition conjuguée d’amines sur des cétones c3-insaturées a également menée à un bon contrôle de la diastéréosélectivité. On peut citer en exemple la cyclisation intramoléculaire d’une chaîne hautement fonctionnalisée par le professeur Armstrong dans
TBSO ÇO2Ph AICI3, CH2CI2 puis NaHCO3 80%
I
5-6 étapes TBSO” 45% M e SO2Ph 6731
le cadre de la synthèse d’une guanidine tricyclique utilisée comme modèle de la cylindrospermopsine (Schéma 15).68
OTBS
O TBSO NHCbz
74% TBSO.,,
Cbz
Schéma 15. Addition conjuguée intramoléculaire d’une amine sur une cétone cL,13-insaturée
L’addition conjuguée intramoléculaire de type 6-exo-dig d’amines sur des esters propargyliques est bien documentée.69 Mais, une publication récente rapporte la première addition conjuguée intramoléculaire d’une amine sur une cétone propargylique.7° Ces réactions ne produisent cependant aucun nouveau centre chiral. D’autre part, les observations expérimentales de cette dernière publication ont montré que le mécanisme n’impliquait pas réellement une addition de Michael intramoléculaire. En effet, l’addition de méthanol à la triple liaison formerait un f3-cétoacétale qui condenserait ensuite avec l’amine pour générer la 4-pipéridone (Tableau 6).
I. J. McAlpine, R. W. Armstrong, Tetmhedron Lett. 2000,41. 1849-1853. D. Ma, W. Zhu, Org. Lett. 2001. 3, 3927-3929.
Tableau 6. Conversion de cétones propargyliques 13-aminés en tétrahydropyridines Q 1.4NHCI dioxane 2. K2C03 Boc R2 MeOH cétone propargylique Boc R Boc R Ph BocHN R
o
NMe H’ Boc R=H R =Me R =Ph R=H R=Me R Ph R=H R =Me R= rir R= —H R2 I1 rendement toi /0 87 87 91 89 87 89 92 96 99 951.3.2.2.3 C’ycÏisation par addition de Mannich
L’ addition de Mannich intramoléculaire est une méthode efficace et stéréosélective pour synthétiser des 4-pipéridones hautement fonctionnalisées. Cette réaction a été utilisée pour accéder à plusieurs composés alcaloïdes isolés de peaux de grenouilles dendrobates
tétrahydropyridine
HMe
°
33
pumiuo.7’ Une approche très élégante utilise des cétales f3-aminés comme précurseurs, qui après condensation avec un aldéhyde cyclisent par réaction de Mannich pour générer des 4-pipéridone-2,6-cis-disubstituées. Cette façon de faire a d’abord été décrite par le professeur Troin,72 puis réutilisée quelques années plus tard par le professeur Davis qui a développé une méthodologie permettant la synthèse des précurseurs avec des rendements supérieurs.73 Davis a d’abord procédé à la synthèse de produits naturels comportant des pipéridines 2,6-cis-disubstituées,74’ puis il s’est ensuite attaqué à la synthèse de composés plus complexes. Comme dans le cas des pipéridone-2,6-cis-disubstituées, l’addition de Mannich intramoléculaire menant à l’alcaloïde (—)-indolizidine 209B a été très sélective ne générant qu’un seul diastéréoisomère (Schéma 16).76
M. Arend, B. Westermann, N. Riscli, Angew. Citent.hit. EU. 1998, 37, 1044-1070. 72
S. Carbonnel, Y. Troin, Heterocyctes 2002, 57, 1807-1830.
Pour une revue de la méthodologie de Davis employant les sulfinimines, voir: P. Zhou, B.-C. Chen, F. A. Davis, Tetmhedron 2004, 60, 8003-8030.
Pour la synthèse de l’alcaloïde (+)-241D, voir: F. A. Davis, B. Chao, A. Rao, Orgi Lett. 2001, 3, 3169-3171.
Pour la synthèse de l’alcaloïde (—)-épimyrtine, voir: F. A. Davis, Y. Zhang, G. Anilkumar, J. Org. Citent. 2003, 68, 8061-$064.
76
9
4 étapes H2N ToI NH2 58% OBn MgSO4 2. TsOH 75°C 61% Me 6 étapes Me C5H1” N (—)-indolizidine 209B OBnSchéma 16. Synthèse de la (—)-indolizidine 209B par une réaction de Mannich intramoléculaire
L’addition de Mannich fut beaucoup moins sélective lorsqu’il procéda à la cyclisation conduisant aux 4-pipéridone-2, 3,5 ,6-tétrasubstituées.77 Effectivement dans ce casci, la chaîne éthyle en position pseudo axiale déstabilise l’état de transition chaise et la contribution de l’état de transition bateau croisé explique la formation du diastéréoisomère minoritaire. Cette approche permet néanmoins la préparation de l’alcaloïde (—)-indolizidine 223A avec un nombre d’étapes record de dix et avec 10% de rendement global (Schéma
17).
35
TsOH benzène 40 h, t.a.
Schéma 17. Synthèse de l’indolizidine (—)-223A par une réaction de Mannich intramoléculaire
1.3.2.3 Cyclisation radicalaire
Les cyclisations radicalaires pour former des pipéridines sont peu exploitées. Les cyclisations utilisant l’hydrure d’étain se sont avérées très peu sélectives.78’79’80 L’utilisation
S. E. Yoo, K. Y. Yi, S. H. Lee, N. Jeong, Synlett 1990, 575-576. ‘
A. F. Parson, R. M. Pettifer, Tetrahedron Leu. 1997, 38, 5907-5910.
3 étapes 73%