Rôle du récepteur 1 de la sphingosine-1-phosphate
dans les dysfonctions épithéliales observées dans un
modèle d’asthme
Mémoire
Michel Taillefer
Maîtrise en médecine expérimentale
Maître ès sciences (M.Sc.)
Québec, Canada
III
Résumé
L’asthme est en progression et 5 à 10 % des asthmatiques sont réfractaires aux traitements. L’administration d’agonistes spécifiques du récepteur 1 de la sphingosine-1-phosphate (S1PR1) dans le poumon inhibe l’inflammation pulmonaire allergique dans un modèle murin d’asthme. Cependant, les mécanismes et les cellules cibles sont inconnus. Puisque les altérations des cellules épithéliales (CE) bronchiques contribuent à la pathogenèse de l’asthme, l’activation de S1PR1 a été évaluée dans l’atténuation des dysfonctions que présentent les CE bronchiques d’un modèle d’asthme de rats et humaines. La surexpression de S1PR1 par les CE bronchiques de rats asthmatiques et humaines semble bénéfique dans la réduction des dysfonctions épithéliales puisque l’activation spécifique par CYM-5442 augmente l’étanchéité paracellulaire et réduit la libération d’une chimiokine, CCL2 (MCP-1), dans un contexte inflammatoire. Donc, il semble que la protéine S1PR1 soit impliquée dans le maintien de l’homéostasie pulmonaire. Cette voie métabolique pourrait être approfondie afin de contrôler l’asthme réfractaire.
V
Abstract
Asthma is in progression and 5 to 10 % of asthmatics are refractory to current interventions. In the lung, activation of sphingosine-1-phosphate receptor 1 (S1PR1) by specific agonists inhibits allergic airway inflammation in a murine model of asthma. However, cellular mechanisms and targeted cells are unknown. Since dysfunctions of bronchial epithelial cells (BEC) are central in asthma pathogenesis, activation of S1PR1 was evaluated in the reversal of BEC dysfunctions in a model of asthma and in human cells. Upregulation of S1PR1 in BEC of rats with experimental asthma and in human cells reversed epithelial cell dysfunctions. Indeed activation of S1PR1 by the specific agonist CYM-5442 decreases paracellular permeability and reduces the release of chemokine, under proinflammatory conditions. Therefore, S1PR1 seems to be involved in maintaining pulmonary homeostasis. This metabolic pathway could be of interest for controlling refractory asthma.
VII
Remerciements
Ce mémoire est un aboutissement que je dois aux nombreuses personnes qui m’ont aidé et accompagné pendant la réalisation de mes études. Tout d’abord, je tiens à remercier Dr David Marsolais, mon directeur de recherche, de m’avoir donné le privilège d’être le premier étudiant dans son laboratoire. Grâce à lui, j’ai pu étudier un sujet qui me passionne depuis mon tout jeune âge. Je tiens à lui témoigner toute ma reconnaissance pour sa patience, sa grande disponibilité, ses bons conseils et ses encouragements.
J’aimerais remercier Dre Anick Langlois pour son support indispensable. Excellente pédagogue, elle a fait l’impossible pour m’offrir l’aide technique, les conseils et l’écoute dont j’avais besoin tout au long de mon projet. Je tiens à remercier Dre Anne-Marie Lemay pour son assistance technique, sa rigueur et ses commentaires toujours très pertinents.
J’aimerais remercier les équipes du Dre Élyse Bissonnette, du Dre Jamila Chakir et du Dre Marie-Renée Blanchet pour leur expertise et leurs ressources techniques lors de l’exécution de ce projet.
Je voudrais remercier les étudiants qui m’ont accompagné pendant ma maîtrise. Jeff (à qui je dois plus d’une bière) pour son enseignement et sa précieuse aide avec les animaux. Je remercie également mes matelots : Sara-Mélissa, Émilie et David Gendron qui m’ont donné un gros coup de main quand j’étais débordé.
Depuis toujours, je bénéficie du support indéfectible de mes parents. Leurs efforts et encouragements m’ont permis de me dépasser et de me concentrer sur l’atteinte de mes rêves. « Merci à vous deux, du plus profond de mon cœur ». Finalement, je tiens à remercier ma copine qui m’a soutenu dans cette aventure. « Merci pour tout mon amour. Merci pour ta compréhension, ta patience et ton support. Sans tes nombreux sourires et ta joie de vivre, la vie ne serait pas aussi agréable. Je t’aime ».
IX
Table des matières
Résumé ... III Abstract ... V Remerciements ... VII Table des matières ... IX Liste des tableaux ... XI Liste des figures ... XI Liste des abréviations ... XII
Chapitre 1 : Introduction ... 1
Section I : L’asthme ... 2
1.1 L’asthme : Généralités et définition... 2
1.2 La pathogenèse de l’asthme ... 6
1.2.1 La cascade asthmatique-sensibilisation ... 6
1.2.2 La cascade asthmatique-exacerbation ... 7
1.3 Les cellules structurales pulmonaires impliquées dans l’asthme... 8
1.3.1 Implication des muscles lisses, de l’endothélium et des fibroblastes dans l’asthme ... 8
1.3.2 Rôle central de l’épithélium bronchique dans l’asthme ... 9
1.3.2.1 Les altérations structurales des CE bronchiques ... 10
1.3.2.2 La production des médiateurs solubles par les CE bronchiques est modulée dans l’asthme ... 11
Section II : Les sphingolipides ... 14
1.4 La biosynthèse de la sphingosine-1-phosphate ... 14
1.5 Effets biologiques de la modulation pharmacologique de la voie de signalisation de la S1P ... 16
1.5.1 Analogues de la sphingosine ... 16
1.5.2 Le récepteur S1PR1 ... 17
1.6 Modulation de la voie de la sphingosine-1-phosphate et l’asthme ... 19
Chapitre 2 : Hypothèse et objectifs ... 23
Chapitre 3 : Matériel et méthodes ... 25
3.1 Isolement et culture des CE bronchiques ... 26
3.2 Immunobuvardage de western ... 28
3.3 Immunofluorescence ... 29
3.4 Perméabilité paracellulaire ... 31
3.5 Dosage de CCL2 par ELISA ... 32
3.6 Analyses statistiques ... 33
Chapitre 4 : Résultats ... 35
4.1 La protéine S1PR1 est exprimée dans des CE bronchiques de rats ... 36
4.1.1 Expression de la protéine S1PR1 en immunobuvardage de western ... 36
4.1.2 Expression de la protéine S1PR1 en immunofluorescence ... 37
4.2 S1PR1 est exprimée dans des CE bronchiques humaines ... 39
4.3 L’activation de S1PR1 tend à diminuer la perméabilité des ABEC ... 40
4.5 L’activation de S1PR1 réduit la libération de CCL2 des CE bronchiques d’un
modèle d’asthme ... 44
4.5.1 Le TNF tend à augmenter la libération de CCL2 des ABEC ... 44
4.5.2 La libération de CCL2 induite par le TNF est réduite par l’activation de S1PR1 ... 46
4.6 L’activation de S1PR1 réduit la libération de CCL2 dans des CE humaines ... 48
4.7 CYM-5442 inhibe fortement la libération de CCL2 induit par le TNF pendant la croissance des ABEC ... 50
Chapitre 5: Discussion ... 53
Conclusion ... 58
Liste des tableaux
Tableau 1. Niveau de contrôle de l’asthme. ... 4
Tableau 2. Des analogues et modulateurs pharmacologiques de la voie de signalisation de la S1P mentionnés dans ce mémoire. ... 19
Liste des figures
Figure 1. Relation entre le contrôle et la sévérité de l’asthme. ... 5Figure 2. La pathogenèse de l’asthme ... 8
Figure 3. L’épithélium bronchique est central dans l’asthme. ... 13
Figure 4. Synthèse de la sphingosine-1-phosphate. ... 15
Figure 5. La protéine S1PR1 semble être surexprimée dans les ABEC. ... 37
Figure 6. Détection par immunofluorescence de S1PR1 dans les ABEC et les NBEC. ... 38
Figure 7. La protéine S1PR1 semble être surexprimée dans les CE bronchiques d’asthmatiques. ... 40
Figure 8. L’activation spécifique de S1PR1 module la perméabilité des ABEC. ... 41
Figure 9. CYM-5442 induit la formation d’anneaux corticaux d’actine dans les BEAS-2B. ... 43
Figure 10. La libération de CCL2 tend à augmenter en fonction de la dose de TNF dans les ABEC. ... 45
Figure 11. CYM-5442 réduit la libération de CCL2 par les ABEC. ... 47
Figure 12. CYM-5442 réduit significativement la libération de CCL2 induite par le TNF dans des lignées de CE humaines. ... 49
Figure 13. CYM-5442 inhibe la libération de CCL2 induite par le TNF lors de la croissance des ABEC. ... 51
Liste des abréviations
ABECapr. J-C :
Bronchial Epithelial Cells isolated from rats with experimental asthma Après Jésus-Christ
ATCC : American Type Culture Collection BAE : Bronchial asthmatic epithelial cells BEC Bronchial epithelial cells
BCA : Bicinchoninic acid
BN Brown Norway
BNE : Bronchial normal epithelial cells BSA : Albumine de sérum de bovin CCL : Chemokine (C-C motif) ligand CCR : Chemokine (C-C motif) receptor CD : Cluster of differentiation
CE : Cellules épithéliales
DAPI : 4',6'-diamidino-2-phénylindole DC : Cellules dendritiques
Derp 1 : Dermatophagoides pteronyssinus 1
DMEM/F12 : Dulbecco's Modified Eagle Medium: Nutrient Mixture F-12 DMSO : Diméthylsulfoxyde
DTT : Dithiothréitol
ECL : Enhanced Chemioluminescence EDG-1 : Endothelial differentiation gene 1 EDTA : Ethylenediaminetetraacetic acid EGF : Epidermal growth factor
ELISA : Enzyme Linkage Immunosorbent Assay FBS : Fetal bovine serum
FGF : Fibroblast growth factor FITC : Fluorescein isothiocyanate GINA : Global Initiative for Asthma HDL : Lipoprotéines de hautes densités
HEPES : 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid HRP : Horseradish peroxidase IgE : Immunoglobuline E IL : Interleukine i.n. : Intranasal i.p. : Intrapéritonéal i.t. : Intratrachéal JS : Jonctions serrées
MCP : Monocyte chimoattractant protein
NBEC : Normal bronchial epithelial cells NF-κB : Nuclear Factor-κB
NHBE : Normal Human Bronchial /Tracheal Epithelial Cells NIH National Institute of Health
OVA : Ovalbumine
PBS : Phosphate buffered saline
PC20 : Dose de méthacoline requis pour diminuer la FEV1 de 20 % PECAM-1 : Platelet endothelial cell adhesion molecule 1
RANTES : Regulated and normal T cell expressed and secreted RIPA : Radio Immuno Precipitation Assay buffer
S1P : Sphingosine-1-phosphate
S1PR : Récepteur de la sphingosine-1-phosphate SCF Stem-cell factor
SDS : Sodium Dodecyl Sulfate SEM : Standard error of the mean SPHK : Sphingosine kinase
TARC : Thymus and activation regulated chemokine TBST : Tris-buffered saline-tween
TEM : Transition épithélio-mésanchymateuse TGF : Transformation growth factor
Th : T helper
TNF : Tumor necrosis factor
TRITC : Tetramethylrhodamine B isothiocyanate TSLP : Thymic stromal lymphopoietin
VCAM-1 : Vascular cell adhesion protein 1 VEGF : Vascular endothelial growth factor
VEF1 : Volume expiratoire forcé durant la première seconde VIH : Virus de l'immunodéficience humaine
1
2
Préambule
L’asthme est une maladie inflammatoire chronique très répandue, dont le nombre de personnes atteintes ne cesse d’augmenter et pour laquelle aucun traitement curatif n’existe. L’administration dans le poumon d’un agoniste spécifique du récepteur 1 de la sphingosine-1-phosphate (S1PR1), CYM-5442, inhibe l’inflammation pulmonaire allergique dans un modèle d’asthme expérimental murin. Ce projet de recherche consistait à identifier les mécanismes et les cibles de cet agoniste.
Section I : L’asthme
1.1 L’asthme : Généralités et définition
GénéralitésL’asthme est une maladie inflammatoire chronique des voies aériennes dont la prévalence est en constante progression. La prévalence de cette maladie augmente de 50 % à chaque décennie [1], affectant maintenant près de 235 millions de personnes dans le monde [2]. Si la tendance se maintient, le nombre de cas d’asthme pourrait atteindre environ 335 millions avant 2025 [3]. L’asthme fait environ 180 000 victimes par année dans le monde [4]. Au Canada, 8,6 % de la population en souffre [5]. Les coûts associés à cette maladie sont très importants et dépassent d’ailleurs ceux reliés à la tuberculose et au VIH combinés [1], de sorte que l’asthme mobilise, à lui seul, 1 à 2 % du budget total pour les soins de santé des pays industrialisés [3]. L’asthme n’est pas seulement très répandu, il est également un syndrome complexe difficile à diagnostiquer.
Définition et étiologie de l’asthme
Nous devons la première description de la maladie aux Grecs vers l’an 100 apr. J.-C. où l’asthme était alors défini comme une difficulté à respirer à l’effort [6]. Vers la fin du 19e
siècle, on parlait alors d’un essoufflement accompagné par un gonflement des muqueuses bronchiques et d’une surproduction de mucus [1]. En 1962, l’asthme était décrit comme un rétrécissement diffus des voies aériennes caractérisé par une contraction excessive de la trachée et des bronches suite à une exposition à divers stimuli [1]. Ce phénomène est nommé hyperréactivité bronchique [1]. Maintenant, l’American Thoracic Society définit l’asthme comme un syndrome clinique se caractérisant par un épaississement de la trachée et des bronches provoqué par des stimuli et qui mène à des symptômes comme de la dyspnée accompagnée d’une respiration sifflante, des serrements dans la poitrine et de la toux [7].
Ainsi, les manifestations cliniques de l’asthme sont relativement bien caractérisées, mais ses causes sont encore mal comprises. D’un point de vue génétique, plus de 100 gènes sont associés au développement de l’asthme [8]. Plus spécifiquement, le gène qui code pour le récepteur S1PR1 a été associé au développement de l’asthme [9] de même que des gènes qui codent pour des chimiokines (CCL5, CCL11 et CCL26) exprimées par les cellules épithéliales (CE) des voies aériennes [8]. Un autre facteur génétique qui est fortement associé au développement de l’asthme est l’atopie [8]. L’atopie se définit par une prédisposition génétique à développer de l’hypersensibilité spécifique à des allergènes communs (pollens, acariens, poils d’animaux) qui se traduit par une surproduction d’IgE spécifiques aux allergènes [10]. Cependant, l’asthme n’est pas toujours associé à de l’atopie, ou bien déclenché par des allergènes. En fait, les symptômes de l’asthme peuvent également être causés par des polluants, des virus, le stress ou même l’activité physique [4]. En somme, l’étiologie de l’asthme est complexe et se veut le fruit des interactions entre divers facteurs génétiques et environnementaux.
Le contrôle de l’asthme
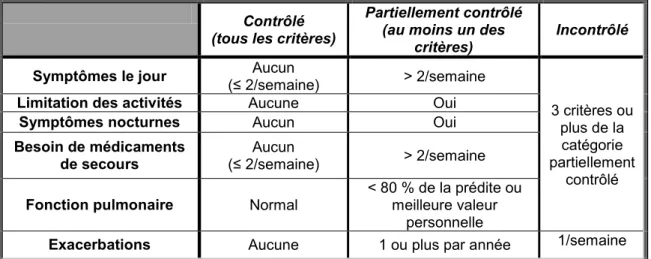
Les traitements actuels de l’asthme permettent de contrôler l’apparition des symptômes. Pour bien caractériser l’effet des thérapies sur le contrôle de l’asthme, l’organisme Global Initiative for Asthma (GINA) classe les niveaux de contrôle de l’asthme en se basant sur des critères de fréquence et de gravité des symptômes (Tableau 1) [4]. Par la suite, la nature
de la thérapie est déterminée en fonction de l’atteinte d’un contrôle optimal avec une dose minimale de traitements pharmacologiques comme mentionné par l’organisme GINA :
The patient’s current level of asthma control and current treatment determine the selection of pharmacologic treatment. For example, if asthma is not controlled on the current treatment regimen, treatment should be stepped up until control is achieved. If control has been maintained for at least three months, treatment can be stepped down with the aim of establishing the lowest step and dose of treatment that maintains control.
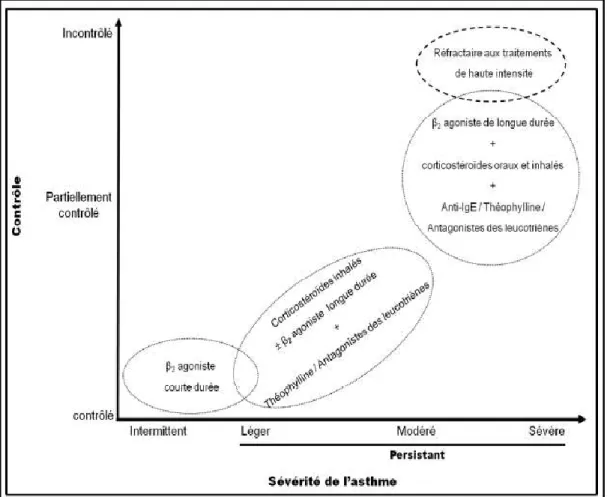
Les organismes GINA et le National Institute of Health des États-Unis (NIH) s’entendent sur l’existence d’une corrélation entre la réponse aux traitements et la sévérité de la maladie [4, 11]. Ainsi, la nature du traitement utilisé dépend de la sévérité de la maladie et vise l’atteinte du contrôle des symptômes (fig. 1).
Contrôlé (tous les critères)
Partiellement contrôlé (au moins un des
critères)
Incontrôlé
Symptômes le jour (≤ 2/semaine) Aucun ˃ 2/semaine
3 critères ou plus de la catégorie partiellement
contrôlé Limitation des activités Aucune Oui
Symptômes nocturnes Aucun Oui
Besoin de médicaments
de secours (≤ 2/semaine) Aucun ˃ 2/semaine Fonction pulmonaire Normal < 80 % de la prédite ou meilleure valeur
personnelle
Exacerbations Aucune 1 ou plus par année 1/semaine
Tableau 1. Niveau de contrôle de l’asthme.
Adapté de Bateman 2008 [4] avec la permission de le reproduire.
Selon le NIH, le stade le moins sévère de l’asthme est appelé intermittent [12]. Les asthmatiques intermittents sont facilement contrôlés avec des bronchodilatateurs tels que les β2 agonistes qui sont utilisés au besoin seulement [12]. En contrepartie, les asthmatiques
persistants doivent être traités quotidiennement avec des doses de corticostéroïdes et/ou d’antagonistes des leucotriènes pour contrôler l’inflammation des voies aériennes [4]. Pour
les cas plus sévères, une combinaison de traitements doit être utilisée pour contrôler les symptômes (fig.1) [13]. Nonobstant les asthmatiques non contrôlés par manque d’adhérence aux traitements, il existe des asthmatiques (5 % -10 %) qui résistent à l’arsenal thérapeutique actuel. Ces personnes souffrent d’asthme réfractaire [13]. Le terme asthme réfractaire ne s’applique pas seulement aux asthmatiques ayant besoin de doses très élevées de corticostéroïdes pour être contrôlés, mais aussi aux cas d’asthme très sévère, voire fatal. Actuellement, il n’existe pas d’alternative de traitement adéquate pour ces asthmatiques. L’arrivée des nouvelles thérapies pourrait non seulement améliorer la condition de la plupart des asthmatiques, mais elle pourrait également contribuer à contrôler l’asthme réfractaire.
Figure 1. Relation entre le contrôle et la sévérité de l’asthme. Adapté d’Ito 2009 [13], avec la permission de le reproduire.
1.2 La pathogenèse de l’asthme
Les symptômes de l’asthme résultent de l’hyperréactivité bronchique et d’une obstruction partielle qui se développent suite à l’exposition à divers stimuli [14]. Des changements dans les voies aériennes mènent à de la détresse respiratoire qui est conséquente à une diminution de la lumière des bronches. La diminution de la lumière des bronches est causée par l’épaississement de leurs parois résultant de l’accumulation des cellules inflammatoires, d’une hyperplasie des cellules épithéliales et des cellules musculaires lisses, de la présence de fibrose et de la surproduction de mucus [15]. Cette diminution est également due à la présence de stimuli qui provoquent de l’hyperréactivité bronchique [15]. Ces altérations sont le fruit de mécanismes cellulaires complexes décrits comme étant la « cascade asthmatique ».
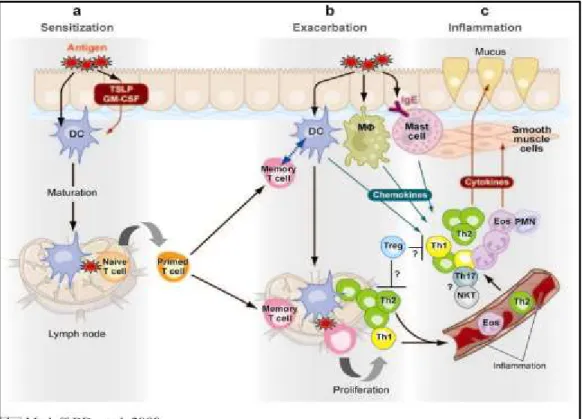
1.2.1 La cascade asthmatique-sensibilisation
Dans l’asthme allergique, la première étape de la cascade asthmatique est appelée la sensibilisation (fig. 2). À cette étape, des cellules présentatrices de l’antigène, tout particulièrement les cellules dendritiques (DCs), captent l’allergène [16] et entament leur processus de maturation et de migration vers les ganglions lymphatiques. Une fois dans les ganglions lymphatiques, les DCs présentent les peptides allergéniques aux lymphocytes T naïfs qui s’activent et, sous l’effet de cytokines telles qu’IL-4, se différencient en lymphocytes Th2 [17]. Ensuite, dans les ganglions, les lymphocytes Th2 activent les lymphocytes B en libérant IL-13 [18]. Ceux-ci se transforment alors en plasmocytes producteurs d’IgE spécifiques à l’allergène [15]. Ainsi, suite à cette phase de sensibilisation, l’individu possède des lymphocytes Th2 et des lymphocytes B activés et/ou mémoires et des immunoglobulines IgE spécifiques à l’allergène qui seront impliqués dans l’amplification de la réponse immune lors de la réexposition à l’allergène. Ces cellules se retrouvent tant dans les organes lymphoïdes que dans les voies respiratoires.
1.2.2 La cascade asthmatique-exacerbation
Lorsque les voies aériennes sont réexposées aux allergènes, la seconde phase de la cascade asthmatique débute, la phase d’exacerbation (fig. 2). Lorsque l’allergène est capté par les IgE présents sur les mastocytes, il provoque la libération de médiateurs par ces derniers [19]. Cette libération de divers médiateurs génère la réponse immédiate qui se produit 30 minutes suivant l’exposition à l’allergène et qui se traduit par la formation d’œdème dans les muqueuses, de la bronchoconstriction et l’initiation du recrutement des cellules inflammatoires [20].
L’histamine contribue à la réponse immédiate en induisant rapidement de la bronchoconstriction. Les produits des mastocytes contribueront également à induire une réponse inflammatoire qui mènera à une réponse de détresse respiratoire tardive. Ainsi seront libérées des molécules qui sont impliquées dans le recrutement de DCs supplémentaires, de mastocytes, d’éosinophiles et de lymphocytes. La conséquence de ce recrutement massif de cellules inflammatoires est l’augmentation de la libération de cytokines proinflammatoires, comme le TNF, non seulement par ces cellules, mais aussi par l’épithélium bronchique et par l’endothélium vasculaire [21]. Des protéases entraînent, quant à elles, des dommages à différents tissus des voies aériennes [22]. Sous l’effet de ces médiateurs, les cellules inflammatoires s’accumulent et provoquent une seconde vague d’inflammation appelée la réaction retardée. Cette réaction se produit généralement 6 à 9 h après l’exposition à un allergène et se présente par une recrudescence des symptômes respiratoires [14]. Par ailleurs, cette phase n’est retrouvée que chez une portion d’individus (60 %) [23].
Ainsi, des épisodes répétés seraient à l’origine d’une inflammation dite chronique. Cette inflammation est associée à divers mécanismes sous-jacents à la pathogenèse de l’asthme. Parmi ces mécanismes, on retrouve une altération des cellules structurales, dont les cellules épithéliales ; le développement de l’hyperplasie et de l’hypersensibilité des muscles lisses ainsi que l’hypersécrétion de mucus par l’épithélium. Bien que les cellules immunitaires jouent un rôle important dans l’asthme, la prochaine section mettra en évidence une
littérature émergente suggérant que les cellules structurales pulmonaires puissent également y jouer un rôle central.
Figure 2. La pathogenèse de l’asthme.
Tiré de Medoff 2008 [16] avec la permission de le reproduire.
1.3 Les cellules structurales pulmonaires impliquées dans l’asthme
Les cellules structurales pulmonaires tout comme les cellules immunitaires libèrent des médiateurs solubles qui sont impliqués dans le remodelage, l’hyperréactivité bronchique et l’inflammation.
1.3.1 Implication des muscles lisses, de l’endothélium et des fibroblastes dans l’asthme Dans les biopsies de poumon de patients asthmatiques, des changements structuraux sont observés au niveau des cellules musculaires lisses, des cellules endothéliales, des
myofibroblastes et des fibroblastes. Tout d’abord, les cellules musculaires lisses sont hypertrophiées, hyperplasiées et se contractent excessivement [14, 24]. La fibrose sous-épithéliale présente dans l’asthme est caractérisée par une forte production des protéines de la matrice extracellulaire ainsi que par le déséquilibre entre l’expression des enzymes qui sont responsables de leur destruction, les métalloprotéinases, et leurs inhibiteurs, les TIMP (Tissue inhibitors of metalloproteinases) [25]. Cette fibrose est également caractérisée par une accumulation de fibroblastes et de myofibroblastes activés ainsi que la destruction des fibres élastiques, ce qui a pour résultat d’augmenter la rigidité des bronches [25].
L’endothélium vasculaire joue également un rôle dans l’inflammation et le remodelage dans l’asthme. Les cellules endothéliales participent au recrutement de cellules inflammatoires en surexprimant des molécules d’adhésion tel que CD31 (PECAM-1), CD106 (VCAM-1) et CD62P (P-sélectine) [22]. L’augmentation des molécules d’adhésion des cellules endothéliales permet ainsi d’accroître l’affinité des leucocytes pour l’endothélium, ce qui facilite leur recrutement massif dans les voies aériennes pendant la phase inflammatoire [22]. L’augmentation de l’angiogenèse et de la perméabilité de l’endothélium observée dans les poumons des asthmatiques est, quant à elle, associée à l’effet de facteurs de croissance tel que le facteur de croissance de l’endothélium vasculaire (VEGF) produit par les mastocytes, les éosinophiles et les cellules structurales des voies aériennes [26, 27].
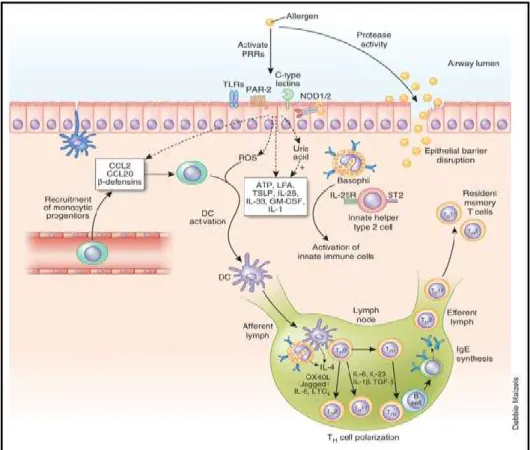
1.3.2 Rôle central de l’épithélium bronchique dans l’asthme
Au cours des dernières années, des études ont révélé que l’épithélium bronchique n’était pas seulement une barrière physique, mais que les CE jouaient un rôle actif dans la régulation de l’inflammation des voies aériennes et dans le remodelage (fig. 3). D’ailleurs, dans l’asthme, on dénote des altérations histologiques et fonctionnelles notables au niveau de l’épithélium. Dans les biopsies de sujets asthmatiques, il y a ainsi une desquamation de l’épithélium et une augmentation de l’espace entre les CE ainsi qu’un profil inflammatoire anormalement augmenté [28].
1.3.2.1 Les altérations structurales des CE bronchiques
Dans des biopsies de sujets sains, l’épithélium des voies aériennes forme une barrière presque étanche par la formation de jonctions serrées (JS) [29]. Dans l’épithélium bronchique des asthmatiques, des protéines comme la zonula occludens-1 (ZO-1), la E-cadhérine et la β-caténine sont sous-exprimées, ce qui a pour conséquence de désorganiser les JS, rendant la barrière épithéliale plus vulnérable aux menaces extérieures [30-32]. Ces dysfonctions ont également été remarquées dans des cultures de CE bronchiques de modèles d’asthme chez la souris [33] et chez les rats Brown Norway (BN) [communication personnelle, Dre. Bissonnette]. De plus, il est connu que des allergènes peuvent désorganiser la barrière épithéliale en altérant les protéines des JS. C’est, par exemple, le cas de l’extrait d’acariens qui possède une enzyme protéolytique, Derp 1, capable de détruire des protéines des JS entre les CE et ainsi contribuer à l’apparition de l’asthme infantile [34-36]. La désorganisation des JS augmente également la perméabilité transépithéliale et la diffusion de protéines vers les couches sous-épithéliales [32]. Ainsi, la diminution de l’étanchéité de la barrière épithéliale des asthmatiques pourrait impliquer une augmentation des échanges de médiateurs proinflammatoires entre le sang et la lumière des voies aériennes. Cette diminution de l’étanchéité pourrait, aussi, faciliter la sensibilisation aux allergènes.
Dans un autre ordre d’idée, l’infiltration des cellules inflammatoires et la désorganisation des JS activent les mécanismes de réparation de l’épithélium bronchique. La réparation des tissus des voies aériennes partage des similarités avec la morphogenèse du poumon chez le fœtus pendant la gastrulation [37]. Pendant la morphogenèse du poumon, la modulation des facteurs de croissance et de transition qui transforment les CE en fibroblastes et les fibroblastes en CE jouent un rôle clé dans l’induction des structures périphériques du poumon et la différentiation des CE [38]. Après la naissance, les CE peuvent effectuer de la transition épithélio-mésanchymateuse (TEM), qui se définit alors comme la transformation des CE en fibroblastes. L’expression des facteurs tel que le facteur de croissance des fibroblastes (FGF) et du facteur de croissance de transformation (TGF-β) sécrétés par les CE pendant leur réparation dans l’asthme seraient à l’origine de la TEM [39]. Lorsque des
CE sont exposées à ces facteurs de croissance, elles perdent graduellement leurs marqueurs comme l’E-cadhérine pour acquérir les marqueurs des fibroblastes tels que la vimentine [40, 41]. En plus de contribuer à diminuer la fonction de barrière de l’épithélium, ces nouveaux fibroblastes contribuent au remodelage en produisant des protéines telles que du collagène et de la fibronectine qui s’accumulent dans la membrane basale et participent à l’épaississement des voies aériennes [39]. Ainsi, dans l’asthme, le processus de réparation « chronique » de l’épithélium pourrait contribuer au remodelage en induisant la formation de fibroblastes à partir des CE bronchiques. Cette transition mène alors à la diminution de la quantité de CE fonctionnelles dans le poumon et augmente du même coup le nombre de cellules libérant de la matrice extracellulaire.
Les CE des voies aériennes des asthmatiques participent donc activement aux mécanismes immunopathologiques de la maladie par le biais d’une diminution de leur étanchéité et par la présence de TEM.
1.3.2.2 La production des médiateurs solubles par les CE bronchiques est modulée dans l’asthme
L’exposition aux différents stimuli et les dommages causés par ceux-ci mènent les CE bronchiques à produire une grande quantité de cytokines et de médiateurs solubles. Ces molécules possèdent des fonctions importantes dans l’initiation et le maintien de la réponse inflammatoire. La surproduction de médiateurs solubles a été remarquée, notamment, dans les CE issues de modèles d’asthme induit avec de l’ovalbumine (OVA). Dans ce modèle, l’expression de médiateurs inflammatoires comme TSLP (Thymic stromal lymphopoietin) et CCL20 chez la souris BALB/c ou CCL2 (MCP-1) chez le rat BN, est plus élevée chez les animaux « asthmatiques » comparativement aux naïfs [42-44]. Dans l’asthme allergique, on a également observé que les CE produisaient du SCF (stem-cell factor) qui attire les mastocytes à la surface de l’épithélium bronchique pendant la réponse immédiate [18]. En libérant la chimiokine CCL2 pendant la réponse immédiate, les CE contribuent au recrutement de monocytes, des progéniteurs des DCs, ce qui accroît le nombre de cellules présentatrices d’antigène dans les voies aériennes [45]. Plusieurs autres cytokines libérées
par les cellules épithéliales comme IL-33, IL-1β et TSLP permettent notamment de recruter, de polariser et d’activer les DCs de manière à amplifier la réponse Th2 [46]. Le recrutement des lymphocytes Th2 vers les tissus des voies aériennes s’effectue sous l’effet d’autres cytokines et chimiokines libérées par les CE comme IL-13, IL-25, RANTES, CCL17/TARC et CCL22 [32]. Les CE des voies aériennes, en relâchant la chimiokine CCL11, sont impliquées aussi dans le recrutement des éosinophiles en liant leur récepteur CCR3 [18].
Finalement, des études faites à l’aide de lignées cellulaires supportent l’idée que des drogues ayant des effets anti-inflammatoires in vivo peuvent interférer avec la libération de médiateurs inflammatoires en ciblant directement les CE. Par exemple, la thiazolidinedione peut réduire la libération de CCL2 induit par le IL-β et TNF dans les cellules A549 [47]. Le budésonide, quant à lui, peut inhiber la libération de CCL13 (MCP-4) et RANTES, dans les cellules BEAS-2B [48, 49]. L’utilisation de glucocorticoïdes peut également inhiber l’éotaxine, une cytokine impliquée dans le recrutement des éosinophiles, exprimée dans ces deux lignées cellulaires [50].
Figure 3. L’épithélium bronchique est central dans l’asthme. Tiré de Lambrecht, 2012 [45] avec la permission de le reproduire.
En résumé, les CE bronchiques « asthmatiques » possèdent des altérations dans l’expression de leurs protéines de jonctions. Ces altérations exposent le poumon aux dommages des allergènes tout en augmentant les échanges de médiateurs solubles entre la surface du poumon et le sang. Le profil inflammatoire élevé des CE bronchiques contribue, de son côté, au recrutement des cellules inflammatoires et à la polarisation des lymphocytes Th2, donc à la pérennisation de l’inflammation dans l’asthme. En contrôlant ces dysfonctions dans l’asthme avec des nouvelles voies thérapeutiques, il serait peut-être possible de réduire les symptômes de l’asthme ou d’interférer avec sa pathogenèse.
14
Section II : Les sphingolipides
Les sphingolipides sont des lipides peu connus, mais indispensables à la vie des mammifères. Ces lipides sont ubiquitaires chez les eucaryotes. Ils sont des composés résultant de l’amidification d’une longue chaîne de carbone (base sphingoïde) et peuvent posséder un groupement de tête. La classe des sphingolipides comprend notamment la sphingomyéline, la céramide, la sphingosine et la sphingosine-1-phosphate (S1P). Ils ont été décrits pour la première fois en 1884 par J.L.W. Thudicum [51] et nommés ainsi car ils possédaient, comme le sphinx, des propriétés énigmatiques [52]. Les sphingolipides ont été décrits, au départ, comme des lipides relativement inertes ayant un rôle structural régulant la perméabilité péricellulaire [51]. Depuis ce temps, les sphingolipides ont émergé comme des régulateurs de plusieurs mécanismes cellulaires.
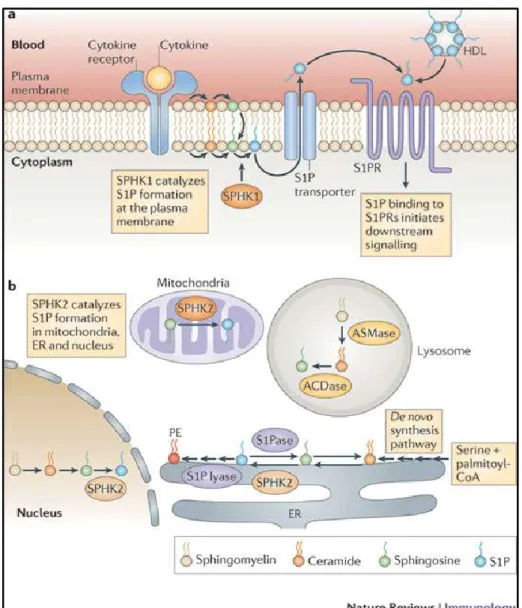
1.4 La biosynthèse de la sphingosine-1-phosphate
La S1P se retrouve principalement dans le plasma. Les érythrocytes constituent la principale source des hauts niveaux de S1P dans le plasma où elle est fixée à l’albumine et aux lipoprotéines de hautes densités (HDL) [53]. Dans les canaux lymphatiques, l’endothélium constitue une source de S1P [54]. La S1P extracellulaire est rapidement déphosphorylée par plusieurs phosphatases dont la S1P phosphatase et d’autres lipides phosphate phosphatases, ce qui résulte en une faible concentration de S1P dans les tissus [55].
La S1P est produite par le recyclage/dégradation de la sphingomyéline présente au noyau et à la membrane plasmique. La S1P est également produite de novo dans le réticulum endoplasmique et dans différents compartiments subcellulaires (fig. 4). Ainsi, en général, sous l’effet d’enzymes, et de manière réversible, la sphingomyéline est convertie en céramide puis en sphingosine. Ensuite, la sphingosine est phosphorylée en S1P par la sphingosine kinase 2 (SPHK2) dans le noyau [56] et les mitochondries. À la membrane plasmique, c’est plutôt la sphingosine kinase 1 (SPHK1) qui phosphoryle la sphingosine [56]. La S1P peut rester intracellulaire ou être externalisée par des transporteurs
transmembranaires, ce qui lui confère des effets extracellulaires en liant 5 récepteurs : S1PR1 à S1PR5 [57].
Figure 4. Synthèse de la sphingosine-1-phosphate.
Tiré de Spiegel 2012 [56] avec la permission de le reproduire.
Les S1PRs sont des récepteurs couplés à une protéine G possédant 7 domaines transmembranaires [58]. L’expression du premier récepteur de la S1P (S1PR1) a d’abord été caractérisée dans les cellules endothéliales in vitro [59]. Aujourd’hui, il est connu que les S1PRs sont exprimés différentiellement dans l’ensemble du corps humain. Les récepteurs S1PR1 à S1PR3 sont exprimés notamment dans le cerveau, les poumons, le
cœur, les reins et la rate [60]. S1PR4 se retrouve surtout dans le poumon et les organes lymphoïdes et S1PR5 dans le cerveau, la peau et la rate [60]. Dans ces organes, la liaison entre la S1P et ses récepteurs régulent de nombreux mécanismes cellulaires.
En homéostasie, la concentration de S1P est élevée dans le plasma et faible dans les tissus. Dans plusieurs pathologies, la concentration de S1P augmente dans les tissus [61-63], et l’expression des récepteurs de la S1P est modulée. Ces modulations supportent que la voie de signalisation de la S1P pourrait moduler diverses conditions pathologiques.
1.5 Effets biologiques de la modulation pharmacologique de la voie de
signalisation de la S1P
L’utilisation de modulateurs pharmacologiques et d’outils génétiques a permis de préciser davantage le rôle de la S1P et de la modulation de ses récepteurs dans plusieurs processus biologiques.
1.5.1 Analogues de la sphingosine
Vers le début des années 90, le premier analogue synthétique de la sphingosine, FTY720, a été découvert suite à des études sur ISP-1 (myoricine), un immunosuppresseur naturel capable de supprimer la prolifération des lymphocytes [64]. La toxicité et la faible solubilité d’ISP-1 ont mené à la simplification de sa structure chimique et à la synthèse de FTY720 [64]. Les cibles de FTY720 se sont révélées différentes de celle d’ISP-1. Donc, au lieu d’inhiber la sérine palmitoyltransférase, FTY720 est plutôt phosphorylé par SPHK2 en FTY720-P et agît comme agoniste de S1PR1, S1PR3, S1PR4 et S1PR5 [65, 66].
L’effet immunosuppresseur de FTY720 est principalement associé à une séquestration des lymphocytes dans les ganglions lymphatiques [67]. FTY720 cible également d’autres cellules et mécanismes de l’immunité. In vivo, il diminue le nombre de DCs dans les organes lymphoïdes chez la souris [68]. Il diminue également la perméabilité des cellules endothéliales [69, 70]. Dans la sclérose en plaques, son utilisation diminue l’infiltration des
lymphocytes dans le système nerveux central tout en aidant la réparation des tissus [71]. Ces effets bénéfiques dans le traitement de cette pathologie ont mené à son approbation par la FDA en 2010 [72]. Il existe également un analogue de FTY720, AAL-R, qui est de plus en plus utilisé pour les études in vivo puisqu’il présente certaines caractéristiques avantageuses comparativement à FTY720. Dans la souris, AAL-R est phosphorylé pour devenir AFD-R, un analogue actif de la S1P, 8 fois plus rapidement que le FTY720 [73]. L’équipe du Dr Marsolais a d’ailleurs démontré son efficacité à inhiber la réponse immune pulmonaire dans au moins 2 modèles de maladies pulmonaires [74, 75].
Les analogues de la sphingosine phosphorylés (FTY720-P ou AFD-R) possèdent de nombreux effets spécifiques lorsqu’ils lient les S1PRs, notamment le renforcement de la barrière endothéliale et la séquestration des lymphocytes via le récepteur S1PR1 [69, 75, 76]. Cependant, les analogues de la sphingosine comme FTY720 et AAL-R possèdent des effets non spécifiques en interagissant avec plusieurs protéines de la voie de signalisation de la S1P comme la SPHK1, la S1P lyase et la phospholipase cytosolique A2 [77-79]. L’utilisation de FTY720 peut entraîner des effets négatifs sur la santé tels que de la bradycardie (via l’activation de S1PR3), de l’oedème maculaire et une augmentation des infections dont les mécanismes sont encore mal compris [80]. Dans le but de comprendre les effets et mécanismes d’action de cette nouvelle classe de drogues, des modulateurs pharmacologiques spécifiques des S1PRs ont été développés. L’utilisation de ces modulateurs combinée aux outils génétiques a permis de caractériser davantage l’effet spécifique des S1PRs. D'ailleurs, le récepteur S1PR1 s’est révélé être un récepteur important, voire suffisant, pour expliquer l’effet immunomodulateur des analogues de la sphingosine.
1.5.2 Le récepteur S1PR1
L’activation de S1PR1 est essentielle à la maturation vasculaire durant l’embryogenèse puisque l’invalidation génique de S1PR1 provoque la mort embryonnaire chez la souris [81]. In vivo, l’activation de S1PR1 par des agonistes spécifiques augmente l’étanchéité de la barrière endothéliale [82-84]. In vitro, son activation est impliquée dans la
réorganisation du cytosquelette et la formation de jonctions intercellulaires [85, 86]. De plus, comme mentionné précédemment, l’activation spécifique de S1PR1 régule des mécanismes de la réponse immune. L’activation spécifique de S1PR1 est suffisante pour induire la séquestration des lymphocytes dans les organes lymphoïdes [67]. D’ailleurs, un agoniste spécifique de S1PR1 démontre des effets bénéfiques importants dans un modèle murin de sclérose en plaques [87]. De manière importante, l’activation locale de S1PR1 court-circuite la réponse immune dans divers modèles d’immunité pulmonaire [75, 89, 90].
En résumé, les outils pharmacologiques ont permis de caractériser le rôle spécifique des acteurs de la voie de signalisation de la S1P. La modulation de la voie de signalisation de la S1P régule des mécanismes de la réponse immune. Parmi ces mécanismes, on retrouve l’inhibition de la libération de cytokines et chimiokines, la séquestration des lymphocytes dans les ganglions lymphatiques (S1PR1) et la modulation de l’étanchéité des barrières. Les effets bénéfiques des analogues et des modulateurs pharmacologiques de la voie de signalisation de la S1P dans l’immunité pulmonaire ont mené à l’étude de cette voie dans l’asthme.
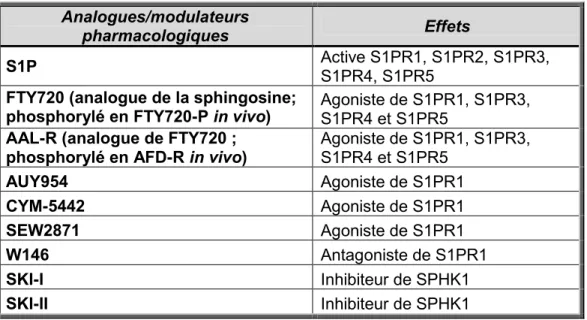
Analogues/modulateurs
pharmacologiques Effets
S1P Active S1PR1, S1PR2, S1PR3,
S1PR4, S1PR5 FTY720 (analogue de la sphingosine;
phosphorylé en FTY720-P in vivo) Agoniste de S1PR1, S1PR3, S1PR4 et S1PR5 AAL-R (analogue de FTY720 ;
phosphorylé en AFD-R in vivo) Agoniste de S1PR1, S1PR3, S1PR4 et S1PR5
AUY954 Agoniste de S1PR1 CYM-5442 Agoniste de S1PR1 SEW2871 Agoniste de S1PR1 W146 Antagoniste de S1PR1 SKI-I Inhibiteur de SPHK1 SKI-II Inhibiteur de SPHK1
Tableau 2. Des analogues et modulateurs pharmacologiques de la voie de signalisation de la S1P mentionnés dans ce mémoire.
1.6 Modulation de la voie de la sphingosine-1-phosphate et l’asthme
Le rôle de la voie de signalisation de la S1P est controversé dans l’asthme. Cette controverse provient des résultats divergents obtenus en utilisant des outils pharmacologiques ayant des effets cellulaires multiples. Les premiers indices de l’implication dans l’asthme des acteurs de la voie de signalisation de la S1P sont apparus avec une étude montrant que le niveau de S1P était significativement plus élevé dans les lavages bronchoalvéolaires de patients asthmatiques comparativement aux sujets sains après une stimulation avec un allergène [63].
Certaines données supportent un rôle délétère de la S1P dans l’asthme. L’administration intranasale (i.n.) de S1P aggrave l’inflammation des voies aériennes dans un modèle d’asthme à l’OVA chez la souris [91]. Aussi, l’inhalation de SKI-I (inhibiteur de SPHK1) chez un modèle d’asthme à l’OVA réduit l’inflammation allergique [92, 93]. Dans le même modèle, l’administration i.n. de SKI-I inhibe également l’activation des mastocytes par l’antigène, l’inflammation pulmonaire et l’hyperréactivité bronchique [93]. De plus, SKI-I inhibe l’activation du facteur de transcription NF-κB qui est associée à une réduction de cytokines retrouvées dans les lavages bronchoalvéolaires du modèle [93]. Par contre,
l’administration intrapéritonéale (i.p.) de SKI-II (inhibiteur de SPHK1) ne réduit pas l’inflammation dans le même modèle [94].
D’autres études supportent un rôle protecteur de la voie de signalisation de la S1P dans l’asthme. L’administration orale de l’analogue de FTY720 diminue la réponse Th2 dans un modèle d’asthme [95]. L’administration intratrachéale (i.t.) de FTY720 inhibe le développement de l’asthme expérimental en altérant les fonctions des DCs, ce qui a pour effet de diminuer la génération de la réponse lymphocytaire [96]. De plus, FTY720 inhibe le remodelage bronchique dans un modèle d’asthme chez le rat [97]. Plus spécifiquement, l’administration i.n. d’un agoniste spécifique de S1PR1 est suffisante pour diminuer l’infiltration de liquide induite par l’allergène dans les poumons d’un modèle d’asthme à l’OVA chez la souris [83]. La modulation locale de S1PR1 par l’administration i.t. d’un agoniste spécifique de S1PR1, CYM-5442 est, quant à elle, suffisante pour inhiber l’inflammation allergique dans un modèle d’asthme allergique comparativement à AAL-R, l’analogue de FTY720, qui inhibe l’inflammation en activant 4 des 5 récepteurs. Cette inhibition de l’inflammation par l’activation spécifique de S1PR1 est associée à une diminution de la libération de cytokines comme TARC et RANTES par les voies aériennes plutôt qu’à une séquestration des lymphocytes dans les ganglions associée à l’effet d’AAL-R [89].
Plusieurs raisons peuvent expliquer les divergences des résultats obtenus par la modulation de la voie de signalisation de la S1P dans l’asthme. La balance entre la formation de sphingosine-1-phosphate et celle de produits de dégradation comme la céramide pourrait expliquer les résultats divergents observés avec l’administration de doses non physiologiques de S1P ou d’inhibiteurs de kinase dans l’asthme. Par exemple, l’administration de doses massives de S1P ou d’inhibiteurs de kinases peut modifier le sentier métabolique et augmenter rapidement les concentrations de sphingosine et de céramide qui sont reconnus comme étant respectivement apoptotiques [98] et pro-inflammatoires [91]. Aussi, un nouveau paradigme veut que la S1P intracellulaire ait des cibles et des actions différentes de la S1P extracellulaire qui agirait seulement sur les récepteurs [99]. Ce phénomène pourrait ainsi expliquer les résultats divergents entre les
études effectuées avec les inhibiteurs de kinases et les études où des agonistes ont été utilisés.
En résumé, la littérature supporte que la voie de signalisation de la S1P soit impliquée dans les mécanismes sous-jacents à l’asthme et que la modulation locale des S1PRs, particulièrement S1PR1, interfère avec le développement de l’asthme expérimental. Par contre, les mécanismes et les cellules impliquées par une modulation locale de la voie de signalisation de la S1P dans l’asthme sont encore inconnus.
23
24
L’épithélium bronchique joue un rôle central dans l’asthme. Chez les individus non asthmatiques, l’épithélium des voies aériennes forme une barrière physique qui protège le poumon du non-soi. Dans l’asthme, les CE bronchiques sont altérées et perméables aux particules extérieures. Elles participent à l’inflammation pulmonaire en libérant des cytokines et chimiokines responsables du recrutement de cellules inflammatoires. Le contrôle des altérations épithéliales rencontrées dans l’asthme serait donc susceptible d’interférer avec le développement de cette maladie.
Dans des modèles d’asthme, l’administration locale d’analogues de la sphingosine et de modulateurs pharmacologiques de S1PR1 tels que CYM-5442 inhibe l’inflammation pulmonaire allergique. Cependant, les cibles de ces drogues et leurs mécanismes d’action dans le poumon sont inconnus.
Il est connu que le récepteur S1PR1 est exprimé dans des lignées de CE [100]. Puisque les CE bronchiques sont centrales dans l’asthme et que les modulateurs pharmacologiques de S1PR1 inhibent l’inflammation lorsqu’administrés localement, notre hypothèse est que l’activation du récepteur 1 de la S1P contribue à la réduction des dysfonctions épithéliales dans l’asthme.
Le but de cette étude est ainsi de caractériser l’effet d’un agoniste spécifique de S1PR1, CYM-5442, sur les dysfonctions des CE bronchiques. L’étude est divisée en trois objectifs spécifiques :
1- Caractériser l’expression de S1PR1 dans des CE bronchiques.
2- Démontrer que l’activation de S1PR1 réduit les dysfonctions des CE bronchiques dans un modèle d’asthme.
25
26
3.1 Isolement et culture des CE bronchiques
L’asthme a été induit à des rats BN tel que décrit par Careau et al. [101]. Les rats ont été sensibilisés de manière intra-péritonéale avec de l’OVA (1 mg/ml) dans du gel d’hydroxyde d’aluminium (10 mg/ml) ou de la saline, puis, 21 jours plus tard, les rats ont été « challengés » avec de l’OVA ou de la saline aérosolisée pendant 5 minutes [101]. 24 h suivant l’aérosolisation, les rats ont été euthanasiés et les CE bronchiques de rats ont été isolées selon la méthode de Dobbs et al. [102] à partir de rats naïfs (Normal Bronchial Epithelial Cells, NBEC) et de rats challengés à l’OVA (Bronchial Epithelial Cells isolated from rats with experimental asthma, ABEC). Après avoir rempli les poumons des rats avec 160 U/rat d’élastase (4 U/ml) (Worthington Biochemical, NJ, États-Unis) dans un tampon isotonique (154 nM de NaCl, 5,55 mM de glucose, 200 uL de septra, 5,3 mM de KCl, 1,9 mM de CaCl2, 1,3 mM de MgSO4, 10 mM d’HEPES, 2,65 mM de
NaPO4, H2O) pendant 15 minutes, leur arbre trachéo-bronchique a été isolé. La trachée et
les bronches ont été émincées dans un tube contenant 5 ml de 250 μg/ml de DNase (Sigma-Aldrich Canada Ltd, Oakville, ON, Canada) dans le tampon isotonique. Après l’inactivation de la DNase avec 5 ml de sérum foetal de bovin 100 % (FBS) (Wisent, St-Bruno, QC, Canada) pendant 2-4 minutes, les cellules ont été filtrées et leur viabilité a été quantifiée par un décompte au bleu de trypan.
Les cellules ont été mises en culture dans des plaques de 6 puits dans un milieu complet (DMEM/F12 (Wisent) additionné de FBS 10 % (vol/vol) inactivé (Wisent), de 10 mM d’HEPES (Wisent), 1 % d’antibiotiques-antimycotiques (100 U/ml pénicilline, 100 μg/ml streptomycine, 25 ng/ml amphotéricine et 85 mg/L NaCl) (Wisent), de 125 μM d’adénine (Sigma-Aldrich), de 0,1 nM de toxine de choléra (Sigma-Aldrich), de 5 μg/ml d’insuline (Sigma-Aldrich), de 0,4 μg/ml d’hydrocortisone (Calbiochem/EMD Millipore, MA, États-Unis), de 5 μg/ml transferrine (Sigma-Aldrich), de 2 nM de 3,3′,5′, triiodo-L-thyronine (Sigma-Aldrich) et de 10 ng/ml de facteur de croissance épidermique (EGF) de rat (Peprotech, NJ, États-Unis)) [103]. Les fibroblastes contaminants ont été éliminés par des traitements rapides (< 2 min.) à la trypsine-EDTA 0,25 % (Wisent). À confluence, 3E05 CE ont été transférées dans des flacons pour constituer des sous-cultures de CE
27 bronchiques. Après une ou deux sous-cultures, la pureté des cellules a été vérifiée par un marquage à la pan-cytokératine. Les cultures ont été maintenues dans du milieu complet dans un incubateur à 37 °C, 5 % CO2-95 % air.
À confluence, les CE ont été passées dans un autre flacon. Tout d’abord, le milieu des cellules a été jeté et les cellules ont été lavées avec une solution saline tamponnée au phosphate (PBS) stérile pour être ensuite incubées avec 3 ml de trypsine-EDTA 0,25 % à 37 °C. Après 4-5 minutes, les cellules ont été récupérées avec 9 ml de milieu complet. Le milieu contenant les cellules a été centrifugé à 500 g pendant 5 minutes à 4 °C. Le culot cellulaire a ensuite été resuspendu dans 2 ml de milieu complet et la viabilité cellulaire a été évaluée par la méthode d’exclusion au bleu de trypan. Ensuite, 1E06 cellules ont été ajoutées à 10 ml de milieu complet et transférées dans un flacon. Les cellules ont été utilisées pour les expérimentations entre les passages 2 et 8.
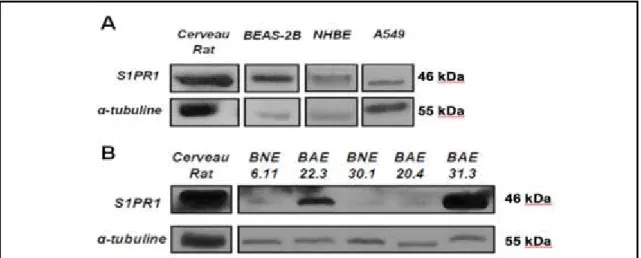
Deux lignées de CE pulmonaires humaines ont été utilisées : les BEAS-2B, des cellules épithéliales bronchiques transformées avec le virus SV-40 (CRL-9609, American Type Culture Collection (ATCC), VA, États-Unis) et les A549, des cellules issues d’une lignée de cellules épithéliales alvéolaires humaines provenant d’un adénocarcinome (CCL-185, ATCC). Les BEAS-2B ont été mises en culture dans du milieu complet contenant de l’EGF humain pour remplacer celui de rat. Les A549 ont été mises en culture dans du milieu DMEM (Wisent) avec 100 U/ml de pénicilline, 100 μg/ml de streptomycine (Wisent) et 10 % de FBS inactivé. Les cultures ont été maintenues dans un incubateur à 37 °C, 5 % CO2-95 % air.
Les CE bronchiques primaires provenaient de sujets sains (Bronchial normal epithelial cells; BNE) ou asthmatiques (Bronchial asthmatic epithelial cells; BAE) (fournies par Dre Chakir) [104]. Les patients normaux possédaient les caractéristiques suivantes : volume expiratoire forcé durant la première seconde (VEF1) moyen = 98,2 ± 5,1 % et une dose de métacholine requise pour diminuer la FEV1 de 20 % (PC20) moyenne = 99,4 ± 1,2 mg/ml, non atopiques et non-fumeurs. Les patients asthmatiques avaient les caractéristiques suivantes : VEF1 moyen = 85 ± 3,1 % et PC20 moyenne = 4,0 ±
2,2 mg/ml, n’utilisaient pas de corticostéroïdes inhalés ou systémiques. Tous les asthmatiques étaient non-fumeurs, atopiques et étaient positifs au test d’allergie aux acariens. Une lignée commerciale de CE bronchiques primaires humaines, des NHBE (Lonza, États-Unis), a été également utilisée. La culture de ces cellules a été effectuée avec les mêmes procédures que celles décrites pour les CE humaines de l’équipe du Dre Chakir [104].
3.2 Immunobuvardage de western
L’expression de S1PR1 par les CE pulmonaires a été mesurée par immunobuvardage de western. Du cerveau et du poumon de rats ont été utilisés comme contrôles positifs. Les lysats protéiques ont été obtenus avec du tampon de lyse Radio Immuno Precipitation Assay buffer (RIPA) (150 mM NaCl, 1 % Igepal, 0,5 % désoxycholate de sodium, 0,1 % SDS, 50 mM tris pH 8, 1x inhibiteur de protéase complet sans EDTA (complete, EDTA free Protease Inhibitor Cocktail Tablets) (Roche Diagnostics, Laval, QC, Canada), H2O).
Pour obtenir les lysats des tissus, du RIPA a été ajouté à environ 0,5 g de tissus. Les échantillons ont été ensuite homogénéisés avec un polytron, centrifugés à 16 000 g pendant 15 minutes à 4 °C et les surnageants ont été recueillis.
Les CE pulmonaires en culture ont été lavées avec du PBS froid et lysées avec du RIPA. Après 5 minutes de lyse avec du RIPA à 4 °C, les lysats ont été récupérés en grattant les flacons ou les plaques avec des grattoirs à cellules (Corning Incorporated, NY, États-Unis). Les lysats ont été transférés dans des tubes de 1,5 ml et homogénéisés à l’aide d’une pipette. Les lysats ont été centrifugés à 16 000 g à 4 °C pendant 15 minutes et le surnageant a été recueilli pour l’expérience [105]. La concentration protéique des lysats a été dosée avec la méthode du BCA (Bicinchoninic acid) (Pierce/Fisher Scientific Company, Ottawa, ON, Canada) [105].
Ensuite, un tampon de chargement dénaturant (250 mM Tris-Cl, 7,25 M β-mercaptoéthanol, 4 % SDS, 0,01 % bleu de bromophénol, 20 % glycérol et 1 mM dithiothréitol (DTT)) a été ajouté aux lysats dosés et les échantillons ont été dénaturés
29 pendant 5 minutes à 100 °C dans un bécher d’eau bouillante. Les protéines des lysats ont été séparées en utilisant la technique du SDS-PAGE sur gels de polyacrylamide 10 % dans du tampon de migration (25 nM tris, 192 nM glycine, 0,1 % SDS, H2O). Les
protéines du gel ont ensuite été transférées sur une membrane de polyfluorure de vinylidène (PVDF) dans un tampon de transfert (25 nM tris, 192 nM glycine, 20 % méthanol, H2O). La membrane contenant les protéines a été bloquée dans du tampon de
blocage contenant 5 % de lait en poudre sans gras dans une solution tampon (Tris 25 mM, 0,05 % tween 20 (TBST 0,05 %)) pendant 1 h.
L’immunobuvardage a été effectué pendant 18h, à 4 °C dans du tampon de blocage avec 100 ng/ml d’anti-α-tubuline (HRP) (ab40742 ; Abcam, Cambridge, MA, États-Unis), utilisé pour détecter la protéine servant de contrôle de chargement, ou 267 ng/ml d’anti-S1PR1 (S1P1 polyclonal antibody; 10005228; Cayman Chemical, MI, États-Unis) pour lier le récepteur [106]. Après 4 lavages de 5 minutes dans le TBST-0,15 %, une autre étape d’immunobuvardage a été effectuée pendant 1 h à la température de la pièce en utilisant 50-80 ng/ml d’un second anticorps, un anti-IgG lapin couplé à la peroxydase du raifort (α rabbit-HRP) (Peroxidase-conjugated AffiniPure Goat Anti-Rabbit IgG (H+L); 111-035-003, Jackson ImmunoResearch Laboratories, Inc., PA, États-Unis) dans du tampon de blocage pour lier l’anti-S1PR1. Les protéines ont été détectées avec la solution d’ECL Plus Detection Reagent (Amersham, Little Chalfont, Buckinghamshire, G-B) ou la solution d’ECL Crescendo (Fisher, Canada) et développées sur des films BioFlex MSI (Mandel Scientific, ON, Canada). L’expression différentielle de la protéine S1PR1 pour les NBEC et les ABEC a été quantifiée en utilisant le logiciel Image J (National Institutes of Health, MA, États-Unis) [106].
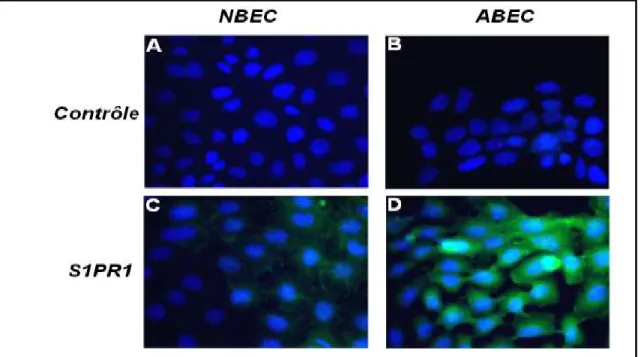
3.3 Immunofluorescence
L’expression de S1PR1 par les NBEC et les ABEC et la polymérisation de l’actine produite par l’activation de S1PR1 ont été observées en microscopie en fluorescence.
Les CE ont été ensemencées sur des lamelles en verre. À confluence, les CE ont été incubées pendant 18 h avec du milieu de stimulation composé de milieu complet sans toxine de choléra pour éviter l’accumulation intracellulaire d’adénylate cyclase responsable de l’activation de protéine G et contenant 1 % de FBS traité avec du charbon activé (Wisent) (pour éliminer les lipides dont la S1P. Ensuite, elles ont été stimulées avec des doses de 1E-08 M (NBEC/ABEC) et 1E-06 M (BEAS-2B) de CYM-5442 couplé à de l’acide tartrique (CYM-5442-tartrique) (fourni par Dr.Rosen, The Scripps Research Institute, CA, États-Unis) ou son véhicule (méthanol 0,1 %) pendant 5 minutes à 37 °C. Par la suite, les cellules ont été lavées avec du PBS pendant 5 minutes. Elles ont été fixées dans une solution de paraformaldéhyde 4 % pendant 10 minutes et perméabilisées avec du triton X-100 0,2 % pendant 5 minutes sur glace [84]. Après 2 lavages avec du PBS, les cellules ont été bloquées dans du tampon de blocage composé de 1 % d’albumine de sérum bovin (BSA) dans du PBS (PBS-BSA 1 %) pendant 1 h dans une chambre humide. Ensuite, les cellules ont été incubées avec 2 ug/ml d’anti-S1PR1 (EDG-1/H-60 ; SC-25489, Santa Cruz Biotechnology, CA, États-Unis) ou avec le véhicule (tampon de blocage) pendant 30 minutes [105]. Après 4 lavages au PBS, les lamelles ont été incubées dans le noir, dans une chambre humide à la température de la pièce pendant 20 minutes avec un anticorps secondaire anti-IgG de lapin couplé au FITC dans du tampon de blocage à une concentration finale de 2,5 μg/ml (FITC Donkey α-rabbit IgG; 406403; Biolegend, CA, États-Unis).
Les filaments d’actine polymérisée ont été détectés par la Phalloïdine-Tetramethylrhodamine B isothiocyanate (Phallaoidin TRITC, SC-301530, Santa Cruz Biotechnology). Pendant 30 minutes, les CE ont été incubées avec 0,165 nM de Phallaoidine couplée au TRITC dans du tampon de blocage à l’abri de la lumière dans une chambre humide. Ensuite, pendant 5 minutes, à l’abri de la lumière et à la température de la pièce, les CE ont été incubées avec 10 μM du 4',6-diamidino-2-phenylindole (DAPI ; D9542, Sigma-Aldrich) (filtré à 0,22 μm) pour détecter les noyaux cellulaires. Après les marquages, les lamelles ont été rincées 3 fois avec du PBS pendant 5 minutes.
31 Après les derniers lavages, les lamelles ont été montées sur des lames de microscope avec du milieu de montage anti-oxydant (CC/Mount Aqueous Mounting Medium, Sigma-Aldrich) et scellées avec du vernis à ongles commercial. Les analyses en immunofluorescence ont été effectuées avec un microscope Nikon E600 (Nikon Canada, Mississauga, ON, Canada) à des objectifs de 20x et 40x. Les photos prises selon les mêmes paramètres d’exposition et la fluorescence a été normalisée en fonction du contrôle où l’anticorps primaire a été omis, avec le logiciel Image J (National Institutes of Health).
3.4 Perméabilité paracellulaire
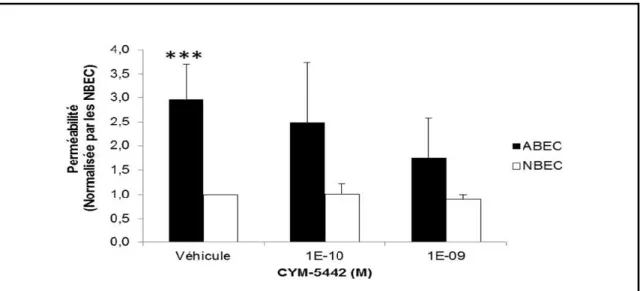
La modulation de la perméabilité paracellulaire des cellules NBEC et ABEC a été évaluée par diffusion au dextran-FITC (40 kDa) (Sigma-Aldrich) au travers de monocouches cellulaires suite à une activation de S1PR1 par l’agoniste CYM-5442-tartrique.
Les cellules ont été ensemencées sur des membranes perméables avec des pores de 0,4 μm fixées dans des cupules (BD Biosciences, San Diego, CA, USA) disposées dans des plaques 24 puits (BD Biosciences). À confluence, les monocouches cellulaires ont été incubées avec du milieu de stimulation. 18 h plus tard, les monocouches ont été lavées avec du PBS. Un volume de 200 μl de Dextran-FITC (4 mg/ml) contenant des doses croissantes de CYM-5442 (1E-10 M à 1E-09 M) ou de véhicule (Diméthylsulfoxyde (DMSO) 0,1 %) dans du milieu de stimulation a été ajouté dans la chambre supérieure sur la monocouche cellulaire et 800 μl de milieu de stimulation sans dextran fluorescent contenant les mêmes doses de CYM-5442 ou de véhicule a été ajouté dans le puits sous la cupule dans la chambre inférieure. 4 h plus tard, les échantillons des chambres inférieures et supérieures ont été prélevés pour quantifier la diffusion. La perméabilité d’une membrane a également été mesurée pour obtenir le niveau de base de diffusion du dextran. La quantité de dextran a été calculée en utilisant une courbe standard. Le dextran-FITC fluorescent prélevé dans les échantillons a été mesuré avec un lecteur de
fluorescence de la marque Fluoroskan Ascent FL (Thermo Fisher Scientific, MA, États-Unis) à une excitation à 520 nm et une émission à 490 nm.
La perméabilité a été estimée par la méthode de Kazakoff [107] où le coefficient de perméabilité (P, cm/h) a été calculé avec la formule suivante :
Où est le flux du dextran (μg/h), la concentration de dextran (μg/ml) et la surface de la membrane (cm2). Le de la monocouche seule a été estimé en séparant le coefficient de perméabilité de la membrane ( ) et celui de la couche cellulaire ( ) où le coefficient de perméabilité totale ( ) s’exprime avec la relation suivante :
3.5 Dosage de CCL2 par ELISA
La libération de CCL2 des CE bronchiques de rats (ABEC) et les CE pulmonaires humaines (BEAS-2B et A549) a été mesurée après l’activation de S1PR1 dans un contexte proinflammatoire. Ces cellules ont été traitées soit par l’analogue de la sphingosine, AAL-R (fourni par Dr.Rosen), soit l’agoniste spécifique de S1PR1, CYM-5442, ou un anti-inflammatoire servant de contrôle positif, la Dexaméthasone (Sigma-Aldrich) [47]. La modulation de la libération de CCL2 a été quantifiée par des essais de dosages immuno-enzymatique (Enzyme Linkage Immunosorbent Assay (ELISA)).
Les cellules ont été ensemencées dans des plaques de 24 puits (BD Biosciences) et les expériences ont été effectuées à 50 % de confluence et à 90-100 % de confluence. Les cellules ont d’abord été incubées pendant 18 h avec du milieu de stimulation, un milieu
33 sans lipide, avant d’être traitées avec des doses croissantes (de 1E-05 M à 1E-09 M) de modulateurs pharmacologiques dont le CYM-5442 (Tocris Biosciences, Bristol, United Kingdom), AAL-R ou la dexaméthasone, ou avec leur véhicule respectif du DMSO. Après 15 minutes de traitement, les cellules ont été stimulées ou non avec 25 ng/ml de TNF (Peprotech) pendant 18 h. La quantification de CCL2 a été effectuée sur les surnageants cellulaires avec des kits ELISA de type sandwich pour CCL2 (BD Biosciences) en suivant les protocoles fournis par le fabricant. La densité optique des échantillons a été mesurée en utilisant un lecteur de microplaque VERSAmax et le logiciel SoftMax Pro V.5.0.1 (Molecular Devices, CA, États-Unis).
3.6 Analyses statistiques
Les données sont exprimées en moyenne ± erreur-type. Les groupes ont été comparés en utilisant une ANOVA à 1 facteur avec des ajustements par le Test de Dunett. Les effets doses-réponses ont été évalués avec un post test de tendance linéaire. Une différence était considérée significative lorsque la valeur p (p-value) ≤ 0,05 bilatéralement. Les analyses statistiques ont été effectuées avec le logiciel GraphPad Prism V.5 (GraphPad Software, CA, États-Unis).
35
36
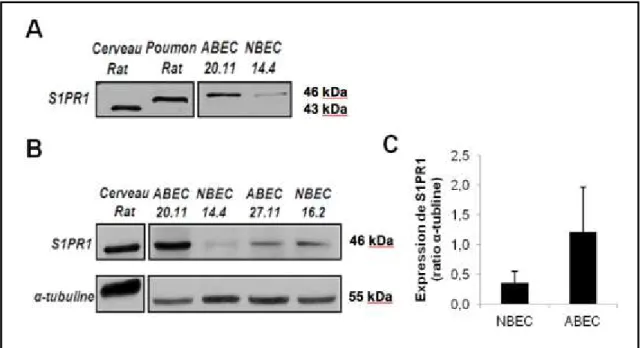
4.1 La protéine S1PR1 est exprimée dans des CE bronchiques de rats
La protéine S1PR1 est exprimée dans des lignées de CE [108] mais son expression dans les CE bronchiques n’avait jamais été caractérisée dans l’asthme. Les rats soumis à un protocole d’asthme induit par l’OVA présentent des analogies avec l’asthme humain [109]. En culture, les CE bronchiques primaires de rats asthmatiques (ABEC), comparativement à celles de rats naïfs (NBEC), présentent des dysfonctions telles qu’une augmentation de perméabilité paracellulaire et une libération importante de médiateurs proinflammatoires [communication personnelle, Dre Bissonnette] [44]. Pour confirmer la présence de S1PR1 dans les CE bronchiques de rats, l’expression de la protéine a été mesurée avec 2 techniques différentes, soit en immunobuvardage de type immunobuvardage de western et en immunofluorescence.4.1.1 Expression de la protéine S1PR1 en immunobuvardage de western
Dans une première série d’expérimentations, nous avons mesuré, par immunobuvardage de de western, l’expression de la protéine S1PR1 dans des lysats d’extraits totaux de poumons de rats naïfs et de CE bronchiques de rats naïfs (NBEC 14.4) et de rats atteints d’asthme expérimental induit par l’OVA (ABEC 20.11). Il est connu que la protéine S1PR1 est grandement exprimée dans le cerveau et dans le poumon [110]. Donc, ces tissus ont été utilisés comme contrôles positifs. Les résultats obtenus montrent que l’anticorps utilisé reconnaît une protéine entre 43 kDa dans le cerveau et 46 kDa dans le poumon qui correspond à S1PR1 (fig. 5A). L’anticorps reconnaît également une protéine correspondant à S1PR1 dans les NBEC et les ABEC. Nous avons ultérieurement mis en culture 2 autres lignées cellulaires provenant de rats normaux et asthmatiques pour quantifier l’expression de S1PR1 dans des quantités égales d’ABEC et de NBEC, relativement à une protéine contrôle, α-tubuline (fig. 5B). En normalisant en fonction de l’immunoréactivité de la tubuline, nous avons déterminé que les ABEC semblent exprimer 3,5 fois plus S1PR1 que les NBEC (fig. 5C). Cependant, la surexpression de S1PR1 varie selon la lignée et il faudra augmenter le nombre de lignées afin d’obtenir une moyenne d’expression fiable. Pour