Cellular immunotherapy in multiple myeloma : lessons from preclinical models
Texte intégral
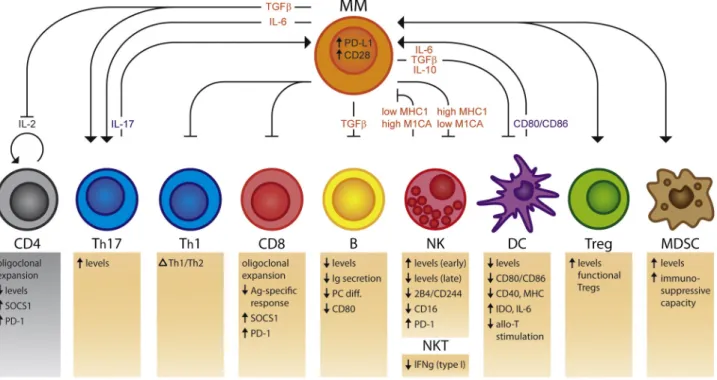
Figure
Documents relatifs
based on parvoviral vectors and hybrid cell vaccination.. Thèse présentée en vue de l’obtention du grade de Docteur en
L’accès à ce site Web et l’utilisation de son contenu sont assujettis aux conditions présentées dans le site LISEZ CES CONDITIONS ATTENTIVEMENT AVANT D’UTILISER CE SITE WEB.
In applications such as the polygonal reconstruction from a digital curve, we sometimes need to associate a representative Euclidean straight line to a preim- age.. In the
The article focuses on the organisation of the Linux traffic controller to investigate, over a classical Ethernet (wired) link, two typical properties of the air interface of an
Every day since 1930, time messages have been broadcast on the radio: Canada’s shortest but longest-running radio program: “At the beginning of the long dash…..” Having
• In comparison to the dynamic test protocol that simulates the fatigue effects on systems, the static test protocol overestimates the wind uplift resistance performance, due to
As mentioned in Fluorescence Quenching of An and Ph in Chloroform Solution section, both collisional and static quenching effects of ODSA on An and Ph in so- lution were much lower
Impeller tip speed: 0.7-2.0 m/s (hydrofoil), 1.0-2.9 m/s (turbine) Gas flow rate: 1.2-7 L/min (5L), 3.5-18 L/min (171L) Foam measurement: • Average of 4 diametrical line of
