Plasticité synaptique dans l’aire tegmentaire ventrale : implication des endocannabinoïdes
Texte intégral
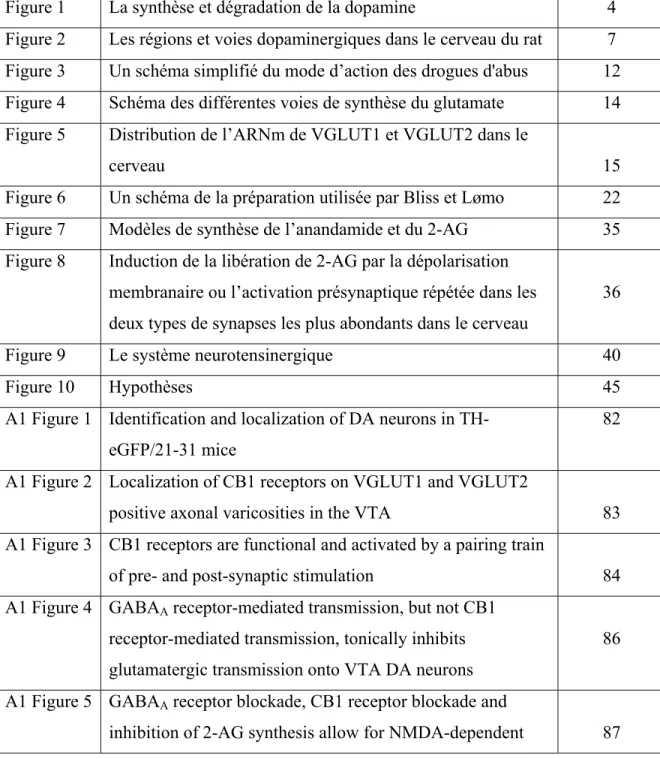
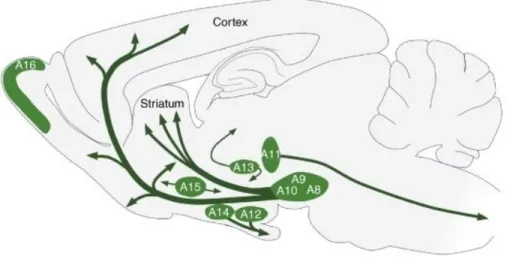
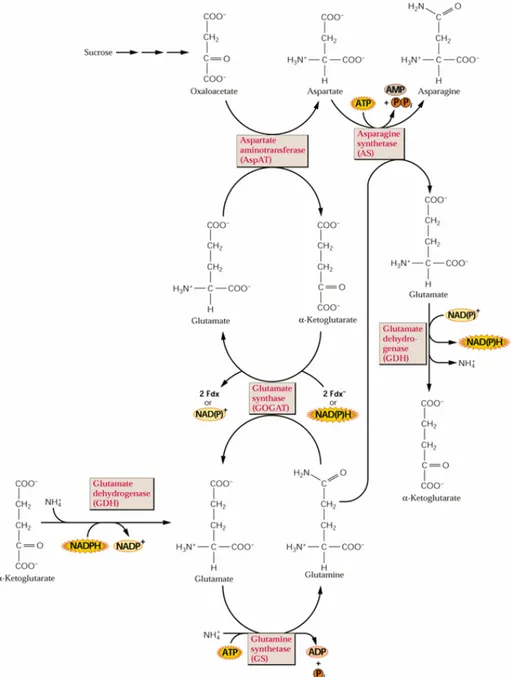
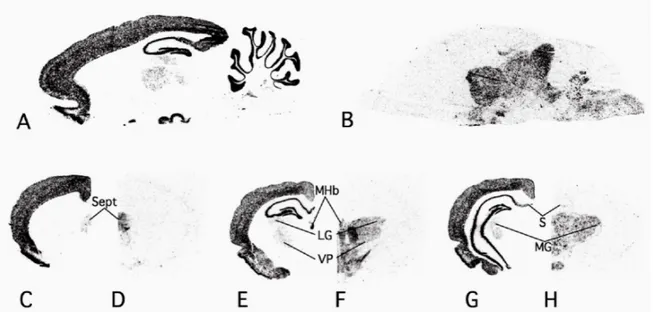
Figure

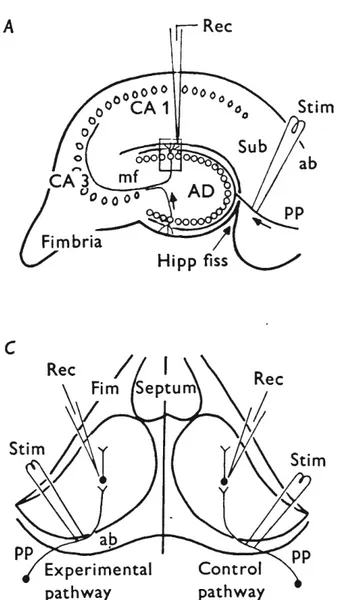

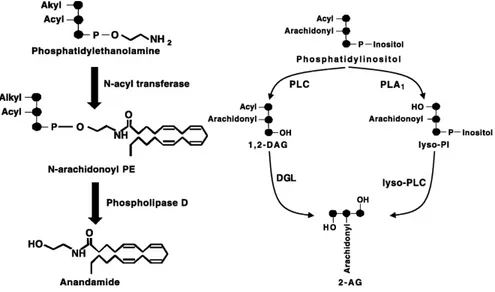
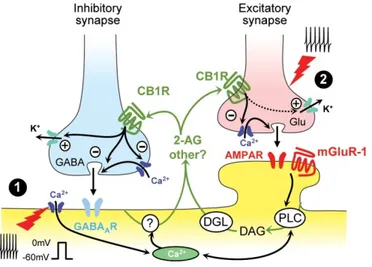
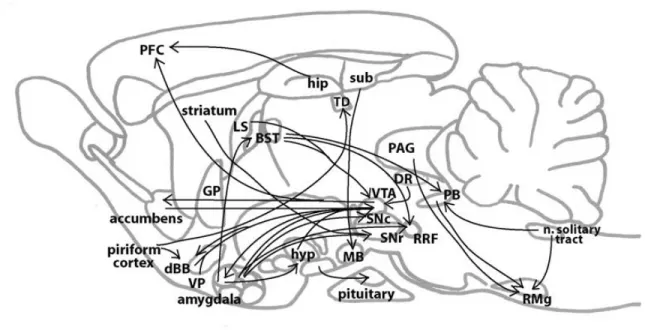
Documents relatifs
295 Ce qui fait que le nombre de chiffres utilisé pour transcrire le nombre oscille de quatre à douze ; un peu comme si le fait que la taille d’un nombre, dans le système
des pertes de recettes 75 , la couverture des dépenses occasionnées par la pandémie (renfort de personnel pour compenser l’absentéisme accru, frais logistiques, petit
L’organisation ainsi mise en place, avec ASL et AFP, a permis par la suite de déve- lopper beaucoup plus rapidement les activi- tés pastorales et de rentrer dans une
La néo-vascularisation des tumeurs cérébrales est directement corrélée avec leur agressivité, leur degré de malignité et la récurrence suite à l’exérèse de la tumeur (1). Les
Dans le sys- tème digestif, le gène Hoxc4 est impliqué dans la formation de l’œsophage, Hoxa5 dans celle de l’estomac, de l’intestin et du côlon, les gènes des groupes 12 et
La myélinisation est assurée par deux types cellulaires , les cellules d e Schwann e n périphérie e t les oligoden drocytes dans le système nerveux
