Université de Sherbrooke
Quantification simultanée des ARN codants et non-codants dans le séquençage d’ARN.
Par Vincent Boivin
Programme de Maîtrise en biochimie
Mémoire présenté à la Faculté de médecine et des sciences de la santé en vue de l’obtention du grade de maitre ès sciences (M. Sc.)
en biochimie
Sherbrooke, Québec, Canada Février 2018
Membres du jury d’évaluation
Michelle Scott, Département de Biochimie, FMSS, Université de Sherbrooke Sherif Abou Elela, Département de Microbiologie, FMSS, Université de
Sherbrooke
François Bachand, Département de Biochimie, FMSS, Université de Sherbrooke Sébastien Rodrigue, Département de Biologie, Faculté des Sciences, Université de
Sherbrooke
Je dédie mon mémoire à mes parents. C’est leur amour qui m’aura porté si loin.
« There is still plenty to learn and much work to be done. This is an exciting time for the study of RNA biology! »
Remerciements
Je tiens à remercier ma directrice Pr. Michelle Scott et mon co-directeur Pr. Sherif Abou Elela pour l’orientation et les conseils offerts au cours de mon projet. Ce projet n’aurait pas pu prendre forme sans l’expertise technique de Sonia Couture du laboratoire Abou Elela ainsi que de l’équipe du Pr. Lambowitz de l’University of Texas grâce auxquels les ensembles de données de séquençage ont pu être produits. Finalement, je tiens spécialement à remercier Gabrielle Deschamps-Francoeur du laboratoire Scott avec qui j’ai eu la chance de travailler sur ce projet pour son expertise et sa camaraderie.
1R
ÉSUMÉQuantification simultanée des ARN codants et non-codants dans le séquençage d’ARN. Par
Vincent Boivin Maîtrise en biochimie
Mémoire présent à la Faculté de médecine et des sciences de la santé en vue de l’obtention du diplôme de maitre ès sciences (M.Sc.) en biochimie, Faculté de médecine et des sciences
de la santé, Université de Sherbrooke, Sherbrooke, Québec, Canada, J1H 5N4
Les ARN sont des molécules aux propriétés diverses interagissant les uns avec les autres dans le but de médier des fonctions spécifiques. L’évaluation de l’abondance relative des ARN est une étape cruciale à la compréhension de la stœchiométrie nécessaire à ces fonctions. Cependant, plusieurs biais et limitations s’imposent avec l’utilisation de différentes techniques d’évaluation, dont le séquençage d’ARN (RNA-Seq), qui sont problématiques dans l’estimation de l’abondance de différents types d’ARN. De récentes études ont exposé les avantages d’un protocole novateur de RNA-Seq qui utilise une rétrotranscriptase thermostable d’intron de groupe II (TGIRT) d’origine bactérienne afin de réduire ces biais. Ce mémoire fait la comparaison entre différentes techniques de RNA-Seq afin d’élucider si l’utilisation de TGIRT en RNA-Seq offre une représentation plus juste du transcriptome. Les comparaisons avec les valeurs d’abondance de différents types d’ARN décrites dans la littérature ainsi qu’obtenues expérimentalement par notre groupe pointent au fait que TGIRT donne une meilleure estimation de l’abondance relative des ARN, et plus particulièrement des ARN hautement structurés. Cette meilleure estimation de l’ensemble de la composition du transcriptome permet de faire des observations sur les rapports d’abondance entre des ARN codants et non-codants fonctionnellement apparentés. Notamment, des ratios d’expression constants entre les ARN non-codants associés à des RNP et les ARNm codants pour leur facteurs protéiques ont été observés. Ceci suggère la présence d’une régulation transcriptionnelle commune nécessaire à la stœchiométrie de ces complexes. Une forte disparité dans l’expression des snoRNA et de leurs gènes hôtes, dépendant du type de snoRNA et de gène hôte a par ailleurs été constatée, corroborant une régulation distincte de la stabilité de ces transcrits. Dans l’ensemble, nos données suggèrent que la méthode TGIRT-Seq est la plus appropriée dans l’évaluation du transcriptome entier et ouvre donc la voie à des analyses plus holistiques par RNA-Seq en donnant une estimation plus juste de l’abondance relative des transcrits d’ARN.
Mots clés : ARN, transcriptomique, RNA-Seq, bio-informatique, snoRNA, ARN non-codants, rétrotranscriptase, TGIRT
2S
UMMARYSimultaneous quantification of coding and non-coding RNA in RNA sequencing.
By Vincent Boivin
M.Sc. in biochemistry Program
Thesis presented at the Faculty of medicine and health sciences for the obtention of Master degree diploma maitre ès sciences (M.Sc.) in biochemistry, Faculty of medicine and health
sciences, Université de Sherbrooke, Sherbrooke, Québec, Canada, J1H 5N4
RNA are molecules with a wide range of properties that can interact with one another to mediate specific function. The evaluation of RNA abundance is a crucial step in understanding the stoichiometry needed for these functions. However, many limitations and biases come with the use of different techniques, including RNA sequencing (RNA-Seq), which affects the estimation of the abundance of different RNA types. Recent studies have exposed the advantages of a new RNA-Seq protocol using a thermostable group II intron reverse transcriptase (TGIRT) of bacterial origin to reduce these biases. This thesis makes the comparison between different RNA-Seq techniques to elucidate if the use of TGIRT in RNA-Seq offers a more representative depiction of the transcriptome. The comparisons with the abundance values given in literature and obtained experimentally by our group agree with the fact that TGIRT gives a better estimation of the relative abundance of RNA, especially highly structured RNA. This better estimation of the transcriptomic landscape allows many observations on the abundance relations between coding and non-coding RNA that are functionally related. Namely, constant expression ratios between RNP associated noncoding RNA and the mRNA that codes for their associated proteins have been observed. This suggests the presence of a common transcriptional regulation which is necessary for the stoichiometry of these complexes. A strong disparity in the expression of snoRNA and their host genes depending on snoRNA and host gene types has also been observed and corroborate a distinct regulation of these transcripts’ stability. In summary, our data suggest that the TGIRT-Seq method is the most appropriate to evaluate the transcriptome and thus opens the way to more holistic RNA-Seq analyses by giving a better estimation of RNA transcripts relative abundance.
Keywords : RNA, transcriptomics, RNA-Seq, bioinformatics, snoRNA, noncoding RNA, reverse transcriptase, TGIRT
3T
ABLE DES MATIÈRES1 Résumé ... vi
2 Summary ... vii
3 Table des matières ... viii
4 Liste des figures ... x
5 Liste des tableaux ... xi
6 Liste des abréviations ... xii
1 Introduction ... 1
Historique transcriptomique ... 1
La diversité de l’ARN ... 2
Les ARN codants ... 4
Les ARN non-codants ... 5
Abondance et impact fonctionnel : connecté mais non-corrélé ... 7
Les petits ARN nucléolaires ... 8
Biogénèse et interacteurs des snoRNA ... 9
Les fonctions non-canoniques ... 11
RNP, abondance relative et fonctionnalité ... 13
Le RNA-Seq : méthode et biais ... 15
Variantes protocolaires du RNA-Seq ... 17
Le désaccord du séquençage et de la littérature ... 19
TGIRT à la rescousse des ARN hautement structurés ... 20
Hypothèse/problématique ... 23 Objectifs ... 23 2 Article ... 24 ABSTRACT... 26 INTRODUCTION ... 27 RESULTS ... 30 DISCUSSION ... 50
MATERIAL & METHODS ... 58
3 Discussion ... 71
Vers une meilleure détection des ARN hautement structurés en RNA-Seq. ... 71
Une ouverture sur l’analyse comparative de l’abondance des snoRNA, transcrits hôtes et transcrits cibles. ... 73
Une régulation élaborée de l’expression de différents ARN pour la formation des RNP. ... 75
Une ère d’analyse transcriptomique holistique ... 75
L’avenir du RNA-Seq : limiter les biais. ... 76
Perspectives sur le TGIRT-Seq et l’étude des snoRNA. ... 80
Conclusion ... 81
4 Liste des références ... 83
4L
ISTE DES FIGURESFigure 1 : Distribution transcriptomique ... 3
Figure 2 : Schématisation des snoRNA ... 9
Figure 3 : Biogénèse des snoRNA ... 10
Figure 4 : Interacteurs des snoRNA ... 11
Figure 5 : Pipeline RNA-Seq; de l’ARN aux analyses. ... 17
Figure 6 : Introns de groupe II. ... 21
5L
ISTE DES TABLEAUX6L
ISTE DES ABRÉVIATIONS ADN ADNc ARN ARNm ARNr ARNt lncRNA miARN NMD PCR piRNA qPCR RNA-Seq RNP RT scaRNA snRNA sncRNA snRNP snoRNA snoRNP TGIRT TGIRT-SeqAcide désoxyribonucléique, Deoxyribonucleic acid ADN complémentaire, complementary DNA Acide ribonucléique, Ribonucleic acid ARN messager, Messenger RNA ARN ribosomal, Ribosomal RNA ARN de transfert, Transfer RNA
Long ARN non-codant, Long non-coding RNA Micro ARN, Micro RNA
Dégradation des ARNm non-sens, Nonsense-mediated-decay Réaction en chaîne par polymérase, Polymerase chain reaction ARN interagissant avec Piwi, Piwi-interacting RNA
PCR quantitative, quantitative PCR Séquençage d’ARN, RNA sequencing Ribonucléoprotéine, Ribonucleoprotein Rétrotranscriptase, Reverse transcriptase
Petit ARN du corps de Cajal, Small Cajal body RNA Petit ARN nucléaire, Small nuclear RNA
ARN non-codant structuré, Structured non-coding RNA Petite RNP nucléaire, small nuclear RNP
Petit ARN nucléolaire, Small nucleolar RNA Petite RNP nucléolaire, Small nucleolar RNP
Rétrotranscriptase thermostable d’intron de groupe II,
Thermostable Group II intron reverse transcriptase
1I
NTRODUCTIONHistorique transcriptomique
Les acides ribonucléiques (ARN) sont une classe de molécules aux propriétés étendues qui sont à la base des mécanismes essentiels des êtres vivants. Que ce soit pour véhiculer l’information codante pour les protéines, faire l’épissage, médier la traduction ou réguler l’expression génique, les différents ARN d’une cellule interagissent et forment des complexes avec d’autres molécules dans le but d’effectuer ces fonctions.
L’ARN devint le centre d’attention d’un nombre croissant de chercheurs après que ces molécules furent démontrées comme étant capables d’encoder de l’information nécessaire à la synthèse de protéines en 1961 (F. Jacob et J. Monod, 1961, F. Gros, 1961). Une vision dichotomique de l’ARN est née à cette époque, présentant des gènes codants contre des gènes non-codants qualifiés de « junk ». Dans le dogme de la biologie moléculaire (Crick, 1970), les ARN étaient vus comme des intermédiaires entre l’information génétique de l’ADN et les protéines considérées comme source des diverses fonctions et structures cellulaires.
Cette vision s’est progressivement estompée par des découvertes qui pointèrent au fait que des ARN peuvent participer à des fonctions cellulaires essentielles seulement par leur séquence nucléotidique. Les ARN de transfert (ARNt) en sont le premier exemple caractérisé avec leur rôle essentiel dans le transport spécifique d’acides aminés (Rich & Rajbhandary, 1976). Ensuite vint la découverte des petits ARN nucléaires (snRNA) (Weinberg & Penman, 1968) qui furent montrés comme étant composantes de complexes ribonucléoprotéiques (RNP), essentiels entre autres à l’épissage des ARN messagers (ARNm) (Busch et al., 1982 ; Lerner et al. 1980). Plus tard, les micro ARN (miARN) furent découverts et exposés comme régulateurs de l’expression des ARNm par un nouveau mécanisme : l’interférence à ARN (Lee et al., 1993 ; Robinson, 2009). Mais la découverte qui donna le coup de grâce au dogme qui plaçait les ARN seulement comme des véhicules d’information génétique fut la découverte que les ARN peuvent, comme les protéines, médier des fonctions catalytiques (ribozymes) (Cech et al., 1981 ; Guerrier-takada 1983). C’est d’ailleurs cette découverte qui vint expliquer la présence d’ARN au sein du ribosome, ce que les biologistes de l’époque arrivaient mal à s’expliquer d’autre façon que par un rôle d’échafaudage (Cech & Steitz,
2014 ; Moore & Steitz, 2002). Avec l’avènement du séquençage et l’annotation progressive des éléments fonctionnels du génome par le consortium ENCODE («ENCyclopedia Of DNA Elements») (Birney et al., 2007), il devint évident que les séquences codantes au sein du génome humain ne représentent qu’une minorité (environ 2%). Le développement de la caractérisation des ARNr, des ARNt, des miARN et autres, démontra des rôles inédits et interreliés avec ceux des ARNm, poussant d’avant l’intérêt de la communauté scientifique pour les ARN non-codants. C’est ainsi qu’une vision plus holistique de l’ARN est née, venant avec un intérêt croissant vers l’interrelation entre ces différents types d’ARN non-codants et codants ainsi que leurs rôles partagés au cœur de différentes voies cellulaires, dans la synthèse et la fonction de complexes RNP et leurs implications dans différentes pathologies.
La diversité de l’ARN
Le transcriptome humain tel qu’il est connu à ce jour est composé d’une multitude d’ARN de types, de représentations génomiques ainsi que d’abondances très diverses (Figure 1, Tableau 1) (Palazzo & Lee, 2015). Le transcriptome est historiquement divisé en deux portions, soit les ARN codants pour des protéines (ARNm) et les ARN non-codants qui eux ont des fonctions variées, allant de la régulation de l’expression génique jusqu’à la traduction des protéines. Le niveau de transcription varie beaucoup pour ces différents ARN, passant de plusieurs millions de transcrits à moins d’une centaine par cellules ce qui peut en soit être un indicateur de leur relation avec d’autres composantes cellulaires ainsi que leur fonction.
Figure 1 : Distribution transcriptomique
Distribution de la masse d’ARN totale et du nombre de molécules d’ARN par classes d’ARN (en moyenne) dans les cellules de mammifères. Valeurs tirées de Palazzo & Lee, 2015
Biotype du gène Nombre de gènes annotés
ARN messagers (ARNm) 20286
Longs ARN non-codants (lncRNA) 14665
Pseudogènes 14589
Micro ARN (miARN) 1524
Petits ARN nucléaires (snRNA) 1899
Petits ARN nucléolaires (snoRNA) 943
Petits ARN du corps de Cajal (scaRNA) 53
ARN de transfert (ARNt) 650
ARN 7SK 301
ARN 7SL 850
Autres 2817
Tableau 1 : Distribution génomique
Description des différents types gènes codants pour différents types d’ARN (biotypes) représentés dans l’annotation d’Ensembl (v.87) (Aken et al. 2016) (complémentée par GtRNAdb (Chan & Lowe, 2009) pour les annotations d’ARNt) et du nombre de gènes annotés associés à chaque biotype.
Les ARN codants
Les ARN codants ou ARNm sont sans équivoque la classe d’ARNm la plus étudiée à ce jour. Ils sont caractérisés par une coiffe 7-methylguanosine en 5’ (Shatkin, 1976), une queue poly-A en 3’ (Edmonds et al., 1971) ainsi qu’une région codante pour une protéine débutant habituellement par un codon méthyl AUG et finissant par un codon stop UGA, UAG ou UAA. Chez les eucaryotes, les pré-ARNm doivent subir certaines étapes de maturation afin de devenir fonctionnels. L’addition de la coiffe 5’ et de la queue poly-A sont essentiels pour protéger l’ARNm de la dégradation et servent de signal pour l’export du noyau (Guhaniyogi & Brewer, 2001). L’épissage, soit l’excision des introns et la conservation des exons, est un mécanisme essentiel dans la production de protéines fonctionnelles. Ce processus peut s’effectuer de différentes façons, de sorte à conserver ou exciser différents exons ou introns, influençant ainsi le produit protéique final; c’est l’épissage alternatif (Black, 2003). L’épissage alternatif permet de produire plusieurs isoformes à partir d’un même pré-ARNm et augmente ainsi grandement le nombre de protéines produites par le génome codant. La fonction essentielle des ARNm et de leur séquence codante fait en sorte que les ARNm ont un fort niveau de conservation ainsi qu’un taux d’expression très diversifié mais généralement élevé.
Cependant, malgré tout l’intérêt que ces ARN suscitent, il semble que notre connaissance de ces derniers demeure limitée. En effet, de par la longueur excessive de leurs séquences (plusieurs milliers de nucléotides), il est difficile d’évaluer leur structure secondaire par des méthodes classiques. La structure des ARNm a été suggérée autant comme étant capable de modifier l’initiation de la traduction (Faure et al., 2016 ; Iserentant & Fiers, 1980) que comme contrôle du repliement des protéines (Babendure et al., 2006). Il est donc possible que leurs structures soient associées à des fonctions encore inconnues. En ce sens, malgré le rôle unique communément accepté des ARNm comme véhicule d’information codante, plusieurs études ont montré que les ARNm pouvaient avoir des fonctions autres que la traduction de protéine; ces ARNm sont dits bifonctionnels (Kloc et al., 2011). Ces fonctions non-canoniques des ARNm comprennent entre autres l’assemblage de corps nucléaires (Shevtsov & Dundr, 2011), l’assemblage de filaments de cytokératine et d’actine dans les oocytes de
complexes cytoplasmiques essentiels à la détermination de l’axe antéro-postérieur lors de l’embryogénèse chez Drosohpila melanogaster (Jenny et al., 2006). Ces découvertes ouvrent la voie sur une toute nouvelle perspective d’étude fonctionnelle des ARNm et rendent ainsi plus floue la frontière arbitraire entre les ARN codants et non-codants.
Les ARN non-codants
Les ARN non-codants forment un groupe hétéroclite ayant pour seule caractéristique commune de ne pas encoder de protéines. On reconnaît parmi eux des ARN aux fonctions essentielles et bien connues comme les ARNr, les ARNt, les snRNA, etc. On trouve au sein de ce groupe des ARN aux tailles diverses passant d’une vingtaine de nucléotides pour les microARN à plusieurs milliers pour les long ARN non-codants (lncRNA). Pour ce qui est de l’expression, la plupart ont un taux de transcription faible alors qu’une minorité de transcrits sont fortement exprimés. On observe que le niveau d’expression des ARN non-codants corrèle grossièrement avec leur taux de conservation (Managadze et al., 2011), ce qui peut potentiellement être indicateur d’un impact fonctionnel minime pour plusieurs ARN non-codants faiblement exprimés. Les pseudogènes en sont un bon exemple : ces gènes sont en fait des copies de gènes codants pour des protéines dont la redondance fonctionnelle a ultimement mené à leur dégénérescence et à la réduction sélective de leur transcription (Mighell et al., 2000). Bien que la plupart des pseudogènes n’ont aucune fonction connue et sont faiblement conservés, ils ne sont pas pour autant inutiles en ce qu’ils permettent à l’évolution de nouvelles fonctions. De plus en plus de groupes de recherche exposent ces fonctions nouvelles et distinctes des pseudogènes par rapport au transcrit codant d’origine, incluant de servir de leurres pour les microARN ciblant leur transcrit codant correspondant et de produire de petits ARN interférents (Tutar, 2012 ; Pink et al., 2011 ; Balakirev & Ayala, 2003).
Une autre explication du manque de conservation de la séquence de plusieurs types d’ARN non-codants peut provenir du fait que la structure est l’élément fonctionnel principal de ces ARN. En effet, la structure des ARN est un aspect particulièrement important dans l’étude fonctionnelle des ARN. Les structures secondaires et tertiaires sont au cœur de mécanismes
essentiels comme la formation de ribosomes fonctionnels ainsi que le transfert des acides aminées par les ARNt. Ceci dit, les nucléotides de part et d’autre d’un appariement peuvent tous deux muter de manière à rester complémentaires ce qui conserve effectivement la structure sans nécessairement conserver la séquence; c’est la covariation (Parsch et al., 2000).
La covariation est de plus en plus avancée pour expliquer les longs ARN non-codants (lncRNA). Les lncRNA ont des niveaux d’expression et de conservation inférieurs aux ARNm, mais ils démontrent une spécificité cellulaire supérieure à ces derniers et plusieurs lncRNA ont des fonctions essentielles malgré leur rapide évolution au niveau de la séquence (Ulitsky & Bartel, 2013 ; Ulitsky et al., 2011). Notamment, XIST, qui est lncRNA le plus étudié pour son rôle essentiel dans l’inactivation du chromosome X (Plath et al., 2002), possède un fort taux de conservation au niveau de la structure mais un faible taux de conservation au niveau de la séquence (Nesterova et al., 2001).
À l’inverse, certains ARN non-codants sont exprimés de manière opulente et sont conservés à travers tous les domaines du vivant. Parmi ces ARN, on remarque particulièrement les ARN ribosomaux, qui composent à eux seuls la majeure partie de la masse d’ARN au sein d’une cellule (jusqu’à 90% de la masse moléculaire totale d’ARN). Mais en termes de nombre de transcrits, ils se font devancer par une autre classe d’ARN : les ARN de transfert. Ces derniers sont beaucoup plus petits (~75 nts) et composent moins de 10% de la masse moléculaire mais représentent à eux seuls approximativement 90% du transcriptome en nombre de transcrits (Palazzo & Lee, 2015). Un autre ARN non-codant qui figure parmi les plus abondants dans la cellule est l’ARN de la particule de reconnaissance du signal (SRP) ou ARN 7SL qui possède à lui seul 850 copies génomiques chez l’humain. SRP est essentielle à la reconnaissance d’un peptide signal spécifique présent à l’extrémité N-terminale des protéines. Une fois le signal détecté, le complexe SRP arrête la traduction et achemine la protéine et son ribosome associé vers le réticulum endoplasmique, à l’intérieur duquel elle sera maturée (Walter & Blobel, 1982).
Abondance et impact fonctionnel : connecté mais non-corrélé
Malgré le niveau d’expression élevé de certains ARN essentiels comme les ARNr, les ARNt et les ARN 7SL causé par des promoteurs forts ainsi qu’une quantité élevée de copies génomiques, l’abondance n’est pas en soit un paramètre qui devrait être utilisé pour déterminer l’impact fonctionnel d’un ARN dans une cellule. En effet, certains ARN sont particulièrement conservés mais n’expriment qu’une dizaine de molécules par cellules et n’en demeurent pas moins indispensables à la survie des cellules. À titre d’exemple, l’ARNm de la protéine hTERT, la rétrotranscriptase des télomères, a une abondance cellulaire d’environ 1 à 20 molécules chez les cellules non cancéreuses (Cao et al., 2008 ; Yi et al., 2001). En effet, hTERT n’a pas besoin d’être présent en million de molécules dans une cellule, le nombre de protéines de hTERT dans une cellule étant estimé à ~500 molécules et sa composante ARN associé, hTERC, étant estimé à ~1000 molécules par cellules (Xi & Cech, 2014). En effet, la cellule n’a besoin que d’un faible nombre de télomérases fonctionnelles pour allonger ses bouts de chromosomes qui sont peu nombreux (92 télomères pour 46 chromosomes). À l’inverse, les ribosomes doivent être présents en très forte quantité pour pouvoir faire la traduction de la totalité des ARNm dans une cellule. La production d’ARN ribosomaux est donc ahurissante avec une abondance de 3 à 10 millions par cellules et les ARNm codants pour les protéines ribosomales sont régulés de concert et figurent parmi les ARNm les plus abondants dans la cellule (Li et al., 2016 ; Perry, 2005). Les ARN de transfert servent quant à eux de substrat au ribosome, leur abondance est donc environ 10 fois supérieure à celle des ARNr, faisant d’eux la classe d’ARN la plus abondante dans la cellule (Figure 1). L’expression des différentes composantes d’un même complexe comme la télomérase et le ribosome doivent être précisément régulés afin d’établir une stœchiométrie appropriée à leur fonctionnalité et d’éviter une surproduction de composantes qui serait inutilement coûteuse au niveau énergétique pour la cellule (Kafri et al., 2016 ; Wagner, 2005). Le niveau d’abondance d’un ARN, aussi extrême puisse-t-il être, n’est pas révélateur de son importance mais plutôt de la stœchiométrie nécessaire à sa fonction.
Ceci dit, la relation entre l’abondance perçue des différents types d’ARN associés aux RNP et leur fonction attendue n’est pas toujours évidente et peut être révélatrice de notre méconnaissance de certains RNP. Les petits ARN nucléolaires (snoRNA), composantes des
snoRNP, en sont un bon exemple. Malgré la fonction canonique essentielle des snoRNP dans la modification chimique des ARNr, les snoRNA qui les composent ont des niveaux d’expression très hétéroclites qui viennent contre l’intuition que leur seule fonction est de maturer les ARNr. En fait, cette hétérogénéité transcriptionnelle concorde plutôt avec les récentes découvertes de fonctions non-canoniques leurs étant associés (Dupuis-Sandoval et
al., 2015) et pointe au fait qu’il reste beaucoup à découvrir sur le rôle de ces ARN dans la
cellule.
Les petits ARN nucléolaires
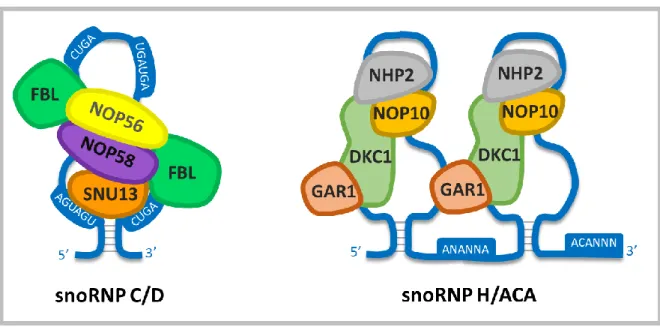
Les snoRNA sont une classe de petits ARN non codants auxquels notre groupe est particulièrement intéressé. Ils sont divisés en deux groupes : Les boîtes C/D qui ont habituellement une taille d’environ 70 nucléotides et les boîtes H/ACA qui eux ont une taille d’environ 120 nucléotides. Les boîtes C/D sont spécialement connus pour médier la méthylation avec un appariement aux séquences guides en amont des boîtes D et D’ (Figure 2) alors que les boîtes H/ACA quant à eux médient plutôt la pseudouridylation avec un appariement bipartite à l’ARN ribosomal (Figure 2). Pour effectuer ces modifications chimiques, les snoRNA ont besoin de plusieurs partenaires protéiques spécifiques qui assurent tant la maturation de ces ARN que leur fonction et leur stabilité.
Figure 2 : Schématisation des snoRNA
Schéma décrivant les différences structurelles principales entre snoRNA C/D et H/ACA. Les séquences cibles sont montrées en rouge avec leur modification chimique associée.
Biogénèse et interacteurs des snoRNA
Les snoRNA sont conservés à travers tous les règnes eucaryotes ainsi que chez les Archéobactéries (Omer et al., 2000). Cependant, leur mode d’expression à travers ces différentes lignées est très diversifié. Chez la levure, les snoRNA sont surtout liés à des promoteurs dépendants de la Pol II, alors que chez certaines plantes on les retrouve liés à des promoteurs dépendants de la Pol III (Dieci et al., 2009). Ils peuvent être exprimés de manière individuelle, ou parfois, comme chez D. melanogaster, ils sont exprimés en groupe allant jusqu’à une dizaine de snoRNA (Boivin et al., 2017). Chez l’humain, la vaste majorité des snoRNA sont des gènes qui n’ont aucun promoteur et qui sont individuellement contenus à l’intérieur d’introns de gènes encodant ARNm ou lncRNA (Boivin et al., 2017). Ces gènes contenant des snoRNA sont communément appelés gènes hôtes. Le mode d’expression des snoRNA est donc entièrement dépendant de la transcription du gène hôte. Le snoRNA est séparé du transcrit hôte lors de l’épissage de l’intron contenant le snoRNA, générant donc un transcrit distinct de celui du transcrit hôte (Figure 3). Ceci dit, l’abondance cellulaire du snoRNA et de son transcrit hôte diffèrent par des régulations sur la stabilité des transcrits. Par exemple, les transcrits hôtes de plusieurs snoRNA sont dégradés par la voie du
régulation derrière cette distinction entre l’abondance du transcrit hôte et du transcrit imbriqué demeure mal comprise et est pourtant essentielle considérant que ces gènes peuvent avoir des fonctions totalement distinctes (revu dans Boivin et al., 2017).
Figure 3 : Biogénèse des snoRNA
Schéma montrant l’excision d’un snoRNA comme intron de son transcrit hôte (1) menant ultimement à sa maturation par liaison de protéines spécifiques aux snoRNP (2) ainsi qu’à la dégradation des séquences flanquantes par des exonucléases (3).
Différentes protéines spécifiques au type de snoRNA viennent protéger le snoRNA afin d’empêcher qu’il soit dégradé comme le reste de l’intron par des exonucléases (Lafontaine et Tollervey, 1999). C’est l’ensemble du snoRNA et de ses protéines liées qui forme un snoRNP fonctionnel. Le complexe snoRNP de type C/D est composé de SNU13, NOP56, NOP58 et de la fibrillarine (FBL), cette dernière est responsable de la méthylation de la cible par son action méthyltransférase (Figure 4) (Newman et al., 2000). Pour les H/ACA, deux amalgames protéiques composés de NHP2, NOP10, GAR1 et de la dyskerine DKC1 se forment sur chaque tige boucle de la structure du snoRNA (Figure 4) (Khanna et al., 2006). DKC1 est responsable de la pseudouridylation de l’ARN ciblé. La composition protéique des snoRNP n’a seulement été validée pour une faible quantité de snoRNA. Une étude de notre groupe montre que cette composition pourrait être variable et spécifique à chaque snoRNA et être révélatrice de fonctions non-canoniques (Deschamps-Francoeur et al., 2014).
Figure 4 : Interacteurs des snoRNA
Schéma montrant les différentes composantes protéiques associées aux snoRNP C/D et H/ACA.
En effet, alors que la majorité des snoRNA servent à la modification des ARN ribosomaux, plusieurs centaines de snoRNA chez l’humain n’ont aucune cible connue et sont donc dits orphelins. Certaines publications récentes montrent que des snoRNA orphelins ou même des snoRNA avec des cibles sur l’ARNr, auraient des fonctions inédites dites « non-canoniques ».
Les fonctions non-canoniques
Parmi les fonctions non-canoniques des snoRNA (décrites dans Dupuis-Sandoval et al., 2015), on retrouve des modifications de l’épissage, l’altération de la stabilité de transcrits cibles ou des modifications chimique d’autres types d’ARN. Une de ces premières fonctions non-canoniques a été montrée chez les snoRNA de la famille des SNORD115. Les SNORD115 ont été montrés comme étant capables de modifier l’épissage alternatif du récepteur de sérotonine 5-HT2CR (Kishore et Stamm, 2006). Un déficit de l’expression du locus exprimant les snoRNA SNORD115 et SNORD116 serait au cœur du syndrome de Prader-Willi (Sahoo et al., 2008 ; Bieth et al., 2015). Cette condition génétique est caractérisée par une hyperphagie et un retard mental, c’est aussi la première cause génétique
d’obésité morbide chez les enfants. Un autre exemple d’une telle altération provient du snoRNA SNORD88C qui modifierait, quant à lui, la rétention des exons 8 à 10 du récepteur de facteur de croissance de fibroblastes FGFR3 (Scott et al., 2012). La forme Δ8-10 de FGFR3 serait un facteur anti-oncogène limitant la prolifération cellulaire et dont l’expression serait fortement diminuée dans des cellules de carcinome agressif de la vessie (Tomlinson et
al., 2005).
Pour ce qui est d’altérer la stabilité, ou du moins, l’abondance de transcrits spécifiques, d’autres études ont montré que certains snoRNA pourraient potentiellement créer des fragments de petites tailles. Ces fragments, similaires à des miARN, cibleraient des transcrits spécifiques et mèneraient à leur dégradation. D’une autre part, une étude a montré que le snoRNA SNORD63 produit un fragment de type piRNA (ARN intéragissant avec les protéines Piwi), qui donc, plutôt que de faire son effet dans le cytosol par l’intermédiaire de la machinerie d’ARN interférence comme un miARN (Agrawal et al., 2003) fera plutôt son effet dans le noyau à l’aide des protéines Piwi (Seto et al., 2007). Les piRNA sont également plus longs que les miARN avec une taille entre 26 et 31 nucléotides plutôt que 21 à 24. Les piRNA ont donc la particularité de cibler des transcrits nucléaires et servent entre autres à limiter la propagation des rétrotransposons (Weick & Miska, 2014 ; Siomi et al., 2011). Dans ce cas-ci, le piRNA (piR30840) produit par SNORD63 ciblerait un intron du transcrit prémature de l’interleukine-4. L’étude pousse même la chose plus loin en démontrant une corrélation inverse entre l’expression de piR30840 et de l’interleukin-4 dans les patients atteints de l’asthme, offrant une potentielle cible thérapeutique pour diminuer l’inflammation chez les asthmatiques (Zhong et al., 2015).
D’autres groupes s’intéressent plutôt à la capacité des snoRNA à modifier des ARN ribosomaux, cette fonction pouvant être directement reliée aux autres fonctions non-canoniques. Une étude s’est penchée sur l’identification de l’ensemble des méthylations causées par des snoRNA C/D en analysant des données de RiboMeth-Seq et de CLIP sur les protéines cœurs des snoRNP C/D (Gumienny et al., 2017). Cette étude a relevé plusieurs centaines de sites de liaisons des snoRNA sur des ARNm qui ne sont associés à aucune
méthylation. Ceci pointe donc au fait qu’il reste beaucoup à explorer en ce qui a trait aux liaisons des snoRNA aux ARNm et sur les effets reliés.
D’autres fonctions particulières ont par ailleurs été démontrées, certaines étant connues depuis longtemps comme les snoRNA SNORD3 qui médient le clivage de l’ARNr prémature (Hughes et Ares, 1991 ; Borovjagin et Gerbi, 1999). Certains snoRNA C/D sont plutôt reconnus comme étant capables de médier la méthylation du petit ARN nucléaire U6, composante essentielle du spliceosome (Tycowski, You et al., 1998 ; Ganot et al., 1999). Les petits ARN du corps de Cajal (scaRNA) qui sont proches des snoRNA et possèdent tant les boîtes C/D que H/ACA sont d’ailleurs spécialisés dans la modification des snRNA (Darzacq
et al., 2002). Une plus récente étude a également montré que deux snoRNA orphelins
pouvaient guider l’acétylation de l’ARNr 18S par l’intermédiaire de la protéine Kre33 (Sharma et al., 2017).
Beaucoup de snoRNA restent orphelins jusqu’à ce jour et le potentiel d’implication de ces derniers dans d’autres pathologies dont le cancer (Nallar et Kalvakolanu, 2013) (Patterson et
al., 2017) est en soit une motivation suffisante à la continuation de leur caractérisation. Les
snoRNA ne sont qu’un exemple de groupe d’ARN non-codant dont nos connaissances demeurent limitées. D’autres groupes d’ARN dont les longs ARN non-codants (lncRNA) sont très diversifiés dans leur structure et leur niveau de conservation et la majorité de leurs fonctions demeurent inconnues (Kung et al., 2013). La découverte et la caractérisation de nouvelles fonctions des différents ARN non-codants sont toutefois grandement limitées dû au fait que certains de ces ARN sont hautement structurés et voient leur abondance fortement sous-évaluée en séquençage d’ARN (RNA-Seq) et autres méthodes nécessitant une rétrotranscription des ARN (Mohr et al., 2013).
RNP, abondance relative et fonctionnalité
Plusieurs RNP essentiels autres que la télomérase, les ribosomes et les snoRNP existent et mettent en jeu la régulation de l’expression d’une multitude d’ARN non-codants et d’ARNm. Pour n’en mentionner que quelques exemples, le spliceosome fait appel à une multitude de
RNP dont entre autres le RNP nucléaire U1 qui fonctionne comme initiateur de l’assemblage du spliceosome sur un site d’épissage (Nagai et al., 2001). Le RNP 7SK, quant à lui, régule le niveau de transcription de certains gènes par son action sur le facteur d’élongation P-TEFb (Peterlin et al., 2012). La RNAse MRP elle est essentielle dans l’initiation de la réplication d’ADN chez la mitochondrie ainsi que dans le clivage des ARNr immatures (Chang et al., 1989 ; Lygerou et al., 1996). La RNAse P, de son côté, contient un ribozyme qui sert à faire le clivage des pré-ARNt afin d’en faire leur maturation (Guerrier-Takada et al., 1983). Enfin, la liste de RNP est longue et leurs fonctions sont tout autant diverses qu’essentielles aux cellules.
L’abondance relative des différents ARN reliés à ces complexes est en soit un paramètre particulièrement important dans l’étude de la fonction des RNP. La régulation de l’abondance, qu’elle soit au niveau de l’expression génique ou au niveau de la stabilité des transcrits, est un outil indispensable pour la cellule afin de contrôler ses différents processus. C’est d’ailleurs pour cette raison que les protocoles de surexpression par vecteurs plasmidiques et de déplétion par interférence à ARN sont fortement prisés par la communauté scientifique pour des études fonctionnelles. Plusieurs études ont tenté d’évaluer l’abondance relative des ARN de différents groupes comme les miARN, snoRNA, ARNt, etc. (Lu et Tsourkas, 2011 ; Ro et al., 2006 ; Zhang et al., 2016 ; Dittmar et al., 2006) dans plusieurs conditions et types cellulaires mais la vraie abondance relative entre ces groupes demeure toutefois mal comprise malgré les efforts d’agglomération des connaissances sur l’expression transcriptomique (Figure 1) (Palazzo & Lee, 2015). Ceci est donc limitant à l’étude de complexes, comme les RNP, mettant en jeu l’expression de plusieurs ARN de différents types. Le séquençage d’ARN à haut-débit (RNA-Seq) (Chu et Corey, 2012 ; Wang et al., 2009) se voulait être une solution à ce problème en offrant la capacité d’évaluer l’abondance de l’ensemble des transcrits et même de pouvoir trouver de nouveaux transcrits dans un échantillon donné. Pour cette raison, le RNA-Seq et ses nombreuses variantes sont de plus en plus utilisées dans l’optique d’évaluer l’abondance de l’ensemble des ARN dans des conditions données. Par contre, cette promesse vient avec son lot de biais causant la surévaluation et la sous-évaluation des différentes classes d’ARN ce qui nuit aux études holistiques portant sur l’abondance des différents types d’ARN.
Le RNA-Seq : méthode et biais
Le RNA-Seq est une méthode actuellement en pleine expansion de popularité, totalisant 2807 publications en faisant mention sur PubMed en 2016 (Calculé avec Medline trend (Dan Corlan, 2004)). Un nombre croissant de biologistes cherchent à produire des ensembles de données dans différents types cellulaires, en exposition à des conditions spécifiques, en déplétion ou en surexpression de transcrits spécifiques. Bien que plusieurs variations protocolaires existent (ex : l’approche Duplex-Specific Nuclease (Zhulidov et al., 2004)), la préparation de librairie de séquençage d’ARN se fait généralement en une série d’étapes standards. La première étape consiste à extraire l’ARN de l’échantillon cellulaire. Ensuite vient la sélection ou la déplétion d’ARN spécifiques de l’extrait afin de concentrer l’échantillon en ARN d’intérêt et retirer des ARN indésirables (ex : ARN ribosomal). Après vient la reverse-transcription des ARN en ADN complémentaires et finalement l’ajout d’adaptateurs appropriés au séquençage (Figure 5A).
Dépendamment du type de séquenceur et de la profondeur recherchée, le nombre de lectures séquencées varie de quelques millions pour des analyses plus superficielles ou pour des organismes avec une plus faible complexité transcriptomique (ex : analyse d’expression différentielle chez la levure), à plusieurs centaines de millions pour des analyses demandant plus de précision et sur des organismes plus complexes (ex : une étude d’épissage alternatif différentielle sur des cellules humaines). Ces lectures de séquençage, une fois produites et informatisées par séquençage, sont prêtes à être analysées. Cependant, le séquençage n’étant pas parfait, certaines bases sont séquencées avec un plus faible taux de confiance, exprimé par le score Phred (Ewig et al.,1998), sur les lectures de séquençage. Pour remédier à ce problème, une étape de rognage des lectures (communément appelée trimming) est nécessaire afin d’enlever les bases de mauvaise qualité ainsi que les potentiels restants d’adaptateurs dirigeant le séquençage aux extrémités des séquences de lectures (Figure 5 B). Une fois traitées, les lectures peuvent être alignées au génome d’intérêt à l’aide d’un aligneur prenant en considération l’épissage (sans quoi les lectures chevauchant les jonctions exon-exon ne pourraient être alignées) (Figure 5 C). Les lectures alignées sont représentées en format standardisé « Sequence Alignment Map » (SAM) (Li et al., 2009). Ces fichiers d’alignement
peuvent par la suite être analysés et comparés avec d’autres fichiers d’alignement représentant d’autres ensembles et recoupés avec d’autres sources d’informations afin de répondre à différentes questions de recherche (Figure 5 D). Dans les analyses les plus couramment effectuées sur le séquençage on peut compter la visualisation des alignements, l’évaluation de l’expression différentielle entre différentes conditions et l’évaluation des changements au niveau de l’épissage des transcrits entre différentes conditions.
Figure 5 : Pipeline RNA-Seq; de l’ARN aux analyses.
(A) Méthodologie résumée du RNA-Seq menant à l’obtention des lectures de séquençage (B) Évaluation de la qualité des lectures de séquençage et «trimming» approprié. (C) Alignement des lectures trimmées sur un génome de référence en prenant compte l’épissage afin de produire un fichier .SAM pour «Sequence Alignment Map». (D) Analyses potentielles à effectuer sur les fichiers .SAM.
Variantes protocolaires du RNA-Seq
Ces différentes étapes protocolaires, tant au niveau de la méthodologie utilisée au laboratoire que dans le traitement et l’analyse des données ont plusieurs variations. La nature du protocole expérimental et la forme de l’analyse peuvent influencer grandement la réutilisation des données produites, leur pertinence ou même la véracité des conclusions qui peuvent en être tirées, constituant un défi majeur pour les groupes de recherche intéressés au séquençage.
L’extraction d’ARN est à la base du RNAseq et les ARN obtenus doivent normalement être filtrés à cause de la trop forte abondance des ARN ribosomaux en termes de poids de l’échantillon (jusqu’à 95% du poids moléculaire total). En effet, à cause du nombre limité de lectures produites lors d’un séquençage, plus il y a de lectures associées à une espèce d’ARN, moins il y en aura pour d’autres espèces d’ARN. Ceci résulterait normalement en une profondeur de séquençage diminuée pour les ARN non ribosomaux et limiterait donc les analyses pouvant être portées sur ces derniers (Sims et al., 2014 ; Malone & Oliver, 2011). L’abondance perçue d’une espèce d’ARN en RNA-Seq est ainsi toujours relative au reste du transcriptome analysé. Pour obtenir suffisamment de lectures de séquençage provenant des espèces d’ARN d’intérêt avec une profondeur de séquençage économiquement et techniquement raisonnable il faut faire une sélection des ARN d’intérêts ou une déplétion des ARN ribosomaux (à moins, bien sûr, qu’ils soient le centre d’intérêt du séquençage en question) (Conesa et al., 2016).
Traditionnellement, la méthode de sélection des queues poly-A était utilisée chez les eucaryotes afin de ne conserver que les ARN messagers (et une faible proportion de lncRNA poly-adénylés). Les chercheurs étant intéressés aux miARN en sont plutôt venus qu’à utiliser
des méthodes de sélection de taille par gel ou par colonne permettant d’isoler seulement les ARN en dessous d’un certain seuil de taille, ne conservant que les plus petits ARN dont les miARN. Ces deux méthodes de sélection parviennent efficacement à se débarrasser des ARN ribosomaux, mais elles ont pour défaut de n’offrir qu’une vision du transcriptome centrée aux intérêts du chercheur. Un ensemble de données de séquençage produit dans une condition spécifique par un chercheur intéressé à l’ARNm par sélection poly-A ne peut donc pas être réutilisé par un chercheur intéressé aux miARN par exemple, limitant largement les conclusions pouvant être tirées d’une expérience. L’intérêt exclusif de l’époque envers les ARNm et les miARN et les deux techniques de sélection pour isoler ces types spécifiques ont sans doute créé un fossé dans l’étude fonctionnelle des ARN. En effet, l’isolation des ARN sous un seuil de 200 nucléotides ainsi que la sélection des ARN par queue poly-A laissèrent tout un territoire transcriptomique inexploré par le séquençage surnommé « le trou noir de la biologie de l’ARN » (Steitz, 2015) ciblant les snoRNA, snRNA, ARNt et plusieurs autres types d’ARN non-codants inclus dans un intervalle de 50 à 300 nucléotides.
Éventuellement, les protocoles de déplétion des ARN ribosomaux apportèrent une solution efficace au problème en n’enlevant que les ARN ribosomaux et en conservant le reste de l’échantillon intact. Avec cette méthode de déplétion plutôt que de sélection, le RNA-Seq put offrir une vision non-biaisée du transcriptome, ouvrant donc la possibilité que ces ensembles de données puissent être évalués pour des études fonctionnelles touchant autant les ARN codants que non-codants.
Les variations des conclusions portées sur une expérience de RNA-Seq ne proviennent pas que des choix en termes de protocole au laboratoire mais peuvent aussi provenir des choix en termes de traitement et d’analyse des données informatiques de séquençage. Le répertoire d’outil bio-informatique OMICtools (Henry et al., 2014) illustre bien les difficultés qui s’imposent aux chercheurs voulant faire l’analyse du RNA-Seq, avec, par exemple, 148 différents outils d’alignement de lectures pouvant être utilisés. Ces différents programmes sont variés dans leurs algorithmes, tant dans leur stratégie de calcul que dans leur optimalité, pouvant résulter en des résultats drastiquement différents. L’exemple le plus extrême de cette disparité est le désaccord entre les outils les plus populaires pour évaluer l’épissage
différentiel entre ensembles de données. Une étude expose que 5 des programmes les plus utilisés à cet effet n’ont aucun évènement différentiellement épissé commun et que la corrélation entre ces programmes est particulièrement mauvaise, avec un maximum de corrélation de Spearman de 0,52 entre deux méthodes (Liu et al., 2014). Plusieurs autres groupes se consacrent à la comparaison des différents programmes offerts afin de conscientiser les bio-informaticiens sur les meilleurs choix à faire dépendamment du contexte expérimental (Conesa et al., 2016 ; Grüning et al., 2017).
Néanmoins, même avec les meilleures mesures expérimentales et bio-informatiques il est évident que le séquençage et les méthodes de quantification d’ARN classiques précédant l’utilisation de la rétrotranscription donnent des résultats divergents.
Le désaccord du séquençage et de la littérature
Pour plusieurs types d’ARN, l’abondance perçue par séquençage diverge significativement des abondances déterminées par méthodes de in vivo labelling ou par micropuces. Par exemple, les ARNt sont connus pour être la classe d’ARN la plus abondante en termes de nombre de molécules (Figure 1) et pourtant, les méthodes de RNAseq classiques décrites précédemment ne détectent qu’une portion négligeable de lectures associées aux ARNt (Zheng et al., 2015 ; Pang et al., 2014). Également, différentes études d’évaluation de l’expression générale des snoRNA montrent que les snoRNA ont un niveau d’abondance total s’approchant du niveau d’abondance total des ARNm (décrit dans Palazzo & Lee, 2015) alors que par séquençage, la proportion des lectures associées aux snoRNA est très faible, avec plusieurs snoRNA supposés être très abondants étant complètement oubliés par RNA-Seq. Ceci indique que le RNA-Seq est biaisé dans sa détection de différents types d’ARN.
La caractéristique commune des ARNt et plusieurs snoRNA est que la plupart de ces ARN possèdent une structure très stable ce qui est en soit un obstacle pour en faire la rétrotranscription puisque la rétrotranscriptase (RT) doit utiliser un substrat d’ARN simple brin. Une solution à ce biais a été apportée par le groupe d’Alan Lambowitz de University of
Texas, soit l’utilisation d’une nouvelle RT thermostable d’intron de groupe II (TGIRT) (Mohr et al., 2013).
TGIRT à la rescousse des ARN hautement structurés
La TGIRT utilisée par l’équipe de Alan Lambowitz à l’University of Texas provient de la cyanobactérie thermophile Thermosynechococcus elongatus (Mohr et al., 2010) et possède un mécanisme de rétrotranscription propre aux RT d’introns de groupe II et distinct de celui de la RT rétrovirale classique qui elle provient du virus de leucémie murine (M-MLV). Les introns de groupes II sont une classe d’introns autocatalytiques (Figure 6A) surtout présents chez les procaryotes et qui sont probablement ancêtres des introns spliceosomaux typiques des eucaryotes (Lambowitz & Zimmerly, 2011). L’ARN de l’intron de groupe II est constitué d’une portion intronique autocatalytique (ribozyme) ainsi que d’une portion encodant une RT. L’ARN intronique et la RT s’associent sous forme de complexe RNP. Ce complexe lie un site d’ADN spécifique, le clive et utilise l’ADN clivé comme amorce pour effectuer la rétrotranscription de l’intron, aidant ainsi à la mobilité des introns de groupe II (Lambowitz & Zimmerly, 2011) (Figure 6B).
Figure 6 : Introns de groupe II.
Schématisation du mécanisme d’épissage et de rétrotranscription des introns de groupe II. (A) Le mécanisme d’auto-excision de l’intron de groupe II (B) Illustration de la mobilité des introns de groupe II par épissage inverse et rétrotranscription.
Ces RT d’intron de groupe II sont connus pour avoir une meilleure processivité et une meilleure fiabilité, avec un taux d’erreur avoisinant 1*10^5 (Conlan et al., 2005) ce qui est une amélioration significative par rapport aux RT rétrovirales qui elles ont un taux d’erreur entre 4,5 * 10^5 et 1 * 10^4 (Potter et al. 2003 ; Baranauskas et al. 2012). Mais le véritable avantage de la TGIRT vient du fait que son activité optimale soit à 60°C (pouvant être fonctionnelle jusqu’à 81 °C) plutôt que 37°C comme la RT MLV (et qui se dénature au-delà de 57°C). À plus haute température, les ARN se dénaturent, rendant ainsi la structure de l’ARN moins encombrante pour la rétrotranscription. Ainsi, TGIRT parvient à rétrotranscrire des ARN qui sont normalement hautement structurés et donc difficiles à rétrotranscrire. Avec tous ces avantages, le groupe de Lambowitz a donc proposé en 2013 (Mohr et al., 2013)
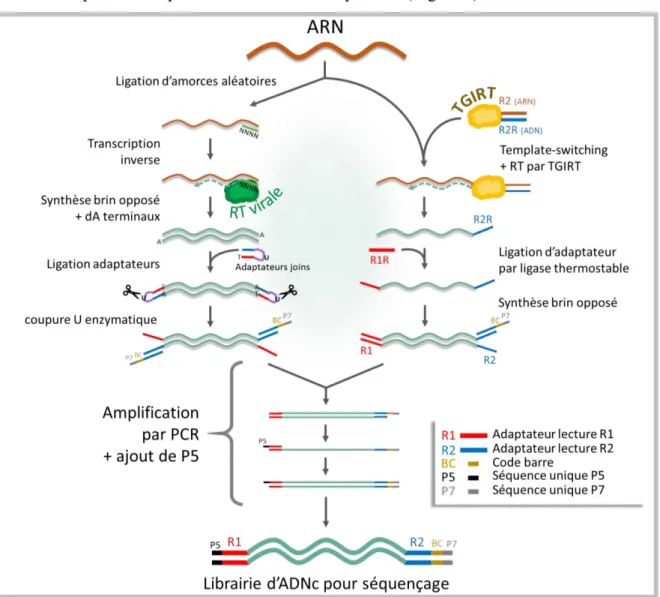
l’utilisation de TGIRT dans la synthèse d’ADN complémentaire (ADNc) pour réduire le biais de structure associé au RNA-Seq. Le TGIRT-Seq promet non seulement de mieux représenter les ARN hautement structurés mais aussi de simplifier les méthodologies de RNA-Seq. En effet, l’équipe de Lambowitz utilise la capacité de TGIRT à utiliser sa propre amorce d’ADN (l’activité « template-switching ») pour initier la rétrotranscription à leur avantage en utilisant la séquence complémentaire de l’adaptateur R2 comme amorce à TGIRT. Ceci enlève le besoin d’utiliser des amorces aléatoires pour initier la rétrotranscription et simplifie l’inclusion des adaptateurs (Figure 7).
Figure 7 : RNA-Seq classique vs TGIRT-Seq
Schématisation des différences protocolaires entre (A) la méthode classique de préparation de librairie pour RNA-Seq utilisant la rétro-transcriptase rétrovirale classique (protocole du « NEBNext Ultra directional RNA library Prep Kit for Illumina ») et (B) la méthode du «Template-switching » utilisant la retro-transcriptase thermostable d’intron de groupe II (TGIRT).
Hypothèse/problématique
Le RNA-Seq ribodéplété avec la RT rétrovirale reflète mal la composition du transcriptome à cause de son biais de structure. La sous-estimation d’un grand nombre d’espèces d’ARN hautement structurés, dont les snoRNA, empêche l’étude fonctionnelle appropriée de ces derniers. La grande majorité des ensembles de données de RNA-Seq qui ont été produits avec une optique de recherche axée sur les ARN codants ne peuvent être réutilisés par la communauté intéressée aux ARN non-codants hautement structurés. Ceci limite grandement l’impact scientifique que ces expériences de RNA-Seq pourraient avoir. L’hypothèse de ce projet de recherche est que l’utilisation de TGIRT en RNA-Seq permettra une évaluation plus juste du transcriptome et devrait devenir un standard pour l’ensemble des groupes de recherche intéressés au séquençage d’ARN.
Objectifs Objectif #1
Déterminer si l’utilisation de TGIRT améliore l’évaluation de l’abondance relative des ARN hautement structurés dont les snoRNA et les ARNt.
Objectif #2
Déterminer la meilleure méthode de séquençage pour l’évaluation de l’abondance relative des snoRNA par rapport à d’autres espèces d’ARN. Cette méthodologie de RNA-Seq nous permettra de perpétuer nos études fonctionnelles sur les snoRNA, incluant la découverte de cibles canoniques ainsi que l’identification des partenaires protéiques des snoRNA non-canoniques.
Objectif #3
Établir la composition transcriptomique générale de notre principal type cellulaire d’intérêt, les cellules cancéreuses ovariennes SKOV3ip1. Tirer ensuite des conclusions stœchiométriques fonctionnelles sur des ARN de différents types ayant des fonctions synergiques et en faire parallèle avec la littérature.
2A
RTICLERNA-seq quantification of coding and non-coding RNA abundance
Auteurs de l’article: Vincent Boivin, Gabrielle Deschamps-Francoeur, Sonia Couture, Ryan M. Nottingham, Philippe Bouchard-Bourelle, Alan M. Lambowitz, Michelle S Scott, et Sherif Abou-Elela.
Statut de l’article: soumis dans RNA
Avant-propos:
Ma participation aux expériences est exclusivement bio-informatique, comprenant le traitement des données de séquençage, l’élaboration d’un pipeline d’assignement des lectures (CoCo) ainsi que l’analyse bio-informatique subséquente. J’ai participé à la rédaction de la méthodologie reliée à mes analyses, à l’élaboration des figures et de la description des résultats en plus d’avoir mis mon apport dans la rédaction de la discussion.
Résumé:
La comparaison de l’abondance d’une molécule d’ARN par rapport à une autre est une étape cruciale dans la compréhension des fonctions cellulaires. Cependant, la plupart des techniques de séquençage ne peuvent cibler qu’un sous-groupe spécifique d’ARN à la fois. Nous utilisons une nouvelle méthodologie de RNA-Seq qui utilise une rétrotranscriptase thermostable d’intron de groupe II (TGIRT) pour générer un portrait du transcriptome humain montrant les relations quantitatives entre toutes les classes d’ARN non-ribosomal dépassant 60 nucléotides de longueur. L’évaluation de l’abondance des ARN dans un modèle de cellules cancéreuses ovariennes humaines utilisant cette méthode corrèle avec les estimés biochimiques, confirmant les ARNt comme le type d’ARN non ribosomal le plus abondant. Cependant, le transcrit le plus abondant est celui de l’ARN 7SL, une composante de la particule de reconnaissance du signal. Les ARN non-codants structurés (sncRNA) associés avec les mêmes processus biologiques sont uniformément exprimés avec l’exception d’ARN aux fonctions multiples dont le snRNA U1. En général, les sncRNA formant des RNP sont des milliers de fois plus abondants que leur ARNm associé. Étonnamment, seulement 50 gènes de sncRNA produisent la moitié des transcrits non ribosomaux dans les cellules humaines. Ensemble, les résultats suggèrent que le transcriptome humain est dominé par une petite quantité de gènes de sncRNA hautement exprimés et spécialisés dans des fonctions reliées à la traduction ou l’épissage.
Simultaneous detection and quantification of coding and non-coding RNAs using
a single sequencing reaction
Vincent Boivin1, Gabrielle Deschamps-Francoeur1, Sonia Couture2, Ryan M. Nottingham3,
Philia Bourelle-Bouchard 1, Alan M. Lambowitz3, Michelle S Scott1*, and Sherif Abou-Elela2*.
1Département de biochimie, Faculté de médecine et des sciences de la santé, Université de
Sherbrooke, Sherbrooke, Québec J1E 4K8, Canada.
2Département de microbiologie et d’infectiologie, Faculté de médecine et des sciences de
la santé, Université de Sherbrooke, Sherbrooke, Québec J1E 4K8, Canada.
3Institute for Cellular and Molecular Biology and Department of Molecular Biosciences,
University of Texas at Austin, Austin, Texas 78712
V. B. and G. D. contributed equally to this work and thus considered first co-first authors. Running Title: RNA-seq quantification of coding and non-coding RNA abundance
Keyword: snoRNA, Non-coding RNA, High-throughput sequencing, RNA detection, Thermostable Group II Intron Reverse Transcriptase, Transcriptome analysis
ABSTRACT
Comparing the abundance of one RNA molecule to another is crucial for understanding cell functions but most sequencing techniques can target only specific subsets of RNA at a time. We used a new RNA-seq method employing a thermostable group II intron reverse transcriptase (TGIRT) to generate a portrait of the human transcriptome depicting the quantitative relationship of all classes of non-ribosomal RNA longer than sixty nucleotides. Measurements of RNA abundance in a model human ovarian cancer cell line using this method correlate with biochemical estimates, confirming tRNA as the most abundant non-ribosomal RNA biotype. However, the single most abundant transcript is 7SL RNA, a component of signal recognition particle. Structured non-coding RNAs (sncRNAs) associated with the same biological process are uniformly expressed with the exception of RNAs with multiple functions like U1 snRNA. In general, sncRNA forming RNPs are hundreds to thousands of times more abundant than their mRNAs counterparts. Surprisingly, only 50 sncRNA genes produce half of the non-rRNA transcripts in the human cell. Together the results indicate that the human transcriptome is dominated by a small number of highly expressed sncRNA genes specialized in functions related to translation and splicing.
INTRODUCTION
The study of RNA expression is a rapidly growing area of genomic research (Ozsolak and Milos 2011; Jiang et al. 2015). The amount of RNA sequencing is expected to surge as the need for understanding gene function and the identification of new biomarkers increases (Rabbani et al. 2016). However, despite the increase in RNA sequencing in the last five years, established methods for the detection of midsize structured non-coding RNAs remain elusive (Veneziano et al. 2016). Indeed, RNA ranging in size between 50 and 300 nucleotides was termed the “black hole” of RNA biology due to the lack of sequencing information (Steitz 2015). Most standard sequencing methods are focused on the detection of polyadenylated messenger RNAs with sizes typically larger than 1 kb (Costa et al. 2010; Liang and Zeng 2016). As such, these methods are not useful for the detection of non-polyadenylated transcripts shorter than 500 base pairs (e.g. snRNA, snoRNA, tRNA and many lncRNA) (Veneziano et al. 2016). In addition, selection of polyadenylated RNA prevents the detection of RNA processing and maturation intermediates. Current approaches for the sequencing of sncRNAs depend on selection techniques that enrich RNAs based on their size or nuclear localization (Deschamps-Francoeur et al. 2014; Bai and Laiho 2016). Recent studies suggest that most of these techniques introduce bias in the relative representation of non-coding RNAs even for those with similar sizes (Deschamps-Francoeur et al. 2014; Nottingham et al. 2016).
Classical estimates of RNA abundance are usually generated by using targeted in
vivo labelling experiments as in the case of rRNA, tRNA, snRNA and snoRNA or by using
microarrays as in the case of mRNA and miRNA (Waldron and Lacroute 1975; Wolf and Schlessinger 1977; Bissels et al. 2009). These class-specific estimates of RNA abundance indicate that ~90% of the human transcriptome by mass is composed of rRNA, while the highest number of molecules per cell is attributed to tRNA (Waldron and Lacroute 1975; Wolf and Schlessinger 1977). Cross experiment comparison between protein-coding and non-coding RNAs suggested that mRNA is more abundant both in terms of mass and number of molecules than are snRNA, snoRNA and miRNA (Kiss and Filipowicz 1992; Bissels et al. 2009). In general, these techniques are efficient in comparing the relative abundance of transcripts within the same class of RNA but they cannot directly compare abundances between different classes of RNA (e.g. between coding and non-coding RNA). Therefore, while the abundance of the different RNA classes might be established, measurements of their true relative abundance remain to be verified.
Recently, a new sequencing method using a thermostable group II intron reverse transcriptase (TGIRT) was developed, which exploits the ability of this highly processive enzyme to reverse transcribe full-length, highly structured RNAs (Nottingham et al. 2016; Qin et al. 2016). This method of sequencing (TGIRT-seq) showed promise for detecting many different types of RNA in a generic RNA reference sample and as such provides a potentially useful tool for direct comparison of the abundance of different classes of RNA. In this study, we compare the capacity of different sequencing methods including TGIRT-based methods
to faithfully depict the landscape of the human transcriptome. The results indicate that sequencing of fragmented ribodepleted RNA using TGIRT provides the most complete and experimentally supported portrait of the human transcriptome. Using this method we were able to confirm the overall conclusions of previous estimates of RNA abundance showing that tRNA is the most abundant RNA species in terms of number of molecules. However, unlike previous estimates our results show that snRNAs are actually more abundant than mRNAs and snoRNAs and that non-coding RNAs are generally at least 1000 times more abundant than mRNAs encoding proteins functioning in the same biological process. Interestingly, direct comparisons between the coding and non-coding RNA participating in the assembly of ribonucleoprotein complexes permitted the identification of specific components with either regulatory or multiple functions. Together our results indicate that simultaneous detection of both coding and non-coding RNA by TGIRT-seq not only increases the number of transcript types analyzed but also improves the precision of RNA ranking and estimates of abundance within each class of RNAs.
RESULTS
Comparison between the capacities of different sequencing methods to quantify
different components of the human transcriptome
Most sequencing methods deal with coding and non-coding RNAs separately (Figure 1A) providing little information about the overall landscape of the human transcriptome. To identify the best approach for an integrated analysis of the human transcriptome, we evaluated the capacity of different sequencing methods to simultaneously quantify different classes of RNAs. We chose five sequencing protocols: 1) Size-selected viral reverse transcriptase sequencing (abbreviated SSV), 2) TGIRT-seq of unfragmented, ribodepleted whole-cell RNA (abbreviated URT), 3) Fragmented poly(A)-selected viral reverse transcriptase sequencing (abbreviated FAV), 4) Fragmented ribodepleted viral reverse transcriptase sequencing (abbreviated FRV), and 5) Fragmented ribodepleted TGIRT-seq (abbreviated FRT). These five different approaches cover the most commonly used methods and test two newly developed techniques (URT and FRT) that use the thermostable group II intron reverse transcriptase, TGIRT-III. TGIRT increases the chance of generating cDNA from short highly structured RNA without size selection (Mohr et al. 2013; Nottingham et al. 2016; Qin et al. 2016). Two of the methods employed RNA selection steps like size selection (SSV) or poly(A) tail selection (FAV), while in the remaining methods (URT, FRT and FRV) rRNAs were ribodepleted and sequenced without size selection. The RNA was extracted from the model ovarian cancer cell-line SKOV3ip1 and two biological replicates for each protocol were sequenced to read depths that vary between 20 and 150 million
reads (Table S1). All comparisons between methods were performed using count per million (CPM) and transcript per million (TPM) to eliminate read depth biases.
The SSV library was prepared using a mirVana kit but without a gel purification step, enabling the consideration of all RNA <200 nucleotides in length (Deschamps-Francoeur et al. 2014). As expected, SSV datasets hardly possessed any reads generated from protein-coding genes and instead were enriched in reads from non-protein-coding RNAs (Figure 1B left panel). However, the reads generated from non-coding RNA were not even appropriately distributed among the different classes of non-coding RNA nor within the same class of RNA. Most reads (61% CPM and 70% TPM) corresponded to snoRNAs, the vast majority of which were mapped to box C/D snoRNA, while 6% of the reads originated from miRNAs and only 3% originated from tRNAs, which are difficult to reverse transcribed by retroviral RTs. Markedly, very little snRNA and 7SL RNA were detected using this method despite all snRNAs being less than 200 nucleotides long. Thus, size selection is inappropriate for surveying the relative abundance of both coding and non-coding RNAs.
In contrast, sequencing of total unfragmented ribodepleted RNA using TGIRT (URT) succeeded in detecting all five classes of the main non-coding RNA with lengths between 60 and 700 nucleotides: tRNA, snoRNA, snRNA, 7SL and 7SK. The majority of the reads (56% CPM and 59% TPM) corresponds to tRNA, followed by snoRNA (32% CPM and 33% TPM), while both long non-coding RNA and miRNA were detected at low levels (Figure 1B second panel). This suggests that direct unfragmented RNA sequencing using TGIRT provides a