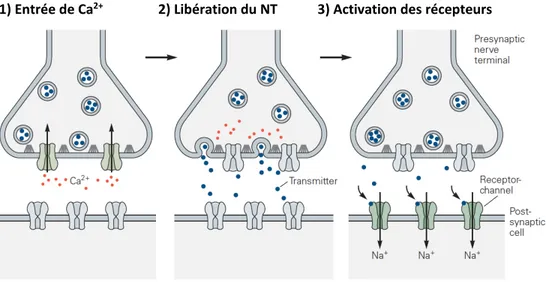
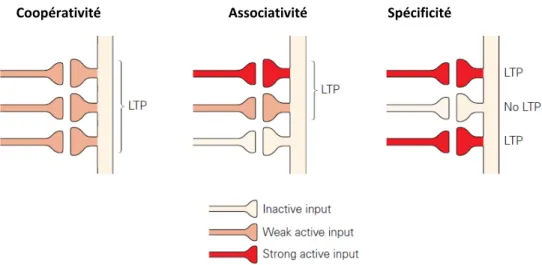
Implication de Syngap1 dans la transmission GABAergique et la plasticité synaptique
Texte intégral
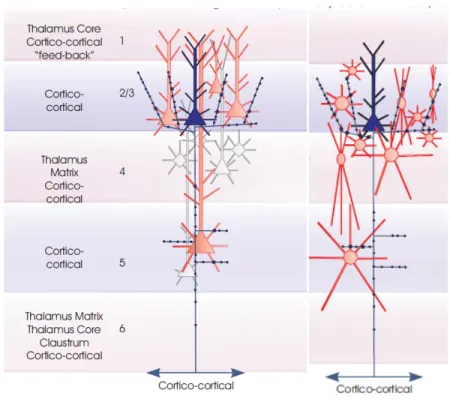
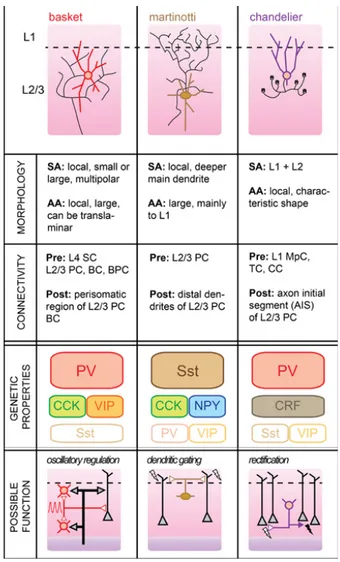
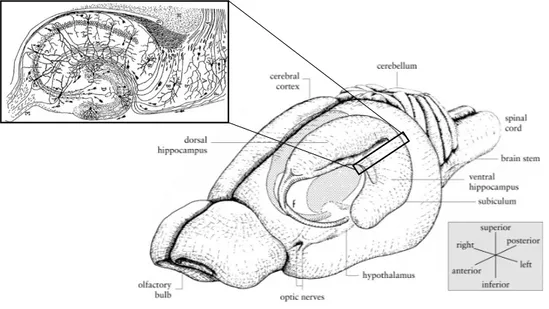
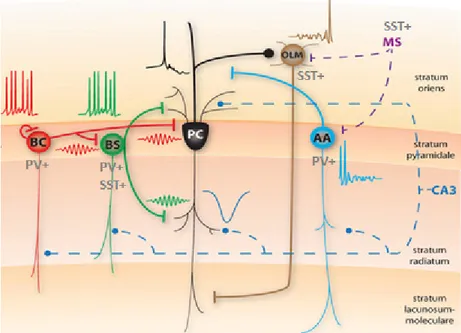
Figure
Documents relatifs
Il est ainsi possible d’associer l’augmentation de l’activation de la voie Notch avec la diminution du nombre de cellules caliciformes chez les souris
Pour évaluer les facteurs qui déterminent la transmission du paludisme dans la région nord-est de Madagascar, des enquêtes entomologiques ont été menées d’avril 1997 à mars
Objectif général : Étudier la synapse aux afférences excitatrices des SOM-INs provenant du rétrocontrôle des collatérales des cellules pyramidales de l’aire CA1 en utilisant
227 Dans notre modèle animal, la présence d’une hyperinsulinémie combinée à une hyperglycémie chez les souris du groupe de la DA nous permet de suspecter la présence
Chez les jeunes souris expérimentales, une diminution dans l'activation du sentier de signalisation MAP kinase ERK1/2 est détectée comparativement aux souris témoins
Dans cet article, en prenant en compte les voies de transmission multiples et la princi- pale source de transmission de la brucellose dans la Région autonome de Mongolie intérieure,
Les souris atteintes avaient, à l’état homozygote, une délétion dans la région 5’ du gène et la réintroduction du gène kl normal dans des embryons de souris homozygotes
Pour définir quelle phase de la mémoire est altérée chez les souris mutantes, nous avons employé une tâche d’identification de nouvel objet dépendant de l’hippocampe et
