/
Anticorps contre des peptides synthétiques sélectionnés
à partir de la séquence d'ADN du gène de la protéinase de !'adénovirus
type 2. Etude innnunologique et biochimique.
par
Lyne Delorme
Département de microbiologie
Thèse présentée
à la Faculté de
médecine en vue de l'obtention
du grade de maître ès sciences (M.Sc.)
REMERCIEMENTS
Je voudrais tout d'abord remercier mon maître de thèse, le docteur Joseph Weber, pour ses conseils, son soutien et son assistance scientifique.
Je tiens aussi à remercier les docteurs Govindranathsing Khittoo, Serge St-Pierre et Brian Talbot pour leur collaboration lors de mon projet. Le docteur Jean Barabé pour m'avoir conseillée et avoir partagé une partie de ses connaissances scientifiques avec moi.
Je remercie aussi tous les gens du laboratoire, en particulier Lise Clavet, pour leur support et leur amitié autant à l'intérieur qu'à l'extérieur du laboratoire.
A toutes les personnes du département et à mes professeurs je dis merci pour l'enrichissement qu'ils m'ont apporté.
TABLE DES MATIERES
REMERCIEMENTS . . . • . . . • . . . • . . . • • . . . •
I
TABLE DES MATIERES . . • . . . • • . . . • . . • . • • . . . . • • • . . . • . . . • . .
II
LISTE DES FIGURES . . . • . . • . . . • . . • . . • . . . . • . . . • . • . . • . • • . • • . . • • .
IV
LISTE DES TABLEAUX . . . • . . . • . . • . . • . • . . . . • . • . . • . . . • • . • . .
V
LISTE DES ABREVIATIONS . . . • . . . • . . • . . • • . • . . • . • . . . • . . • . . . . •
VI
SOMMAIRE ...•.•...•...•.•...••.•..•.•.••..•.••....• VIII
INTRODUCTION . . . • . • . • . . . . • . • . . • . . . . • . . • . . . 1 A) L 'adénovirus . . . 1B) Activité protéolytique .••..••...•.•...•....••.••..••
4
C) D)Autres
Projet
protéinases .•.•.•..•...•....•...••....••....
10 13MATERIEL ET METHODES • . . . . • . . . • . • . . • . • • . . • . . . . • • • • . • . . . . • . . . • . . .
1 7
Cellules
Virus et
infection ... .17
17
Purification du virus . . . . • . . • • . • . . . • . • . • . . . • . . • • . . . . • . . .
17
Marquage des protéines virales •.•....•..••.•..•....•..•...
18
Isolation des noyaux et purification des
matrices nucléaires
18
Réaction de clivage de la protéinase virale ....•..••..••...•
19
Réaction de clivage de la protéinase nucléaire •.••...•••..
20
Electrophorèse . . . 20MATERIEL ET METHODES (suite)
Synthèse et purification des peptides ...••...
22
Formation des anticorps . . . . • . . . • . . . • . • . . • • • . . • . . .
24
Immunofluorescence . • • • . . . . • • . . . • . . . • . . . • . . • . . . • . .
26
Etudes par microscopie électronique .••.••.•..••.•••...•..•..
27
RESULTATS . . . . • . . • . . . • . . . . • . • • . • • • • . • • . . • . • • . . . • . .
2 9
Choix des séquences d'acides
amines ... ..
,29
Synthèse et purification des peptides synthétiques ..••.••.••
33
Formation des anticorps . • . . • . • . . . • . . • . • • . • • • • • . • • • . . • . . • . .
38
Spécificité des anticorps ...••.•...•.•••.•.•••••••••..•.•.•.
41
Mise en évidence de la protéinase par
méthode immunologiques: • • • • • . • . • . . • . . • • . • • . . • . . . • . . • • . • • . • • •
41
a) Immunofluorescence . . . • • • • . • • • • • . • • • • • • • • • • • • • . • • • .
41
b) Microscopie électronique •••••.•.••.•.•.••....•..•..•.
48
Association de la protéinase virale aux matrices
nucléaires
S2
Les jeunes virions sont la source
de l'activité protéinase ...•..•..••....•..•....••.•...••
SS
DISCUSSION . • • . • . . • . . . • • . • . • • • • • . • • . • • • • . • . . . . • . . • • . • • . . •
S7REFERENCES . . • . . . • . • • . . • . . . • • • • • • . . • • . . . • . • . . . • • • • . • • • . • . • • • . • • .
66
LISTE DES FIGURES
Figure 1: Structure de la capside de l'adénovirus Figure 2: 2 modèles de localisation des protéines
Figure 3: Figure 4:
Figure 5: Figure 6:
du virion Ad-2
Représentation schématique des ARN-m de Ad-2 ...•.. Séquence de l'ADN codant pour la protéinase de
Ad-2 avec la mutation de H2ts1 •.•..••••.•..•...••. Structure particulière de la proline ••••..•••••.••.. Technique de synthèse peptidique .••.••••..••••.•••••
2 3 6 7 14 23 Figure 7: Patron d'injection des lapins •••.•••••.•..•.••..•.•• 25 Figure 8: Courbe d'hydrophilicité de la protéinase
Figure 9: Séquences et emplacements des 4 peptides choisis .••• Figure 10: Vérification de la pureté des 4 peptides
synthétiques par analyse des acides aminés
30 32
35
Figure 11: Vérification de la pureté du peptide IV par CLHP 37
Figure 12: Vérification de la spécificité de chacun des
anticorps par buvardage ••..•.••.•.••...•.••••••••• 42 Figure 13: Localisation de la protéinase par
immunof luorescence 46
Figure 14: Localisation de la protéinase par microscopie
électronique 49
Figure 15: Association de l'endoprotéinase virale avec les
matrices nucléaires . • . . . . • • • • . • • . • • . . . • • • • • . . • . . . 54 Figure 16: Instabilité des jeunes virions dans le test
LISTE DES TABLEAUX
Tableau 1: Sites de clivage possibles des protéines précurseurs 9
Tableau 2: Composition de chacun des peptides par analyse
des acides aminés • • . . • . . . • . . . • . . • • . . . . . . . . . . . . • . • . . . . • 36 Tableau 3: Vérification de l'efficacité de couplage ...•..•... 40 Tableau 4: Comparaison de l'immunofluorescence contre la
protéinase et des titres obtenus pour chacun des antisérums contre leu~s peptides respectifs Tableau 5: Représentation de la réponse en microscopie
électronique de l'anticorps antiprotéinase par
44
LISTE DES ABREVIATIONS
Ad-2:
Adénovirus de sérotype-2 humain.
Ad2-tsl (ou H2-tsl): Mutant thermosensible (tsl) de !'adénovirus de
type 2 humain.
BOC:
BSA:
CCM:
CLHP:
Cs Cl:
DEA:
DFP:
DMEM:
EDC:
EDTA:
ELISA:
EMC:
HF:
KLH:MBS:
NP-40:
PBS:
pfu/<t: p. i.:Groupement protecteur.
Albumine bovine de sérum (bovine serum albumin).
Chromatographie sur couche mince.
Chromatographie liquide
àhaute pression (HPLC).
Chlorure de césium.
Diéthylamine.
Diisopropyl fluorophosphate.
Milieu de Eagle modifié par Dulbecco.
1-éthyl-3-(3-diméthylaminopropyl) carbodiimide.
Acide tétraacétique-éthylène diamine.
Enzyme
linked immunosorbant
d'enzyme lié par immunosorbtion.
Encéphalomyocardite.
Acide fluoridrique.
assay ou essai
Keyhole lympet heamocyanin.
N-maléimidobenzoyl-N-hydroxysuccinimide.
Nonidet P-40.
Tampon phosphate salin.
Unité de formation de plaque/cellule ou Plaque
forming unit/cell.
P. I.:
PMSF:
RIA:
SDS:
Sf:
Ta:
TEMED:
TFA:
TLCK:
TPCK:
wt:ZLCK:
Sérum préimmunitaire.
Phénylméthylsulfonylfluoride.
Essai radioimmunologique.
Sulfate dodécyl de sodium.
Sérum immunitaire final.
Température ambiante.
N, N, N', N-tétraméthy(éthylène-diamine).
Acide trifluoridrique
N-a.-P-tosyl-L-lysine chlorométhyl cétone.
L-a.-tosylamine-2-phényl éthyl chlorométhyl cétone.
Type sauvage.
N-CBZ-L-lysine chlorométhyl cétone.
SOMMAIRE
Afin d'obtenir un antisérum contre la protéinase de l'adénovirus de type 2, nous avons fabriqué 4 peptides synthétiques dont les séquences ont été choisies en se basant sur la séquence de l'enzyme
établie
à
partir de sa séquence nucléotidique. Les peptides purifiéscouplés
à
une protéine porteuse ont été injectésà
de jeunes lapins. Les antisérums obtenus ont été utilisés pour des études d'immunofluorescence et de microscopie électronique. En premier lieu, il fut observé que la protéinase était située dans les noyaux de cellules infectées. Aucune différence de reconnaissance des anticorps pour l'enzyme normal et 11 enzyme d'un mutant thermosensible n'a été remarquée. Par microscopieélectronique, il a été établi que l'enzyme était lié
à
la chromatine virale avant l 'encapsidation. Comme ses substrats se retrouvent sous leur forme précurseur; ceci suggère que la protéinase se retrouve tout d'abord sous forme inactive ou de précurseur avant de devenir active pour permettre la maturation des virions. Il fut aussi observé que l'activité protéinasique était concentrée dans les fractions des matrices nucléaires. Ceci soutient 11 idée que les matrices nucléairesINTRODUCTION
A) L'adénovirus
Les adénovirus sont des virus animaux très fréquemment retrouvés (pour revue, Philipson, 1984). Ils furent isolés pour la première fois en 1953 par Rowe et Hubner à partir de tissus adénoïdes; d'où leur nom. Les virus de type a dé no se retrouvent chez une grande étendue de types d'animaux. On en dénombre 37 humains, 23 simiens, 10 bovins, 8 aviaires, 4 porcins, 2 canins, 2 murins, 1 ovin et 1 chez
l'opossum.
Quoique provenant d'origines différentes; tous ces
adénovirus ont une morphologie uniforme. Leur forme est icosahédrique. Ils sont constitués de 252 capsomères dont 240 hexons et 12 pentons postés aux extrémités de l 'icosahèdre et chacun d'eux entouré de cinq hexons péripentonéaux. Sur chaque penton, on retrouve une fibre apicale (cf. fig. 1) (Everitt et al., 1975). Ces éléments constituent la capside,
à
l'intérieur de laquelle on retrouve le "core" nucléaire constitué d'ADN et d'au moins trois protéines virales (cf. fig. 2)(Brown et al., 1975).
Parmi ces trois protéines, on retrouve: premièrement la protéine majeure du "core" que l'on nomme VII, possédant un poids moléculaire de 18 Kd. En deuxième lieu, on a la protéine mineure du "core" V, de 48 Kd (Laver et al., 1968; Prage et al., 1968; Laver, 1970;
a
•
~----PER I PEN T O N AL HE XO NS @
+ - - - F I BER
F.igure 1: Structure de la capside de 1 'adénovirus. (a) Capside et localisation de ses différentes composantes selon Philipson et Petterson, 1973. Prog. Exp. Tumor Res. Vol. 18. (b) et ( c) Virion Ad5 selon Valent i ne et Pereira, 1965. J. Mol. Biol., Vol. 13; Horn e et al., 1959. J . Mol. Biol. Vol. 1.
a
b
Figure 2:
Polypept1d t 505 - gel Structural Und
- Pentonbase _ Peripentonal protein - Fiber ~. - Terminal prote in ... - Core prote1n 1
---
-
·X
__.. XI - - - -XII VIRION POLYPEPTIDES- Hexon -ass oc1 ated prote1n
- Core prot e1n ll
_.,,,.,...Huon assoc1ahd prot e1n
- Pro te in spec 1f1c for:
grou ps of n1ne he.cons
oYo
oo
DISRUPTED VIRION
2 modèles de localisation des proté ines du virion Ad2.
(a) Modèle bas é sur des études de "cross-linkage" selon Philipson, 1985 . Cur . Top ics Microbiol . Immun. Vol. 109. (b) Modèle schématique selon Russell et Precious, 1982. J. Ge n. Virol. Vol . 63
Prage et al., 1970; Prage et Pettersson, '1971; Russell et al., 1971; Everitt et al., 1973; Nermut, 1978). Et en troisième lieu, on retrouveµ d'un poids moléculaire d'environ 4,000 daltons (Hosakawa et Sung, 1976). Ni leur emplacement, ni leur rôle précis ne sont bien établis. Ces trois protéines sont reconnues comme étant riches en arginine.
B) Activité protéolytique
Chez l'adénovirus de type sauvage (wt), un certain nombre de protéines sont retrouvées tout d'abord sous forme de précurseurs (Anderson et al., 1973; Ishibashi et Maizel, 1974; Edvarsson et al., 1976; Stillman et al., 1981, Philipson, 1984). On peut souligner:
la protéine majeure du "core", VII qui provient de PVII; - PVIII; qui lorsque clivée devient VIII, une protéine de la
capside;
PVI; qui est maturée en VI, aussi une protéine de la capside;
- 87 K qui donne une protéine de 55 K;
- et 11 K dont les produits de clivage ne sont pas entièrement établis; mais tout laisse croire qu'il y en ait 3 (Weber, résultats non publiés).
Il semble donc qu'il y ait une activité protéolytique associée aux particules virales. Cette activité enzymatique fut étudiée de plus près en grande partie grâce
à
un mutant thermosensible de l 'adénovirus de type 2 (H2tsl) (Bégin et al., 1975; Weber et al., 1975;Weber, 1976; Bhatti et Weber, 1979a,b; Yeh-Kai et al., 1983). Chez ce mutant, on note une déficience dans une fonction tardive; soit le
clivage polypeptidique qui n'est pas effectué à la température non
permissive (39°c). Les particules virales H2tsl-39°c sont donc immatures et de ce fait non infectieuses. Alors, le clivage protéolytique des précurseurs est une éta~e très importante, voire même essentielle dans la maturation des jeunes virions. Comme il y a un mutant déficient dans cette fonction, il semble bien que cette activité enzymatique soit d'origine virale.
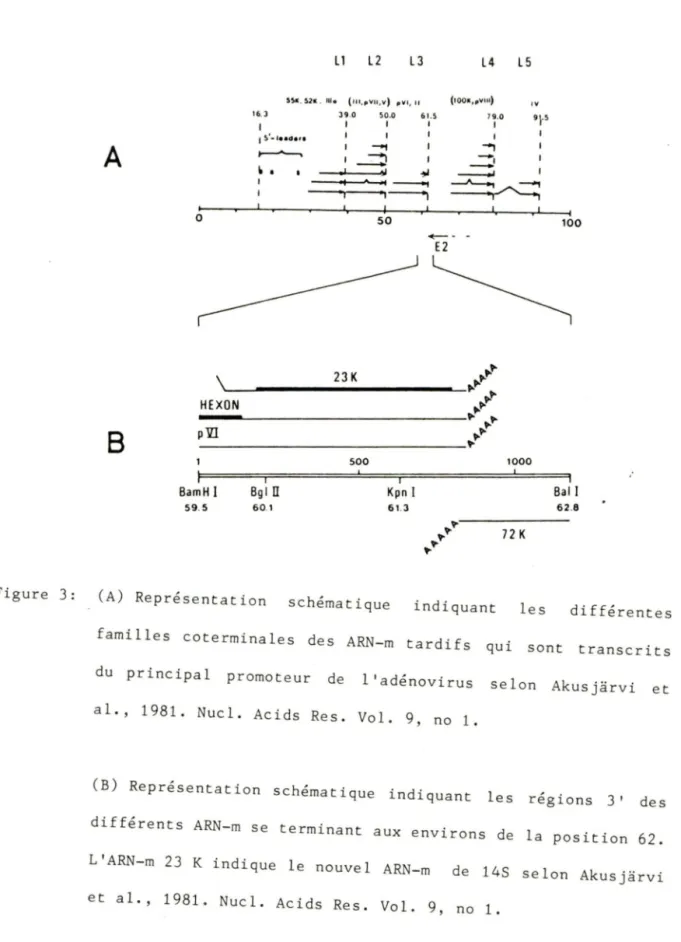
Des études plus poussées furent exécutées sur cette activité protéolytique. En 1981, Akusjarvi en voulant séquencer la région 3' non codante du ARN-messager (ARN-m) de l'hexon découvrit un cadre de lecture ouvert de 609 nucléotides (Akusjarvi et al., 1981). Cette séquence d'ADN était située entre les positions 59.5 et 62.8 du génome de Ad2; soit dans la région tardive L3 (cf. fig. 3). Ce ARN-m pouvait donner naissance à une protéine d'environ 23 K. Par la suite, Yeh-Kai et al. (1983) ont voulu situer la mutation de H2tsl afin de vérifier s'il y avait un lien avec le cadre de lecture ouvert de 609 nucléotides découvert par Akusjarvi. Les méthodes utilisées étaient celles de complémentation et de séquençage de l'ADN. La région nécessaire pour la comp lémenta t ion du mutant était celle espérée. Plus précisément, on situa la mutation
à
la position 61.1 et le changement de nucléotide produit est le remplacement d'un C par un T. La mutation résulte donc en le changement sur la protéine d'une proline par une leucine (cf. fig.Figure 3:
A
B
0 BamH 1 59. 5 L1 L2 LJ L4 L 5 s~ . S2• . 111. ( 111, 111v11,\I ) ,v1 , 11 (1 00•,,v111) IV 16. J 1 I S'·l••der1 ~.
Bg 1 Il 60.1 39 .0 50.0 6 1. 5 1 1 1 1 1 1 .,1 --t---"""-4-"'"4 ~ --..---.... ---i 50 1~. o 91.5 1...,
-,
-
---~
---
.
Kpn 1 61.3 E2 1000 .,..,. 7 2 K.,.
.,.
..
(A) Représentation schématique indiquant les
100
Bal 1 62 .8
différentes familles coterminales des ARN-m tardifs qui sont transcrits du principal promoteur de l 'adénovirus selon Akusjarvi et al., 1981. Nucl . Acids Res. Vol. 9, no 1.
(B) Représentation schématique indiquant les régions 3' des différents ARN-m se terminant aux environs de la position 62. L'ARN-m 23 K indique le nouvel ARN-m de 14S selon Akusjarvi et al., 1981. Nucl. Acids Res . Vol. 9, no 1.
1 O•J TGCGCACGCCCTTCTCGGCCGGCAACGCCACAACATAAAAGAAGCAAGCAAC:ATCAACAACAGCTGCCGCC LeuArqThrProPheSerAlaGlyAsnAlaThrThr+++ 200 Bq! li ~ ATr,r,r,crccAGTGAGCAGGAACTGAAAGCCATTGTCAAAGATCTTGGTTGTGGGCCATATTTTTTGGGCACC '·'e tG l .v Se rSerGl ur.1 nGl uleuly sA 1 a Il eV al Ly sA s ple'uG 1 yCy sG 1 yP roTyrPheleuG 1 yîhr
300 TATG~CAAGCGCTTTCCAGGCTTTt.TTTCTCCACACAAGCTCGCCTGCGCCATAGTCAATACGGCCGGTCGr TyrAsoLysArqPheProGlyPheValSerDroHisLvsLeuAlaCysAlalleValAsnThrAlaGlyArg Gftr.Acrr,r.r;r,r,cr.TACACTGGATGGCl.TTTGCCTGGAACCCGCGCTCAAAAACATGCTACCTCTTTGAGCCC r,luThrr,lvGlvValHisTroMetAlaDheAlaTroAsnProAroSerLysThrCysTyrLeuPheGluPro 40() '
TT v.r;(TT TTC TGACCAAC:G ACTCAAGCAGG TTT ACCAG TTTGAGT-ACGAGTCAC TCC TGCGCC:GTAGCGCC PheGlyPheSerAsor.lnArqLeuLysGlnValTyrGlnPheGluîyrGluSerLeuleuArgArqSerAla ~no ATTGC?Tl.TTCC:CCCGACCGCTGTATAACGCTGGAAAAGTCCACCCAAAGCGTGCAGGGGCCCAACTCGGCC Ile!laSerSerProAspArqCys!lPThrleuGluLysSerThrGlnSerValGlnGlyProAsnSerAla Pro 600 ccc ' GCCTGTGGACTATTCTGCTGCATGTTTCTCCACGCCTTTGCCAACTGG CAAACTCCCATGGATCACAAC AlaCvsGlyLeuPheCysCysMetPheleuHisAlaPheAlaAsnTro GlnThrPro~PtAsoHisAsn CTC KpnI Leu
r-.
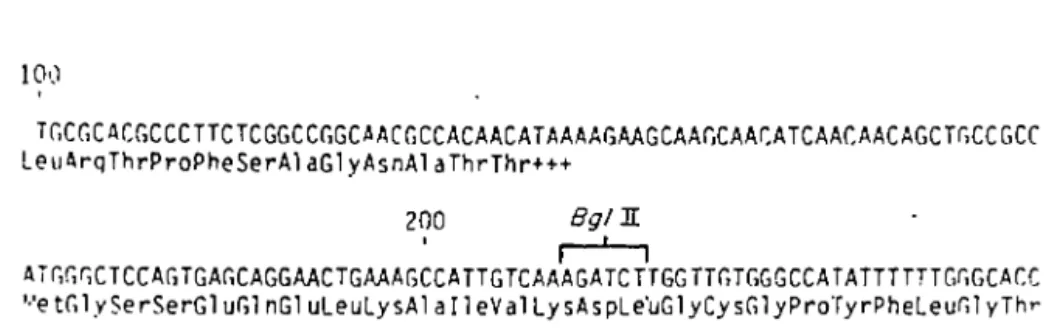
CCCACCATGAACCTTATTACCGGGGTACC:CAACTCCATGCTTAACAGTCCCCAGGTACAGCCCACCCTr.cr,r ProThr~etAsnleulleThrGlvValProAsnSerMetLeuAsnSerProGlnValGlnProThrleuA-a 700 ' Cr.CAACCAGGAACAGCTCTACAGCTTCCTGGAGCGCCACTCGCCCTACTTCCGCAGCCACAGTGCGCAGATT ArqAsnGlnGluGlnLeuTyrSerPheLeuGluArgHisSerProTyrPheArgS~rHisSerAlaGln!le ROO 1 !GGAGCGCCACTTCTTTTTGTCACTTGAAAAACATGTAAAAATAATGTACTAGGAGACACTTTCAATAAAGG Ar~)erAlaThrSerPheCysHisLeuLysAsnMet+++ CAAATr;TTTTTATTTGT-(AAAAAA)Figure 4: Séquence de l 'ADN codant pour la protéinase de l 'adénovirus de type 2 avec la mutation de H2tsl selon Yeh-Kai et al., 1983.
J.
Mol. Biol. Vol. 167.particulière apportée par la présence d'une praline. Donc, on conclut que la protéinase est codée par le virus dans la région L3, et que tsl est muté dans ce gène.
Tremblay et al. (1983) mirent au point deux systèmes d'essai de protéinase in vitro afin d'étudier la spécificité de la protéinase de Ad2 au lien peptidique et au substrat. Pour ce faire, ils utilisèrent une source d'enzyme viral provenant de particules de Ad2 wt
partiellement désintégrées et de substrat issu soit de noyaux de cellules infectées par H2tsl-39°C ou soit de particules virales purifiées du même mutant. H2tsl-39°C fut utilisé car on y retrouve les 5 précurseurs non clivés dû au fait que ce mutant est défectif dans l'activité protéolytique. Ils déterminèrent que la protéinase hydrolysait spécifiquement des liens glycine-alanine (cf. table 1) et de plus qu'elle était spécifique à son substrat naturel puisqu'elle ne coupait pas de protéines étrangères. Cette dernière observation suggère donc que le clivage n'est pas déterminé seulement par la séquence primaire mais aussi par d'autres caractéristiques physiques du substrat.
On sait de plus que 11 enzyme est une protéinase de type sérine car elle est inhibée par le diisopropylfluorophosphate (DFP) (Tremblay et al., 1983). Son activité est aussi bloquée par le phénylméthylsulfonyl fluorure (PMSF); signe d'une activité protéoli-tique. Le L-1-tosylamine-2-phényléthylchlorométhyl cétone (TPCK) a aussi le même effet; ce qui signifie que l'on est en présence d'un enzyme de type chymotrypsine (Bhatti et Weber, 1978). Le EDTA n'affectant pas la protéinase (Bhatti et Weber, 1978), ce n'est donc pas une
SITES DE CLIVAGE POSSIBLES SUR LES PROTEINES PRECURSEURS
Protein M, Location of Gly-Ala residues Frag ments
N-lerm. C-term. PVI 27000 SerAsnMetSer~y~y-~aPheSerTrp~y~r 33 217•
PVI I 20 457 ( 1 ••• Gly3 •••••• 20)-~ alyslysArgSerAsp 20 163 •
PVIII 24 707 ~n MetThrAs nS er~y-~ a~nl e u~a~y~y 105 122 ·
ThrPhe~n.!__l:e~y~ y -~a~yArgSer~rPhe 156 71
Ser~y~a ~nleu~ a-~y~yPheArgHisA rg 109 118•
Met~yleu~a~ a~y-~a~aGLuAspT y rSer 22 205
87K 74833 TyrTh r.!__l:eAsn Thr~ y-~ aT yrHisArg~eVal 116 536•
GluAspleu~aPro~y-~aPro~aThrleuArg 47 605
~ aSerThrThr~ a~a-~y.!__l: eTh rTry Met~r 24 628
LeuArg.!__l:e~n~n~ a-~ y Prol y sAspMetVa l 224 428•
.!__l:e Asp~aSerAsp~a-G lyGLnGLuAsp A L a~u 183 469
Ta bleau 1: Sites de c livage s possible s sur les protéine s pré c urseurs de l 'adénovirus de type 2 . Selon Tremblay et al . , 1983. Biochim . Biophys . Acta 743 : 239-245.
métalloprotéine. Le cycloheximide, inhibiteur de la synthèse de nouvelles protéines n'a aucun effet sur elle (Weber, 1976), ce qui signifie que la synthèse de nouvelles protéines n'est pas néc-essaire pour que le processus de clivage se poursuive à la même vitesse. On sait aussi qu'elle a une activité maximale à un pH de 6.5-7.0 et une activité enzymatique stable jusqu'à 45°C. Elle reconnaît aussi son substrat même si celui-ci est dénaturé.
c) Autres protéinases
Traditionnellement les protéinases étaient considérées comme des enzymes ne servant qu'à couper des protéines en petits peptides et en acides aminés. Donc les rôles qu'on leur attribuait n'étaient que de digérer les protéines nutritives ou de participer au renouvellement des protéines cellulaires. Ceci est vrai pour certaines protéinases comme la trypsine, la chymotrypsine, la pepsine, la cathepsine B et la cathepsine
D.
Mais plus récemment il a été démontré qu'elles avaient des rôles très importants dans une grande variété de processus cellulaires (Holzer et Heinrich, 1980; Holzer et Tschensche, 1979). Elles peuvent effectuer une fonction de régulation comme dans l'activation des
hormones. On leur attribue aussi un rôle dans le transport
cotraductionnel de polypeptides sécréteurs au travers des membranes microsomales et dans le transport posttraductionnel de polypeptides à l'intérieur des organelles (Kreil, 1981). Le renouvellement des protéines cellulaires, rapporté pour la première fois chez les animaux
par Schoenheimer, per~et aux cellules d'éliminer les protéines anormales et d'adapter leurs compléments de protéines plus rapidement en fonction des besoins physiologiques (Schoenheimer, 1942; Pine, 1980).
On retrouve une activité protéinasique dans plus de 150 genres de champignons, de protozoaires et de moisissures (North, 1982). Les protéinases ont généralement des poids moléculaires variant de 18,500 à 35,000 daltons mais le plus fréquemment aux environs de 25,000. Par contre, des enzymes plus lourds ont été rapportés chez des champignons tels que aspergillus niger (Bosmann, 1973), aspergillus nidulans (Stevens et Stevens, 1980), phycomyces blakesleeanus (Fisher et Thompson, 1979), blastocladiella emersonii (Lodi et Sonneborn, 1974) et dans blakeslea trispora (Govind et al., 1981). La dernière citée est celle possédant le plus haut poids moléculaire rapporté; soit 126,000.
On retrouve des protéinases aussi chez les virus. Des évidences montrent qu'elles sont présentes entre autres chez les picornavirus, les togavirus et les virus tumoraux à ARN (Butterworth, 1977). En effet chez les togavirus, la présence de précurseurs clivés par la suite en protéines matures fut observée (Clegg et Kennedy, 1976; Wengler et Wengler, 1976; Snyder et Sreevalsan, 1974; Jones et al., 1974). Par des expériences de cartographie et de marquage suivi d'une chasse ("pulse-chase"), certains auteurs ont déterminé que la protéine virale p-130 (de 130,000 daltons) du virus Sindbis est le précurseur de la protéine L du "core" et des protéines El et E2 de l'enveloppe (Schlesinger et Schlesinger, 1973; Snyder et Streevalsan, 1974). Ceci fut confirmé aussi avec l'aide des mutants thermosensibles ts2, ts5 et
ts13. A température non permissive, ces mutants sont déficients dans la formation de la nucléocapside et dans ces cas on peut voir une accumulation de la protéine p-130 (Strauss et al., 1969; Scheele et Pfefferkorn, 1970).
Chez les picornavirus, on retrouve différents clivages protéolytiques. Des études sur le virus de l 'encéphalomyocardite (EMC) ont démontré que différents précurseurs étaient clivés en polypeptides de plus faible poids moléculaire. En effet, on sait que le polypeptide C est rapidement coupé en D (sa demi-vie est d'environ 10 minutes)
(Butterworth et Ruckert, 1972) et par la suite ce dernier est clivé en E (Butterworth et al., 1971). Pour le virus du polyome, une activité similaire est associée aux particules. Il est dit que chez le virus du polyome; la protéine VP-1 subit des clivages protéolytiques qui donnent des protéines de 43. 5 K et de 40 K (Bowen et al., 1984). Ils ont démontré ceci en mesurant la radioactivité contenue dans VP-1, 43.5 K et 40 K. A l'aide d'un gel, ils ont vu que la somme de 43.5 K et 40 K après 24 heures d'incubation correspondait au taux de radioactivité contenue dans VP-1 au temps O. De plus, tout laissait croire qu'il s'agissait d'une sérine-protéinase puisqu'elle était inhibée par le DFP et le PMSF. Par contre elle n'était pas inactivée par TLCK, TPCK ou ZLCK donc ce n'était pas une protéine de type trypsine ou chymotrypsine. En marquant les protéines virales au [1,3-3H] DFP; ils ont vu que seule VP-1 était marquée de façon significative. Donc tout laisse croire que la protéine VP-1 du virus du polyome est une sérine-protéinase.
D) Projet
Le but premier de ce projet est d'obtenir des anticorps contre la protéinase de l'adénovirus de type 2. Ceci prouvera de façon indépendante que le cadre de lecture ouvert de 609 nucléotides dans L3 code réellement pour la protéine en question. Ces anticorps pourront par la suite permettre d'autres études sur l'enzyme. La protéinase étant présente en faible quantité et jamais vue sur gel, l'approche choisie pour prouver son existence a donc été de faire des anticorps contre des peptides synthétiques.
Donc pour arriver
à
ceci, nous avons tout d'abord choisi quatre séquences d'acides aminés de la protéine contre lesquelles des anticorps polyclonaux seront fabriqués. Dans le choix des séquences il y a certains facteursà
tenir compte; on les appel le des déterminants conformationnels:La présence de prolines; car· vu sa conformation
particulière (cf. fig. 5), la proline forme une sorte de coude. Donc elle a plus de chances d'être exposée
à
la solution. De par sa conformation, elle force les résidusavoisinants
à
se rapprocher età
former ce que l'onappelle des déterminants conformationnels mineurs. Plus le nombre de prolines est élevé dans un peptide, plus les
chances sont grandes que sa structure soit similaire à
0
"
----C-o
On doit aussi avoir une séquence contenant le plus
d'acides aminés hydrophiles possible; car elle aura alors
tendance
à se tenir à la face extérieure de la protéine.
On doit de préférence avoir une longueur minimale de 6
acides
aminés
pour
induire
une
bonne
réponse
immunologique.
L'étape succédant au choix des séquences est alors la
synthèse des peptides sélectionnés. Ceux-ci seront par la suite
fusionnés
à une protéine appelée porteuse (normallement BSA-bovine sérum
albumine ou KLH-keyhole lympet hemocyanin) au moyen d'un agent de
couplage. Les agents de couplage fréquemment utilisés sont:
- MBS (N-maléimidobenzoyl-N-hydroxysuccinimide); qui est un
ester liant la protéine porteuse
àla cystéine du peptide;
- triazine; qui attache le peptide
àla protéine par un
résidu tyrosine;
- glutaraldéhyde;
et les carbodiimides;
surtout le
EDC
(1-éthyl
3(3-diméthylaminopropyl) carbodiimide) qui active les
groupement COOH pour les faire réagir avec les amines.
Lorsque le couplage est effectué on peut procéder
àl'immunisation afin d'obtenir les anticorps en question.
La vérification du titre d'anticorps peut s'effectuer par
RIA (essai radioimmunologique) ou par ELISA (essai d'enzyme lié par
immunosorbtion).
L'étape finale qui est la vérification de la spécificité de
ces anticorps pour chacun des peptides ou pour 1 'enzyme, se fera par
buvardage des peptides synthétiques ("dot blot"), par immunofluorescence
et par microscopie électronique.
On pourra aussi vérifier s'il y a présence de 1 'activité
protéolytique associée aux matrices nucléaires des cellules infectées.
On tentera aussi de vérifier s'il y a une différence de
reconnaissance des anticorps pour l'enzyme de type sauvage et celui du
mutant tsl.
Son emplacement dans les cellules pourra être vérifié par
immunofluorescence et par microscopie électronique.
Tout ceci permettra donc de pouvoir poursuivre l'étude de
cette protéinase qui joue un rôle important voire même essentiel dans la
maturation des virions.
MATERIEL ET METHODES
Cellules
Des cellules de type Hep-2 (Morre et al., 1955) sont cultivées dans du milieu de Eagle modifié par Dulbecco (DMEM) (Dulbeco et Freeman; 1959) contenant 10% de sérum de veau, 1% de fungizone, pénicilline (100 U/ml) et streptomycine (100 µg/ml).
Virus et infection
Un pétri de 9 cm contenant une couche monocellulaire de 6
Hep-2
à
90-95% de confluence (1-2 X 10 cellules) est infecté avec un inoculum viral de 0.2 ml pendant1-1~
heureà
39°C avec agitation toutes les 15 minutes. En général, on utilise une multiplicité d'infection (m.i.) de 10 pfu/cellule (plaque forming unit) (Weber, 1976).Purification du virus
Les cellules infectées sont récoltées avec leur milieu et centrifugées (2,000 RPM, 5 min.). Après centrifugation, on resuspend les cellules dans un faible volume de milieu et on extrait les particules virales par 6 congélations - décongélations successives dans un mélange de glace carbonique et éthanol. Une autre centrifugation (2,000 RPM, 5 min.) permet d'éliminer les débris cellulaires. Le surnageant contenant les particules virales est déposé sur gradients de CsCl (chlorure de
césium) préformés (1.2-1.5 g/ml) et ultracentrifugé à 35 K pour 1~ heure
à
4°C (Weber, 1976; Khittoo et Weber, 1977). La bande viraleà
1.345g/ml est ~écoltée par aspiration, remise sur un gradient de CsCl
autoformé (1.4 g/ml) et ultracentrifugée (27 K, -18 heures, 4°C). La bande virale isolée est dialysée contre un tampon 10 rnM Tris-HCl pH 7.5 et 1 rnM EDTA; afin d'éliminer le CsCl. Le virus purifié peut être congelé (-2o0c) pour utilisations ultérieures.
Marquage des protéines virales
A 20 heures post-infection (p. i.) à 39°C; on effectue un marquage des protéines avec 1 ml de milieu sans méthionine contenant 50 C · d µ i e met ion1ne-• h · ·
s
3 5 pour une per10 e e -• · d d 3 4 h eures avec ag1tat1ons · · régulières (Weber et al., 1977; Khittoo et Weber, 1977). On ajoute ensuite 7 ml de milieu complet (pour un pétri de 9 cm) sans enlever la• h' ·
s
35 d' ·' • · 1 ' 1 f' d 1 1 ·<
48met ion1ne- eJa presente et ce, JUsqu a a in u cyc e yt1que
-heures) pour un marquage continu.
Isolation des noyaux et purification des matrices nucléaires
Les cellules infectées et non-infectées sont récoltées et centrifugées
à
500 g pour 10 minutes. Le culot cellulaire est d'abord lavé dans du PBS (tampon_ phosphate salin), ensuite dans un tampon RBS (0.01 M Tris-HCl pH 7.7, 0.01 M KCl, 0.015 M MgC1 2 , 0.4 rnM CaC12 et 10-3 M DTT) (dithiotreithol) et resuspendu dans 25 volumes du tampon TECK (10 rnM Tris-HCl pH 7.8, 1 rnM EDTA, 3 rnM CaC1 2 et 10 rnM KCl). Après une incubation de 10 minutes sur glace, les cellules sont homogénéiséespendant 30 secondes avec un homogénisateur ultra-turrax (Janke et Kunfel, Stafen, West Germany). Les résidus cytoplasmiques sont éliminés par l'addition de 0.5/o NP-40 suivie d'une agitation de 2 minutes. Les noyaux sont centrifugés à 400 g pendant 5 minutes et lavés plusieurs fois avec le tampon RBS, pour enlever tous les débris cytoplasmiques. A ce stade, les noyaux étaient propres lorsqu'examinés par microscopie en contraste de phase. Les matrices nucléaires sont purifiées comme décrit précédemment (Biber-Hardy et al., 1982) en utilisant la procédure de Pardoll et al. (1980). Les noyaux des cellules normales ou infectées sont resuspendus dans 10 volumes de 10 mM Tris-HCl pH 7 .8, 2 mM MgC1 2
(tampon à faible concentration de sel) et centrifugés à 400 g pendant 10
minutes. Après une deuxième extraction dans le tampon
à
faibleconcentration saline, le culot est resuspendu dans la moitié du volume original de tampon contenant 50 µg/ml de DNAse I (Worthington) et incubé
à la température de la pièce pendant 30 min. Ils sont alors centrifugés
comme ci-haut, resuspendus dans la moitié du tampon à faible
concentration saline, amenés à une concentration finale de 2 M (tampon à
haute concentration saline) par l'addition lente d'une solution 4 M NaCl et centrifugés pendant 20 min.
à
300 g. Après une deuxième extraction dans le tampon à haute concentration saline, les matrices résultantes sont lavées dans le tamponà
faible concentration saline et alors prêtesà être analysées. Toutes ces manipulations sont effectuées sur la glace.
Réaction de clivage de la protéinase virale
Le substrat utilisé pour détecter la présence de la protéinase virale provient de virions tsl marqués à la méthionine-s35 brisés avec 10% pyridine pendant 30 min.
à
la température ambiante, etdyalisés comme décrit plus haut. Des quantités égales (normalement 10 µl) du substrat et de l'enzyme (provenant des cellules, noyaux ou matrices) sont aliquotées dans des tubes Eppendorf. Par contre, nous avons utilisé moins de cellules (1/5 de la quantité utilisée pour préparer les échantillons de noyaux et de matrices)
à
cause du contenu trop élevé en protéines; ce qui rendait difficile le fait d'avoir la même radioactivité sur les gels SDS-polyacrylamide, sans avoir beaucoup de distorsion. Les mélanges réactionnels sont complétés à 50 mM Tris-HCl pH 7. 0, 1 mM EDTA, 5 mM DTT, 25 mM Na Cl, soniqués pendant 5 min. et incubés pour 2 heuresà
37°C. La réaction est arrêtée par l'addition de la solution lysante pour SDS-Page et les échantillons sont bouillis pendant 3 min. La présence de la protéinase virale est suivie par le clivage de PVII en VII sur des gels SDS-polyacrylamide (Bhatti et Weber, 1978).Réaction de clivage de la protéinase nucléaire
Des cellules Hep-2 infectées par wt sont marquées
à
la, h. . s35
met ionine- et les noyaux en sont isolés comme décrit par Tremblay et al. (1983). Ceux-ci sont incubés sur la glace ou
à
37°C pendant 2 heures. Les échantillons sont analysés par gel SDS-polyacrylamide.Electrophorèse
La polymérisation du gel s'effectue entre deux plaques de verre séparées et fermées par des barres d'espacement situées de chaque
d'acrylamide-bis 30-0.24, 3.75 ml 3 M Tris-HCl pH 8.9, 0.3 ml SDS 10% (sodium dodécyl sulphate), 0.1 ml TEMED (N, N, N; N-tétraméthy-léthylène-amine), 13.29 ml H20 distillée et 0.15 ml d'ammonium persulfate 10%. La polymérisation est produite par réaction radicalaire de l'ammonium persulfate et du TEMED et requiert un minimum de 12 heures. La seconde étape est la formation d'un gel concentrateur avec 1.67 ml acrylamide-bis 30-0.8, 1.25 ml 3 M Tris-HCl pH 8.9, 0.1 ml SDS 10%, 5 µl de TEMED, 6.9 ml H2
o
distillée et 0.1 ml d'ammonium persulfate 10%. Sa polymérisation requiert une période minimale d'environ 30 minutes. C'est dans le gel concentrateur que l'on introduit le peigne afin d'y former les puits où on dépose les échantillons (Maizel, 1969; Weber et al., 1977). Les gels utilisés sont de 0.75 cm d'épaisseur.Les échantillons sont traités avec un tampon de lyse contenant 0.05 M Tris-HCl pH 6.8, 1% SDS, 1% mercaptoéthanol, 10% glycérol et du rouge de phénol, ensuite bouillis pendant 3 minutes et mis sur gel. La migration du gel est effectuée dans un tampon d'électrophorèse de 5 mM Tris, 37.5 mM glycine et 0.10% SDS (sodium dodécyl sulfate) en concentration finale. La migration se poursuit à 60
mM pendant environ 2 heures; c'est-à-dire jusqu'à la sortie du rouge de phénol, incorporé dans les échantillons, qui indique le front de migration.
'
Lorsque l'électrophorèse est terminée, le gel est soit coloré au bleu de comassie ou séché sous vide et exposé pour la nuit avec un film XRP-1 pour autoradiographie.
Synthèse et purification des peptides
La synthèse des peptides est effectuée selon la méthode de Merrifie ld (cf. fig. 6) (Merrifield, 1963; Margl in et Merrifie ld, 1970; Houghten et al., 1980; Stutcliffe et al., 1980). Ici l'acide aminé de l'extrémité carboxyle du peptide est attaché à la résine par sa fonction acide. La résine est un copolymère de styrène et divinylbenzène portant habituellement des groupements chlorométhyl sur lesquels on fait réagir le premier acide aminé. Un groupement protecteur, appelé Boc, sert
à
bloquer l'amine des acides aminés afin que seul le carboxyle de l'acide aminé à attacher soit libre pour la réaction de couplage. Le Boc peut facilement être -enlevé lorsque désiré; c'est cette étape que l'on appelle déprotection. Avec le TFA (acide trifluoroacétique), on obtient d'abord un fluorure que l'on neutralise par la suite avec une solution de DEA (diéthylamine) pour obtenir l'amine libre prête à réagir avec le prochain acide aminé.La réaction de couplage se réalise en ajoutant un anhydride symétrique de l'acide aminé voulu. On fait cette étape car lors d'un couplage avec un acide aminé simple, on observe la formation d'urée alors qu'en faisant l'anhydride symétrique on élimine le précipité qu'est l'urée lequel pourrait bloquer le filtre des vaisseaux de synthèse. On vérifie l'efficacité de couplage par un test de Kayser (à
la ninhydrine), si ce dernier se révèle être très positif on refait le couplage; sinon on peut acétyler les quelques sites qui n'ont pas réagi, afin d'éviter l'obtention de peptides de différentes longueurs.
Figure 6: Technique de synthèse pept idique utilisée selon Merrifie ld, 1963.
J.
Amer. Chem. Soc. Vol 85.1 . 1
CH3
H
Boc- aminoacyl polymère
dioxane -TFA
DEA
Déprotection
Neutralisation
aminoacyl polymère
anhydride du
Boc - acide aminé
Couplage
CH3
R2 0
R1
0
1
·
1Il
1 11H3C- C-,O-C-N-C-C-N-C-C-0-CH
1 1 1 1 1 .
2
CH3
H H
H H
Boc -peptide polymère
HF
Clivage
R2 0
R1
1 Il 1 H~-c-c-N-C-COOH+
. 1 1 1H
H H
.peptide
polymère
polymère
polymère
Finalement on procède au clivage au HF (acide fluoridrique) afin de se débarrasser de la résine et des groupements protecteurs des chaînes secondaires.
Pour la purification des peptides, on utilise des colonnes échangeuses d'ions avec un gradient de concentration et de pH (Bittle et al., 1982; Pharmacia, 1980). Les résines utilisées sont: la CM-23 qui est échangeuse de cations et qui est donc utilisée pour les peptides chargés positivement et la DEAE-A-25 pour ceux chargés négativement. La colonne RP-8 (chaîne aliphatique de 8 carbones) a aussi été utilisée pour essayer d'augmenter la pureté du peptide chargé négativement.
Après passage sur colonne, les peptides ont été lyophilisés et congelés.
Formation des anticorps
Les anticorps sont obtenus en immunisant de jeunes lapins avec les peptides synthétiques couplés à une protéine porteuse (BSA ou KLH) (cf. fig. 7) (Dockray, 1980; Green et al., 1982; Bittle, 1982; Johnson-Winegar, 1984). L'agent de couplage utilisé est le EDC. Dans la
majorité des cas, KLH a été utilisée à cause de son pouvoir
immunogénique supérieur à BSA puisqu'elle est très étrangère pour les lapins. L'inoculum est constitué d'adjuvant de Freund's et du mélange peptide-KLH (ou BSA) dans un rapport 1:1. La réaction utilisée pour faire l'attachement du peptide à la protéine porteuse est:
(1 mg peptide + 3 mg KLH (ou BSA) + 3 mg EDC)/0.75 ml H2
o
Jour 1
So
prélèvement pré- immunitaire
1
1injection intradermique (réaction
Jour 29
1 2Jour 43
1
3
Jour 47
Jour 57
1
4
Jour 71
1
s
Jour 75
S
2
de couplage et adjuvant complet)
idem
11
(sauf adj. complet rem placé par
de l'incomplet )
idem
12
prélèvement post- immunitaire
Figure 7: Patron d'injection de jeunes lapins de Nouvelle-Zélande. Les lapins sont injectés (I) avec un mélange de peptide lié à K.LH (ou BSA) par EDC et d'adjuvant de Freund's. Les saignements (S) réguliers sont faits par l'oreille tandis que le saignement final est fait par cannulation de la carotide.
On laisse réagir 6 heures à la température ambiante (Ta) et on rajoute 3 mg EDC; car celui-ci s'hydrolyse très rapidement; puis on continue la réaction pendant 18 heures.
L'efficacité de couplage est vérifiée en utilisant des peptides marqués à l'iode (1125 ) et en passant de mélange de
coupl~ge
sur une colonne de G-50. Donc en comptant la radioactivité qui sort dans les premières fractions on peut déterminer l'efficacité de couplage (Desbuquois et Aurbach, 1971; Marks et al., 1974; Showsky et al., 1974; Lutterodt et al., 1976; Dockray, 1980).
La production d'anticorps a été vérifiée par RIA, cette méthode servant
à
déterminer le titre d'anticorps d'un sérum. Pour un échantillon donné on fait réagir 100 µl de tampon RIA (0.15 M NaCl, 2 mMazide de sodium, 0.1% BSA dans un tampon phosphate), 100 µl de traceur (ici, 10,000 CPM de peptide iodé) et 100 µl du sérum dilué. On laisse à
4°C pour la nuit. L'arrêt de la réaction est effectué avec 100 µ 1 de y-globuline 0.5% et 500 µl de PEG 30% (polyéthylène glycol 6,000-8,000). On centrifuge puis aspire le surnageant et compte le culot. Le titre de l'anticorps est établi par la dil~tion de sérum o~ l'on retrouve 50% de liaison avec le peptide traceur.
Immunof luorescence
Les cellules sont cultivées sur des lamelles rondes. Elles sont ensuite infectées avec 50 pfu de Ad-2 et fixées dans le méthanol
à
lavées avec du PBS sans et contenant 1 fo BSA à
température de la pièce. Elles sont traitées pendant 4 min. avec un
mélange de PBS -triton 0.4% (pour augmenter l'accès aux sites
antigéniques) et ensuite lavées dans le tampon PBS-BSA. Les lamelles sont déposées dans une chambre humide (un pétri contenant un carré de papier Whatman inhibé de PBS ) et incubées pendant 1-2 heures à Ta avec une goutte du sérum
à
tester dilué dans PBS-1% BSA sur la lamelle. Leslamelles sont ensuite lavées au PBS (30 secondes par lavage sans
agitation). Le deuxième anticorps est ensuite ajouté (ici anticorps anti-lapin lié
à
la biotine dans PBS-1% BSA) et laissé pour 1 heureà
Ta. Les lamelles sont ensuite lavées comme précédemment, colorées avec fluorescéine-streptavidine dans PBS-1 % BSA pendant une demi-heure (à l'obscurité) et relavées (Arnersham, 1984). Si on désire conserver la fluorescence plus longtemps on traite les lamelles 10 minutes
à
-2o0c
dans une solution acide-alcool (95% éthanol absolu -5% acide acétique). Après avoir été séchées, les lamelles sont montées sur lame en plaçant la face portant les cellules sur une goutte d'une solution 100 rnMTris-HCl pH 8.9 (pH optimium pour révéler la fluorescence) contenant 90%
de glycérol. Finalement les cellules sont observées au microscope à
fluorescence et photographiées sur du film 400 ASA noir et blanc pendant 1 minute d'exposition.
Etudes par microscopie électronique
Des cellules Hep-2 normales ou infectées avec un inoculum
0
viral de wt 40 pfu sont fixées dans 1% glutaraldéhyde (2 heures, 4 C) à
tester (1/10) contenant 1 mM PMSF est laissée au contact des cellules
pendant 1 heure à la température de la pièce. Ces dernières sont
relavées au PBS. La réaction est révélée par un complexe protéine A-palladium/or pendant 15 min. Les cellules sont finalement relavées au
PBS et
à
l'eau. Les préparations sont observées au microscopeRESULTATS
Comme la protéinase virale n'avait jamais été visualisée en tant que protéine et que ceci serait une preuve encore plus tangible de
son existence, nous avons décidé de nous attaquer à apporter cette
preuve. La méthode choisie a été de faire des anticorps contre l'enzyme.
Sa séquence en acides aminés étant connue (à partir de la séquence nucléotidique); nous avons décidé de faire des peptides synthétiques choisis à partir de cette séquence. C'est avec ces peptides que nous obtiendrons des anticorps reconnaissant la protéinase.
La preuve de cette reconnaissance sera démontrée par immunofluorescence et microscopie électronique.
Choix des séquences d'acides aminés
On veut déterminer quelles séquences en acides aminés seront les plus susceptibles de donner des anticorps qui réagiront aussi contre la protéinase.
La séquence des peptides est choisie en tenant compte de l'idée première qui est d'obtenir une bonne réponse immunitaire. Tout
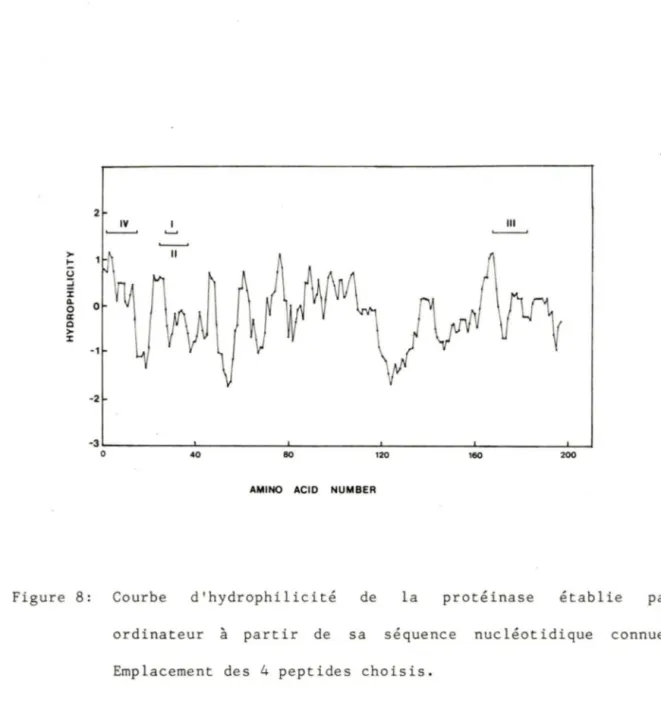
d'abord, la séquence de la protéinase est analysée pour son
hydrophilicité (cf. fig. 8). On peut remarquer que plusieurs régions ont une hydrophilicité suffisante pour être choisies. C'est alors que l'on
2 IV 1 Ill '-' > Il ~ () ~ J: a. 0 0 a: Q > J: - 1 -2 -3 0 40 80 120 160 200
AMINO ACID NUMBER
Figure 8: Courbe d'hydrophilicité de la protéinase établie par ordi nateur
à
partir de sa séquence nucléot idique connue. Emplacement des 4 peptides choisis .peut regarder plus attentivement les séquences en acides aminés. Comme mentionné précédemment, on doit tenir compte de trois principaux
facteurs dans la sélection. En premier lieu, plus les acides aminés des peptides sont hydrophiles (His, Lys, Arg, Asp, Gly), plus grandes sont les chances que cette séquence se trouve à l'extérieur de la protéine et par conséquent les anticorps peuvent y accéder plus facilement. En deuxième lieu, la présence de praline joue aussi un rôle important. Comme elle possède une structure particulière (cf. fig. 5), elle donne donc une conformation plus fixe à la protéine. Dans la séquence du peptide, plus le nombre de pralines est élevé, plus grandes sont les chances que sa structure soit semblable à celle de la protéine et qu 'ainsi les anticorps formés reconnaissent bien les deux. En dernier lieu, les tyrosines jouent aussi un rôle; mais plutôt au point de vue pratique. En effet si une tyrosine est présente dans chacun des peptides; il est donc facile de marquer ceuK-ci à l'iode-125 (I 125 ) par la chloramine T. Mais s'il n'y en a pas, il faut utiliser la méthode de Bolton-Hunter (Bolton et Hunter, 1972,1973). Par cette technique, on attache un ester cyclique sur les amines libres et sur celui-ci on fixe l'I1250
Par ces critères, quatre séquences d'acides aminés sont choisies (cf. fig. 8 et 9). A 26 acides aminés de l'extrémité N-terminale, il y a le peptide I. Celui-ci contient six acides aminés, ce qui est la longueur minimale établie pour avoir une réponse immunologique (Sut cl iffe et a 1., 1983), dont 2 sont hydrophiles (Lys, Arg). Il contient aussi une praline mais pas de tyrosine. Il devra donc être marqué par la méthode de Bolton-Hunter.
1
1 Splice(14S mRNA) 1
GATCCCATGGAC GAGCCCACCCTT CTTTATGTTTTG
AspProMetAsp GluProîhrleu LeuîyrValLeu TTTGAAGTCTTT GACGTGGTCCGT GTGCACCAGCCG PheGluValPhe AspValValArg ValHisGlnPro CACCGCGGCGTC
HisArgGlyVal
i~o
STOPHexonATCGAGACCGTG TACCTGCGCACG CCCTTCTCGGCC GGCAACGCCACA
ACA~GAAG
IleGluîhrVal TyrLeuArgîhr ProPheSerAla GlyAsnAlaîhr Thr3JO Bgl II
1 1
CAAGCAACATCA ACAACAGCTGCC GC~TGGGCTCC AGTGAGCAGGAA CTGAAAGCCATT GTCA.AAGATC
1,MetGlySer SerGluGlnGlu LeulysAlaile ValLysAspleu
IV
GGTTGTGGGCCA TATTTTTTGGGC ACCTATGACfAG CGCTTTCCAGGC !1°\GTTTCTCCA CACAA~CTCGCC
GlyCysGlyPro TyrPheleuGly ThrfyrASJt-YS ArgPheProGly Ph,.,alSerPro Hisly.J-euAla
1
Il
300
1
TGCGCCATAGTC AATACGGCCGGT CGCGAGACTGGG GGCGTACACTGG ATGGCCTTTGCC TGGAACCCGCGC CysAlalleVal AsnîhrAlaGly ArgGluThrGly GlyValHisîrp MetAlaPheAla TrpAsnProArg
400 1
TCAAAAACATGC TACCTCTTTGAG CCCTTTGGCTTT TCTGACCAACGA CTCAAGCAGGTT TACCAGTTTGAG SerlysThrCys TyrleuPheGlu ProPheGlyPhe SerAspGlnArg LeulysGlnVal TyrGlnPheGlu
500 1 TACGAGTCACTC CTGCGCCGTAGC GCCATTGCTTCT TCCCCCGACCGC TGTATAACGCTG GAAAAGTCCACC TyrGluSerleu LeuArgArgSer AlaileAlaSer SerProAspArg Cyslleîhrleu GluLysSerThr CAAAGCGTGCAG GGGCCCAACTCG GCCGCCTGTGGA CTATTCTGCTGC ATGTTTCTCCAC GCCTTTGCCAAC GlnSerValGln GlyProAsnSer AlaAlaCysGly LeuPheCysCys MetPheleuHis AlaPheAlaAsn
600 ~pnl
1
.--1-.
TGGCCCCAAACT CCCATGGATCAC AACCCCACCATG ~ACCTTATTACC GGGGTACCCAAC TCCATGCTTAAC
TrpProGlnîhr ProMetAspHis AsnProîhrMet Asnleulleîhr GlyValProAsn SerMetleuAsn 700
1 '
AGTCCCCAGGTA CAGCCCACCCTG CGTCGCAACCAG GAACAGCTCTAC AGCTTCCTGGAG CGCCACTCGCCC SerProGlnVal GlnProThrleufrgArgAsnGln GluGlnleuTyr SerPheleuGlu ArgHisSerPr~
Ill
TACTTCCGCAGC CACAGTGCGCAG ATTAGGAGCGCC ACTTCTTTTTGT CACTTGAAAAAC ATGTAAAAATAA TyrPheArgSer HisSerAlaGln lleArgSerAla ThrSerPheCys HisLeulysAsn Met
Figure 9: Séquences et emplacements des 4 peptides choisis
à
partir de la séquence d'ADN du cadre de lecture ouvert de 609 nucléotides découvert par Akusjarvi et al., 1981. Nucl. Acids Res. Vol. 9, no. 1.Le peptide II est un allongement du premier; c'est une sécurité car si jamais le peptide I n'est pas immunogénïque, le second devrait l'être. Dans le peptide II, il y a 13 acides aminés donc 5 sont hydrophiles (Arg, Asp, His et 2 Lys). Il contient aussi 2 pralines et 1 tyrosine. Théoriquement, c'est ce peptide qui devrait donner de meilleurs résultats.
Le peptide III, situé
à
21 acides aminés de l'extrémité carboxyle, possède 16 acides aminés dont 6 hydrophiles (3 Arg, His, 2 Glu). Dans sa séquence, on retrouve 1 proline et 1 tyrosine; ce qui devrait donner d'assez bons résultats.Et
finalement, le peptideIV,
qui est l'extrémité N-terminale de la protéinase, contient 15 acides aminés dont 5 sont hydrophiles (Asp, 2 Glu, 2 Lys). Contrairement aux autres peptides, il ne possède ni proline, ni tyrosine. Ceci est dû au fait que pour avoir ces deux acides aminés, une cystéine aurait due être insérée et comme cette dernière est très difficile à coupler lors de la synthèse, il résulte donc une baisse de rendement très importante.Synthèse et purification des peptides synthétiques
Lorsque la séquence des peptides est établie, la prochaine étape est de les fabriquer.
La méthode utilisée pour la synthèse des peptides est celle décrite par Merrifield (voir matériel et méthodes) (cf. fig. 6). L'acide aminé carboxyle est tout d'abord attaché
à
une résine, ce qui permet depouvoir laver et filtrer le peptide sans problème. Les autres acides
aminés sont ajoutés 1 'un après l'autre sous forme d'anhydride
symétrique, afin d'éli~iner l'urée. La synthèse est effectuée au moyen d'un synthétiseur automatique. Lorsque la séquence est terminée, le peptide est traité au HF, afin de le débarrasser de la résine et des groupements protecteurs.
Les peptides sont ensuite purifiés par chromatographie échangeuse d'ions. Ceux chargés positivement soit: I(+2), II(+3) et
III(+2) sont passés sur CM-23 dans l'acétate d'ammonium. Après
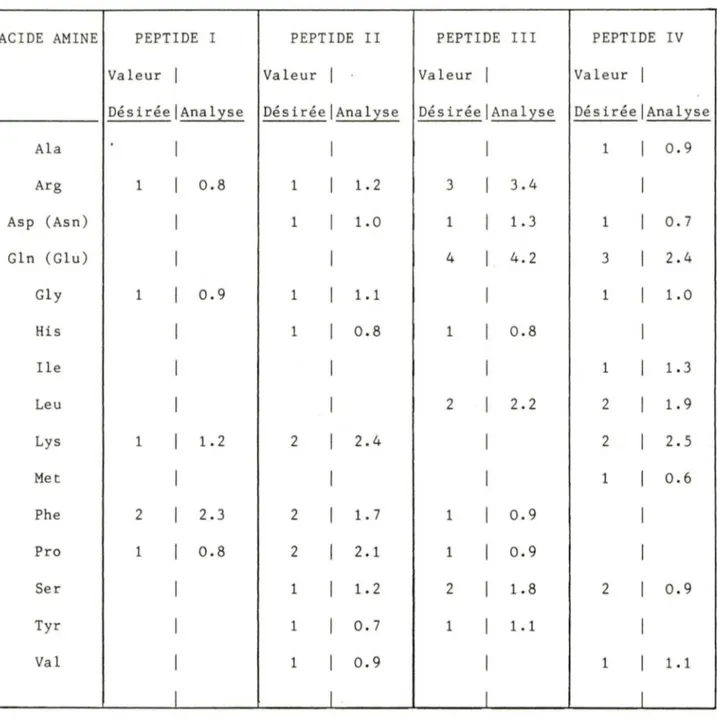
purification et lyophilisation, les rendements obtenus sont: peptide II: 44.7%, III: 39%. Le peptide I a été synthétisé dans le laboratoire du Dr Serge St-Pierre et son rendement est inconnu. La pureté des peptides est vérifiée par CCM (chromatographie en couche mince) et analyse d'acides aminés (cf. fig. 10 a, b, c). Les trois peptides ont donné des résultats, indiquant qu'ils étaient purs. En effet par CCM il n'y a qu'une seule tache, celle du peptide. Par l'analyse des acides aminés, on voit que tous les rapports sont exacts et on arrive aux nombres établis et aux bonnes proportions d'acides aminés (cf. table 2).
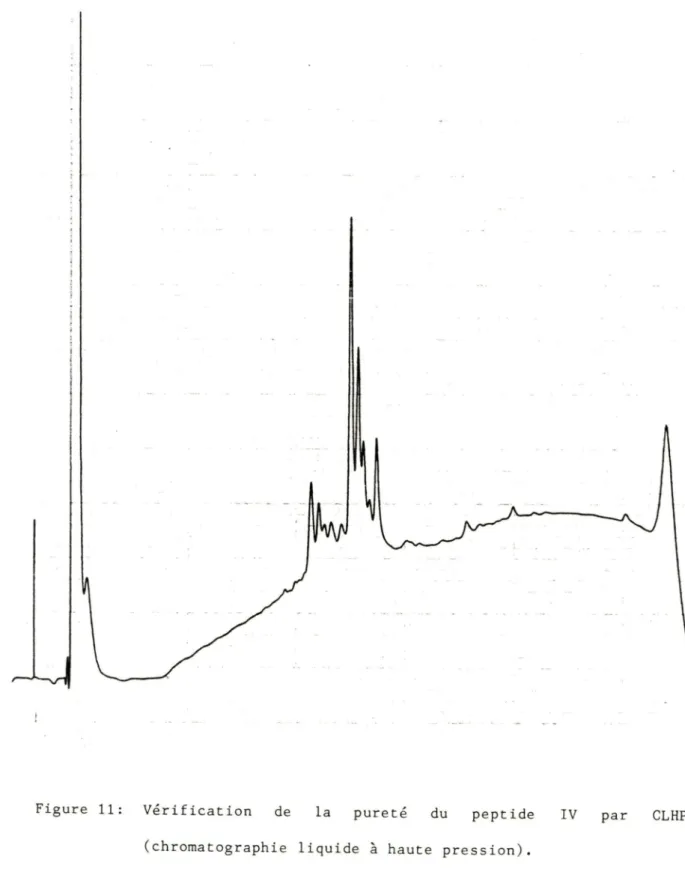
Le peptide IV, contrairement aux trois autres, est chargé négativement (-1). Donc pour sa purification, il est passé sur une colonne contenant de la résine DEAE-A25 dans l'acétate d'ammonium. Même après la chromatographie on voit par CCM, CLHP (chromatographie liquide
à
haute pression) (cf. fig. 11) et analyse d'acides aminés (cf. fig. 10 d); qu'il n'est pas encore pur. En effet sur l'enregistrement du CLHP, on peut voir qu'il y a beaucoup d'impuretés présentes. Par l'analyse des--Figure 10: Vérification de la pureté des 4 peptides synthétiques par analyse des acides aminés. (a) Peptide I (6 acides aminés). (b) Peptide II (13 a.a.). (C) Peptide III (16 a.a.). (d) Peptide IV (15 a.a.).
1 --
,
-' ·--~
COMPOSITION DE CHACUN DES PEPTIDES PAR ANALYSE DES ACIDES AMINES
ACIDE AMINE
PEPTIDE I
PEPTIDE II
PEPTIDE III
PEPTIDE IV
Valeur
1Valeur
1Valeur
1Valeur
1DésiréelAnalyse Dés iréelAnalyse Désirée !Analyse DésiréelAnalyse
Ala
1 1 11
10.9
Arg
1
10 .8
1
11. 2
3
13.4
1Asp (Asn)
11
11.0
1
11.3
1
10.7
Gln (Glu)
1 14
14.2
3
12 . 4
Gly
1
10.9
1
11.1
11
11.0
His
11
10.8
1
10.8
1Ile
1 1 11
11.3
Leu
1 12
12.2
2
11. 9
Lys
1
11. 2
2
12.4
12
12.5
Met
1 1 11
10.6
Phe
2
12.3
2
11. 7
1
10.9
1Pro
1
10.8
2
12.1
1
10.9
1Ser
11
11. 2
2
11.8
2
10 . 9
Tyr
11
1o.
7
1
11.1
1Val
11
10.9
11
11.1
1 1 . 1 1Tableau 2: Composition de chacun des peptides par analyse des acides aminés.
La comparaison entre les valeurs désirées lors de la synthè se et
les données obtenues par l'analyse des acides aminés est établie.
Figure 11: Vérification de la pureté du peptide
IV
parCLHP
(chromatographie liquide à haute pression).acides aminés on remarque que selon les rapports, il manquerait 1 sérine car la valeur obtenue par rapport aux standards est 28 et elle aurait due être le double. La méthionine est très étrange car on retrouve une valeur de 4, ce qui est environ 5-6 fois moins que ce qu'elle aurait due être. Donc tout laisse croire qu'il y a des peptides de différentes longueurs. Si on considère la méthionine comme étant 1, on aurait: 7 Asp (ou Asn), 7 Ser; 11 Glu (ou Gln), 4 Gly, 7 Ala, 4 Val, 6 Ile, 12 Leu et 12 Lys. Il semble que la valine ait mal été couplée ainsi que les 3 acides aminés en N-terminale, soit Met-Gly-Ser. Si tel est le cas il y aura des peptides où il peut manquer: une valine; valine et sérine; valine, sérine et glycine; sérine et glycine; etc. Donc tous les rapports seront faussés.
Une partie du peptide IV est passée sur colonne RP-8 qui est une chaine aliphatique et qui retarde plus ou moins les peptides tout dépendamment de leur longueur. Mais aucun peptide n'est sorti de la colonne, tout est resté accroché, impossible de le récupérer. Ceci laisse donc beaucoup le rendement et comme il est impur, il est par conséquent impossible de l'établir correctement.
Formation des anticorps
Comme les peptides sont synthétisés et purifiés, il s'agit maintenant d'obtenir des anticorps contre ceux-ci.
Les peptides synthétiques sont couplés à une protéine
porteuse par l'intermédiaire du EDC, comme décrit dans matériel et méthodes. Le peptide I est couplé
à
BSA; tandis que II, III et IV sontcouplés
à KLH qui est plus immunogénique que BSA; donc préférable. En
effet, KLH est une protéine provenant d'un crustacé tandis que BSA est
d'origine bovine, donc elle est beaucoup plus étrangère pour les lapins.
L'efficacité de couplage est vérifiée avec un peptide marqué
à I 125
couplé
à KLH (cf. table 3). Un couplage à 92% est obtenu.
De jeunes lapins de Nouvelle-Zélande sont tout d'abord
saignés pour un prélèvement pré-immunitaire (PI) et ensuite injectés
avec un mélange de peptide-BSA (ou KLH) et d'adjuvant de Freund complet,
pour la première injection et d'incomplet par la suite, dans un rapport
1: 1 de façon intradermique sur le dos. Pour le peptide I un lapin est
injecté, le #45; pour le peptide II, il y en a 3; #46, #47 et #48; pour
le III, il
yen a 4: #50, #51, #52 et #53; et pour le peptide IV, quatre
aussi: #54, #55, #56 et #57. Les lapins sont saignés régulièrement afin
de suivre la formation des anticorps par RIA avec leurs peptides
respectifs marqués
à I 125 . Après quelques injections (3-5) tous les
lapins, sauf ceux injectés avec le peptide IV, donnent une réponse
positive. Les lapins 54, 55, 56 et 57 n'ont donc pas été utilisés.
Lorsque les titres d'anticorps (titre: dilution
àlaquelle on observe
50% de liaison du peptide traceur par RIA; donc plus la dilution est
grande, plus le sérum est bon) sont stables, i.e. qu'ils n'augmentent
plus, ou qu'ils commencent
à diminuer, les lapins sont saignés "à blanc"
par cannulation de la carotide.
Les sérums isolés de ces saignements, appelés ici Sf (sérum
final), sont ceux qui serviront pour les expériences suivantes. Lors
d'un saignement, un volume de sérum d'environ 70 ml est isolé. Les
VERIFICATION DE L'EFFICACITE DE COUPLAGE
!Compte avantl
Compte
Compte
lchromatogra- ICompte restantlpassé sur
sortant
IEchant i llonl~~p~h_1_·e~~
dans le tube
G-50
au début 1% liaison!
Sans 2e
addition
24, 724 CPM
1,608 CPM
123,116 CPMl19,904 CPM I
81
%de EDC
Avec 2e
addition
18,650 CPM
1,944 CPM
116,706 CPMl15,438 CPMI
92%
de EDC
Tableau 3: Vérification de l'efficacité de couplage. Le peptide marqué
à
125
l' '
d
'l
,
1
I · est coup e a KLH au moyen u EDC. Le me ange reactionne
est passé sur colonne G-50 (dans 10% acide acétique) . Le
peptide libre reste pris sur la résine tandis que le peptide
lié
à KLH sort immédiatement. L'efficacité de couplage a été
déterminée pour 1 ou 2 additions de EDC. Les résultats pour
les 3 peptides sont similaires .
titres d'anticorps obtenus par RIA varient de 1:100 à 1:3000 et sont les suivants: #45-1:100; 46-1:600; 47-1:140; 48-1:100; 50-1:100; 51-1:2700; 52-1:3000; 53-1:1600.
Spécificité des anticorps
Afin de s'assurer que chacun des sérums est spécifique
à
son peptide, tous les sérums immunitaires sont soumis à un buvardage ("dot blot"). Pour ce faire, 10 µg de chacun des peptides est mis sur un carré de nitrocellulose. Après le filtre est saturé avec une solution 1% BSA lavé et réagit avec chacun des sérums dilués (1/100). La réaction est. ' 'l' '· A I 125 L ' 1 b ( f f'
ensuite reve ee avec proteine - • es resu tats o tenus c • ig.
12) montrent clairement que tous les sérums réagissent de façon spécifique avec leur peptide. En effet le lapin 45 se lie au peptide I, les sérums 46, 47 et 48 avec le peptide II et 50, 51, 52 et 53 avec le peptide III.
MISE EN EVIDENCE DE LA PROTEINASE PAR METHODES IMMUNOLOGIQUES
Le but de cette recherche étant de démontrer l'existence concrète de la protéinase; différentes méthodes furent donc essayées. Deux d'entre elles ont bien fonctionné: l'immunofluorescence et la microscopie électronique.
A) Immunof luorescence
Avec cette méthode, le travail est effectué sur les cellules mêmes, donc l'enzyme ne devrait pas avoir été abimé et les anticorps