MiR-16, un nouveau régulateur du transporteur de glucose dépendant de l’insuline GLUT-4 par Nour El-amine Département de Pharmacologie Faculté de Médecine
Mémoire présenté à la Faculté des études supérieures en vue de l’obtention du grade de Maîtrise en pharmacologie
Décembre 2010
Université de Montréal
Faculté des études supérieures et postdoctorales
Ce mémoire intitulé :
MiR-16, un nouveau régulateur du transporteur de glucose dépendant de l’insuline GLUT-4
présenté par : Nour El-amine
a été évalué par un jury composé des personnes suivantes :
Dr Guy Rousseau Président-rapporteur Dr Éric Thorin Directeur de recherche Dr Zhiguo Wang Co-directeur de recherche Dr Christian Beauséjour Membre du Jury
Résumé
Les microARNs sont des petits ARNs non codants d'environ 22 nucléotides qui régulent négativement la traduction de l'ARN messager cible (ARNm) et ont donc des fonctions cellulaires. Le microARN-16 (miR-16) est connu pour ses effets antiprolifératifs. Nous avons observé que l’expression de miR-16 est diminuée dans les cellules endothéliales humaines sénescentes et quiescentes en comparaison à des cellules prolifératives. Une analyse informatique des sites potentiels de liaison de miR-16 prévoit que GLUT-4, un transporteur du glucose insulinodépendant, pourrait être une cible potentielle du miR-16. Nous avons donc testé l'hypothèse que miR-16 régule négativement le métabolisme du glucose cellulaire. Dans des HUVEC, l'inhibition de miR-16 endogène avec des anti-miRNA oligonucléotides (AMO) augmente les niveaux protéiques de GLUT-4 de 1,7 ± 0,4 fois (p=0,0037 ; n=9). Dans des souris nourries avec un régime alimentaire normal ou riche en graisse et en sucre, l’expression de GLUT-4 dans le muscle squelettique a tendance à corréler négativement avec les niveaux de miR-16 (p=0,0998, r2=0,3866, n=4). Ces résultats suggèrent que miR-16 est un régulateur négatif de GLUT-4 et qu’il pourrait être impliqué dans la régulation du métabolisme cellulaire du glucose.
Mots clés : microARN, Glut-4, miR-16 Subventionné par les IRSC MOP14496
Abstract
MicroRNAs are small noncoding RNAs of approximately 22 nucleotides that negatively regulate translation of the target messenger RNA (mRNA) and therefore have cellular functions. MicroRNA-16 (miR-16) is known to display anti-proliferative effects. We observed that miR-16 was down-regulated in non-proliferative human senescent endothelial cells. Computational analysis of the potential binding sites of miR-16 predicted that GLUT-4, an insulin-dependent glucose transporter, is a potential target of miR-16. We therefore tested the hypothesis that miR-16 down-regulates cellular glucose metabolism. In HUVEC, inhibition of using anti-miRNA oligonucleotides (AMO) endogenous miR-16 up-regulated GLUT-4 protein levels 1,7 ± 0,39 folds (p=0,0037; n=9). In mice fed a regular or high fat diet, skeletal muscle expression of GLUT-4 tended to negatively correlate with miR-16 levels (p=0,0998, r2=0,3866, n=4). These results suggest that miR-16 is a negative regulator of GLUT-4 and may be involved in the regulation of cellular glucose metabolism.
Key words : microRNA, Glut-4, miR-16 Supported by CIHR MOP14496.
Table des matières
Résumé ... iii
Abstract ... iv
Table des matières ... v
Liste des figures ... ix
Liste des abréviations ... xiii
Remerciements ... xvii
1 Introduction ... 1
1.1. ARN non-codant et miRNAs ... 2
1.1.1. Biogenèse des miRNAs ... 2
1.1.2. Fonctions et modes d’action ... 4
1.1.3. Études in silico ... 4
1.1.4. Potentiels thérapeutiques ... 6
1.2. Rôle des miRNAs dans le développement cardiaque et les maladies cardiovasculaires ... 7
1.2.1. Cardiogenèse ... 7
1.2.2. Hypertrophie cardiaque ... 8
1.2.3. Apoptose et prolifération des cardiomyocytes. ... 9
1.2.5. Fibrose ... 10
1.2.6. Angiogenèse ... 13
1.3. Sénescence ... 15
1.3.1. Sénescence Réplicative vs SIPS ... 15
1.3.2. miRNAs impliqués dans la sénescence ... 16
1.4. Transport actif du glucose ... 17
1.4.1. Transporteurs de Glucose ... 18
1.4.2. miRNA et INSR ... 19
1.4.3. miRNA et IRS-1 ... 20
1.4.4. miRNA et GLUT-4 ... 20
1.4.5. Rôle des miRNAs dans le diabète ... 21
1.5. microRNA-16 ... 22
1.5.1. Rôles connus de la famille de miR-15/107 ... 23
1.5.1.1. miR-16 et division cellulaire ... 24
1.5.1.2. miR-16 dans le métabolisme et le stress cellulaire ... 24
1.5.1.3. miR-16 et angiogenèse ... 25
1.5.1.4. miR-16 et cancer ... 25
2 Raisonnement et objectifs de l’étude ... 27
3 Matériel et Méthodes ... 29
3.1. Analyse in silico ... 29
3.2. Microarray ... 30
3.4. Transfection ... 34
3.5. Essai viabilité ... 35
3.6. Extraction protéique ... 36
3.7. Immunobuvardage de type Western ... 37
3.8. Extraction des miRNA et ARN total ... 38
3.9. Transcription inverse (RT-PCR) ... 39
3.10. PCR en temps réelle quantitative (qPCR) ... 40
3.11. Clonage moléculaire ... 44
3.11.1. Digestion du vecteur pmiR-Report-1 ... 44
3.11.2. Réaction PCR 3’UTR GLUT-4 ... 45
3.11.3. Réaction PCR 3’UTR-GLUT-4-muté ... 46
3.11.4. Ligation pmiR-Report et réaction PCR GLUT-4 et 3’UTR-GLUT-4-muté ... 49
3.11.5. Migration des produits PCR sur gel d’agarose ... 50
3.11.6. Purification des bandes séparées par gel d’agarose ... 51
3.11.7. Transformation du pmiR-Report-GLUT-4 et 3’UTR-GLUT-4-muté ... 52
3.12. Essai luciférase ... 54
3.13. Souris ... 54
3.14. Analyse statistique ... 55
4 Résultats ... 56
4.2. Microarray sur des hIMAEC prolifératives et sénescentes ... 57
4.3. Analyse des cibles potentielles de miR-16 ... 61
4.4. Effet de miR-16 sur GLUT-4 et INSR ... 62
4.5. Corrélation entre miR-16 et la glycémie de patients coronariens ... 68
4.6. Corrélation miR-16, GLUT-4 et glycémie souris WD et RD ... 70
5 Discussion ... 78
5.1. Microarray ... 78
5.2. Rôle possible de miR-16 dans la sénescence ... 79
5.3. Effet de miR-16 sur GLUT-4 ... 79
5.4. Relation miR-16 et glycémie ... 81
5.4.1. miR-16 et glycémie des patients coronariens ... 82
5.4.2. miR-16, GLUT-4 et glycémie dans le muscle squelettique de souris ... 83
6 Conclusion ... 85
Liste des figures
Figure 1. Biogenèse des miRNAs.
Figure 2 : Altération de l’expression des miRNAs impliqués dans les maladies reliés aux cardiomyocytes et fibroblastes
Figure 3. Altération de l’expression des miRNAs impliqués dans les maladies reliés au remodelage vasculaire et à l’angiogenèse.
Figure 4. Transport actif du glucose par le GLUT-4.
Figure 5. Séquence de la famille des miR-16.
Figure 6. Effet du traitement simultané au NACet hTERT sur les hIMAEC. Figure 7. Représentation schématique de la méthodologie abordée pour le microarray.
Figure 8. Représentation schématique de la méthode de qPCR des miRNAs.
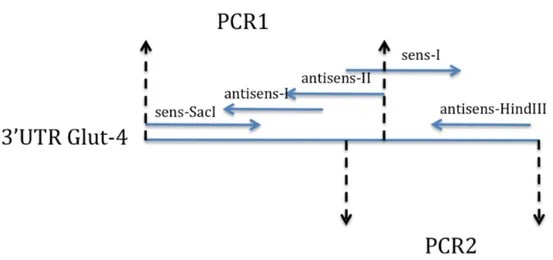
Figure 9. Cartographie du pMIR-REPORT de la compagnie Ambion. Figure 10. Représentation schématique des trois réactions PCR 1, 2.
Figure 11. Niveaux d’expression du miR-34a dans des hIMAEC prolifératives et sénescentes par qPCR
Figure 12. Niveaux d’expression du miR-34a dans des hIMAEC prolifératives et sénescentes.
Figure 13. Expression des miRNAs dans les hIMAEC prolifératives comparée à celle des hIMAEC sénescentes.
Figure 14. Niveaux d’expression du miR-16 dans les cellules endothéliales prolifératives et sénescentes de patients coronariens.
Figure 15. Prédictions des sites de liaison potentiels du miR-16 par le logiciel TargetScan.
Figure 16. Niveaux d’expression protéique de GLUT-4 suite à l’inhibition du miR-16 dans des HUVEC.
Figure 17. Niveaux de miR-16 suite à l’inhibition du miR-16 dans des HUVEC.
Figure 18. Régression linéaire entre miR-16 et GLUT-4 dans des HUVEC. Figure 19. Niveaux d’expression protéique de INSR suite à l’inhibition du miR-16 dans des HUVEC.
Figure 20. Relation entre la glycémie de patients coronariens et les niveaux d’expression relatifs de miR-16 dans des hIMAEC.
Figure 21. Niveaux glycémiques de souris nourries par une diète western comparée à une diète normale.
Figure 22. Corrélation des niveaux de GLUT-4 protéiques avec la glycémie de souris nourries par une diète western et une diète normale.
Figure 23. Corrélation des niveaux d’INSR protéiques avec la glycémie de souris nourries par une diète western et une diète normale.
Figure 24. Expression de miR-16 dans les muscs squelettiques de souris RD et WD.
Figure 25. Corrélation entre l’expression de miR-16 et les niveaux protéiques de GLUT-4 et INSR dans le muscle squelettique de souris.
Liste des tableaux
Tableau I. Séquences d’amorces utilisées pour les réactions qPCR.
Tableau II. Séquences d’amorces utilisées pour les réactions PCR de clonage moléculaire.
Liste des abréviations
3’UTR : 3’ untranslated region ADNc : ADN complémentaireAMO : anti-miRNA oligonucléotides ARNm : ARN messager
BCL2 : B-cell lymphome 2 BSA : bovine serum albumin
CCND1 : cyclin D1
CCNE1 : G1/S-specific cyclin E1
CDC25a : cell division cycle 25 homologues A
CLL : leucémie lymphocytaire chronique ddH2O : H2O bi-distillé
DMEM : Dulbeco’s Modified Eagle Medium EBM 2 : endothelial basal medium 2
EGTA: acide ethylene-glycol-bis (b-aminoéthylether)-N,N,N’,N’-tétraacétique
eNOS : endothelial nitric oxide synthase FBS : fetal bovine serum
FGF : fibroblast growth factor FOXO1: forkhead box protein O1
GAPDH : glycéraldéhyde 3-phosphate déhydrogénase GLUT-1 : glucose transporter-1
GLUT-4 : glucose transporter-4 HDAC4: histone deacétylase 4
HEK293 : Human Embryonic Kidney 293 cells HIF-1β : hypoxia-inducible facteur-1β
hIMAEC : human internal mammary artery endothélial cells
HRP : horseradish peroxidase HSP20: Heat-Shock Protein 20 hTERT : human telomerase
HUVEC : cellules endothéliales de la veine ombilicale humaine IGF2R : insulin growth factor 2 receptor
IL-6 : interleukine-6
IL-8 : interleukine-8
INSR : insulin receptor
IRS-1 : insulin receptor substrat-1
KLF15 : krueppel-like factor 15 miRNA : microRNA
MHC: myosin heavy chain NAC : N-Acétyl-Cystéine NTC : no template control
PBS : phosphate buffered solution PCR : polymerase chain reaction PHD2: propyle hydroxylase 2
PI3K : phosphatidylinositol 3-kinases PI3KR2 : PI3K regulatory subunit 2
qPCR : quantitative real-time PCR RT-PCR : reverse transcription PCR
scnRNA : small non coding RNA
SAPS: senescence-associated-secretory-phenotype SIPS : stress induced premature senescence
SIRT1 : Sirtuine-1
SRF: serum response factor
TBST: tris buffered-saline tween TP : température pièce
UV : Ultra-Violet
VCAM-1 : vascular cell adhesion protein 1 VEGF : vascular endothelial growth factor
RD : diète régulière WD : diète western
Remerciements
Je souhaiterais remercier mon directeur de recherche Dr Éric Thorin. Dr Thorin m’a tendu la main quand ma confiance en moi n’existait presque plus, un attribut que je suis fier de présenter aujourd’hui, comme ma plus grande qualité. Ce que je retiens le plus de mon passage dans son laboratoire c’est que je n’ai jamais cessé d’apprendre tant au plan personnel que professionnel. Dr Thorin n’était pas juste un patron de travail, mais plutôt un mentor qui s’intéressait à développer ses étudiants et non seulement leurs projets de recherche. C’est quelqu’un qui a acquis tout le respect que je peux accorder à une personne.
J’aimerais également remercier mon codirecteur Dr Zhiguo Wang. Un pionnier dans le domaine des miRNAs, sa codirection était essentielle pour ma progression. Il avait toujours sa porte ouverte pour m’aider et j’en ai profité pour acquérir beaucoup de ses connaissances.
Dre Nathalie Trescases-Thorin est quelqu’un qui va certainement me manquer. La proximité de nos bureaux a fait en sorte que nous avons eu des discussions quotidiennes des plus intéressantes. Son aide autant grâce à sa vaste connaissance de sujets diversifiés et son organisation du laboratoire nous a facilité notre travail. Je pense que mon bench et bureau très organisés vont lui manquer, mais je suis sûr quel pourra vivre avec. Merci !
Je remercie, Mme Maya Mammarbachi, une personne sans qui je n’aurais jamais pu effectuer ce travail. C’était un guide durant toute ma maîtrise puisque je n’ai pas passé une journée sans lui poser une question. Mme Mammarbachi m’a non seulement aidé au niveau technique, mais aussi dans l’orientation de mon projet. Je n’ai pas eu la chance de collaborer avec d’autres étudiants, mais je pense que nous avons travaillé, elle et moi, comme une équipe à deux pour réussir ce projet.
Xiaobin Luo était ma référence quotidienne pour les miRNAs. Sans lui j’aurais été comme une personne sans boussole dans le désert. C’est un étudiant qui a fait un travail incroyable durant son doctorat et qui mérite toute la reconnaissance de ses pairs. Merci pour cette personne qui m’a appris toutes les bases du domaine des miRNAs, mais aussi les détails les plus complexes. C’est quelqu’un que j’apprécie énormément et avec qui j’ai développé une belle camaraderie qui ne s’arrêtera pas après ce travail.
J’aimerais également remercier tous les autres membres du laboratoire du Dr Wang qui m’ont accueilli comme un des leurs dans le laboratoire.
Pour continuer, j’aimerais remercier tous les étudiants et employés que j’ai côtoyés quotidiennement durant ma maîtrise. Quand j’ai commencé à travailler à l’ICM, je n’aurais jamais pensé développer autant de relations amicales. C’est cette ambiance spéciale à l’ICM qui me manquera tellement. Un remerciement
aussi aux membres du comité étudiants qui m’ont aidé à développer la vie étudiante durant mon passage.
Ce qui fait de notre laboratoire spécial, c’est l’amitié, l’honnêteté et l’éthique de travail qui existe entre nous les étudiants. J’aimerais donc remercier un par un tous les étudiants du Dr Thorin avec qui j’ai eu le plaisir de partager le même laboratoire :
Merci à Nada Farhat, l’encyclopédie biochimique et technique du laboratoire. C’est une vraie amie que j’ai eu la chance de connaître. Notre relation amicale n’a fait que grandir avec le temps. C’est une personne avec qui j’ai pu partager toutes mes confidences et qui était toujours prête à m’écouter et m’aider. Sur les plans techniques et pratiques, inutile d’expliquer le grand rôle qu’elle a joué dans mon développement. C’est une grande personne que je respecte énormément.
Merci à Albert Nguyen, un ami que je vais continuer de côtoyer malgré mon départ. Jamais je n’aurais cru rencontrer une personne qui partage tant de ressemblance avec moi, tant au niveau de la personnalité, de la manière de penser ou des intérêts personnels. C’est une personne qui a vraiment rendu le travail plus agréable, mais c’est aussi quelqu’un qui était prêt à suivre mon cheminement et qui a d’excellentes connaissances techniques qui m’ont certainement aidé.
Merci à François Leblond, un exemple à suivre dans le laboratoire. Son perfectionnisme, sa discipline et sa structuration le représentent le mieux. Ces caractéristiques sont des points que j’aimerais développer en moi durant mon doctorat et François est le modèle que je veux suivre. Il n'attend pas seulement qu’on lui demande de l’aide, mais il est toujours le premier à l’offrir. C’est agréable de voir comment une personne ayant tant de responsabilités peut accomplir autant et plus que tous les autres étudiants !
Merci à Virginie Bolduc, une personne qui a une éthique de travail irréprochable. Nos connaissances scientifiques ne se ressemblent pas, mais c’est surtout au niveau personnel qu’on a développé une belle complicité au travail. Virginie me fait réaliser à quel point notre groupe était différent, mais comment on s’est bien complété entre nous. Les heures tardives qu’on passait ensemble avec Albert et les multiples belles discussions qu’on a eues ont certainement été une motivation pour travailler plus fort.
Merci au Dre Annick Drouin, une personne très animée qui nous a beaucoup manqué. Nos heures de dîner n’étaient plus jamais les mêmes après son départ. C’était toujours agréable d’assister aux longues argumentations très fréquentes entre elle et Albert. Cela m’a fait plaisir de l’avoir connue pendant mon passage.
Merci à Carol Yu qui est arrivée récemment au laboratoire. J’aurais aimé l’avoir eu comme collègue pour une plus longue durée.
Merci à Shiva Safavi, qui est aussi arrivée dernièrement dans le laboratoire. C’est une personne qui a apporté une nouvelle dynamique au laboratoire.
J’aimerais finalement remercier le président rapporteur, le Dr Guy Rousseau et le membre du jury, le Dr Christian Beauséjour, qui ont accepté de corriger ce mémoire.
1 Introduction
Le dogme central qui propose le transfert d’informations génétiques de l’ADN vers l’ARN pour former des protéines constitue toujours une base solide qui relie la médecine à la génétique. Mais les détails et exceptions de fonctionnement de ces trois composantes ne sont pas encore éclaircis. Un nouveau groupe d’ARN non-codant surnommé les microRNAs (miRNA) a été conservé durant l’évolution; ces miRNAs sont d’importants régulateurs de l’expression des gènes du génome (Boyd, 2008). Ils ont la capacité de réguler plusieurs gènes cibles en même temps, d’où l’importance qui leur a été accordée durant ces dernières années (Boyd, 2008). Les miRNAs agissent en inhibant la transcription de l’ARNm cible. Notre compréhension des miRNAs est encore primitive, mais les études montrent un rôle potentiel dans le développement humain, la différenciation cellulaire, l'adaptation à l'environnement, l'oncogenèse, le métabolisme cellulaire, la neurobiologie et l'interaction des cellules hôtes avec des pathogènes (Esau and Monia, 2007). Les miRNAs constituent un mécanisme de régulation post-transcriptionnel conservé à travers l’évolution, mécanisme qui ajoute un niveau de complexité dans le réseau de régulation génétique (Boyd, 2008; Esau and Monia, 2007).
1.1. ARN non-codant et miRNAs
Les études de séquençage d’ADN et du génome humain ont montré que 50% de l’ADN génomique est transcrit en ARN, dont 2% sont traduits en protéines alors que les 98% restants sont de l’ARN non-codant, de taille moyenne de 18 à 500 nucléotides (Zhang, 2008b). L’ARN non-codant de très petite taille constitue la classe de petits ARN non-codants (sncRNA) (Zhang, 2008b). Les miRNAs font partie de ces sncRNA (Zhang, 2008b). Le premier miRNA, lin-4, a été découvert en 1993. Ce miRNA n’a pas été reconnu comme une classe à part d’ARN non-codant, mais plutôt comme un artéfact (Lee et al., 1993; Wightman et al., 1993). Ce n’est qu’au début des années 2000, lors de la découverte du miRNA let-7 que les miRNAs ont été reconnus comme des importants régulateurs biologiquement actifs (Pasquinelli et al., 2000; Reinhart et al., 2000). Depuis, on estime que le génome humain encode pour près de 1000 miRNAs (Bentwich et al., 2005). Ensemble, ces miRNAs régulent l’expression d’approximativement 30 % du génome humain (Lewis et al., 2005).
1.1.1. Biogenèse des miRNAs
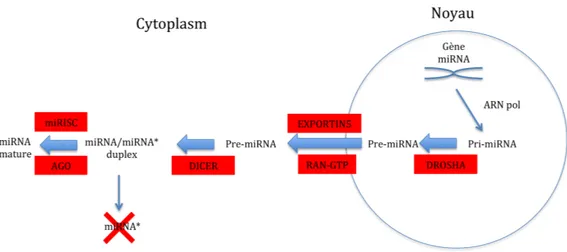
Les miRNAs sont des ARNs simple brin dont la taille moyenne est de 21 à 22 nucléotides. Ils sont transcrits par l’ARN polymérase II ou III dans le noyau sous forme de pri-miRNA (Borchert et al., 2006; Kim, 2005). Cette forme subit
deux étapes majeures de maturation (figure 1), la première dans le noyau par l’enzyme RNase III, Drosha, pour former le miRNA. Ensuite, le pré-miRNA est transporté par RanGTP et Exportin 5 vers le cytoplasme. Puis, une deuxième maturation par une autre RNase III, le Dicer, génère une forme transitoire de miRNA doubles brins. Cette forme transitoire est recrutée sur un complexe associé au miRNA, le miRISC, qui inclut la protéine Argonaute. Ce complexe retient un des deux brins préférentiellement, et devient le miRNA mature. Le brin opposé est éliminé (Kim and Kim, 2007; Ruby et al., 2007; Zhang, 2008a).
Figure 1. Biogenèse des miRNAs. Les miRNAs sont transcripts en pri-miRNA. Le pri-miRNA subit une première étape de maturation à travers le DROSHA pour devenir le pré-miRNA. Ce dernier est transporté vers le cytoplasme où il subira une deuxième étape de maturation par le DICER pour former le miRNA mature double brin. La séquence mature du miRNA sera recrutée sur l’ARNm cible par le complexe miRISC. Adapté de (Kim and Kim, 2007; Ruby et al., 2007).
1.1.2. Fonctions et modes d’action
Les miRNAs agissent en se liant sur des sites complémentaires d’ARNm pour inhiber l’expression génique selon deux manières: (1) les miRNAs qui se lient parfaitement à l’ARNm cible induisent une dégradation de l’ARNm. (2) Les miRNAs qui lient imparfaitement l’ARNm cible bloquent la traduction de l’ARNm sans toutefois dégrader ce dernier. Cette deuxième méthode est la plus fréquente et les mécanismes par lesquels les miRNAs inhibent cette traduction sont encore mal compris (Hutvagner and Zamore, 2002; Zeng et al., 2003; Zhang, 2008a). Une des particularités des miRNAs est de pouvoir réguler l’expression de plusieurs gènes dans une même cellule (Lim et al., 2005). D'autre part, plusieurs miRNAs peuvent réguler le même gène. Les miRNAs peuvent s’organiser en tandem et peuvent être groupés en “cluster’’ très proche dans le génome. Dans plusieurs cas, ces miRNAs ont des séquences très similaires et la cohorte de miRNAs peut réguler de manière additive un ou plusieurs ARNm (Ambros, 2004).
1.1.3. Études in silico
Chez les animaux la plupart des miRNAs ont des sites de liaisons qui ne sont pas parfaitement complémentaires. Pour cette raison, les prédictions de cibles potentielles de miRNAs, par le biais de logiciels de prédictions de miRNAs, sont basées sur plusieurs critères :
• La séquence de miRNA est complémentaire à une région qui se trouve dans le 3’UTR de l’ARNm (Doench and Sharp, 2004; Kim et al., 2006).
• La complémentarité entre les deux séquences doit être parfaite dans la région « seed » qui représente les nucléotides 2 à 8 de la terminaison 5’ du miRNA.) (Doench and Sharp, 2004; Kim et al., 2006).
• Des appariements en dehors de cette région « seed » (c’est à dire dans le 3’ du miRNA) sont aussi requis (Vella et al., 2004).
• Un appariement presque parfait dans la région 3’ n’est pas suffisant sans un appariement parfait de la région « seed » (Ghosh et al., 2007).
• Les liaisons G:U « wobbles » peuvent jouer un rôle dans l’appariement (Brennecke et al., 2005; Doench and Sharp, 2004). • Les coopérations de contrôle traductionnel d’un ARNm cible par
plusieurs miRNAs, ainsi que le potentiel d’un miRNA à cibler plusieurs ARNm sont aussi pris en considération (Doench and Sharp, 2004; Ghosh et al., 2007).
1.1.4. Potentiels thérapeutiques
Malgré notre compréhension encore primitive des miRNAs, plusieurs études montrent que les miRNAs pourraient représenter une nouvelle classe pharmacologique, et ce, dans différents domaines thérapeutiques. Les différentes modulations thérapeutiques des miRNAs seraient basées sur des stratégies d’inhibition des miRNAs ou de remplacement des miRNAs (Esau and Monia, 2007). La technologie la plus efficace pour l’inhibition des miRNAs est l’utilisation d'anti-miRNA oligonucléotides (AMOs). Cette inhibition se fait par des appariements de type Watson-Crick entre les AMOs et son miRNA cible. Des modifications sur certains nucléotides sont essentielles pour la stabilité de l’AMO (Esau and Monia, 2007). Les AMOs sont beaucoup utilisés dans les expériences in vitro de culture cellulaire, mais ils représentent aussi un mode thérapeutique potentiel (Esau and Monia, 2007; Liu et al., 2008). L’effet inverse d’une inhibition est la surexexpression de miRNAs; elle se fait principalement par deux méthodes : par transfection transitoire de miRNA double brins ou par l’utilisation d’un vecteur qui peut surexprimer un miRNA spécifique (Liu et al., 2008). Ainsi, dans un cas pathogène où un miRNA est faiblement exprimé cette technique permettrait de restaurer les niveaux basaux de miRNA. Les méthodes de transfert des AMOs seraient similaires à ceux des siRNAs, puisque les molécules sont de tailles et de natures très similaires (Liu et al., 2008). Le transfert des siRNAs locaux a déjà été réussi dans plusieurs organes comme les yeux et les poumons (Soifer et al., 2007). Le problème
majeur reste le transfert de ces molécules dans des cellules et tissus spécifiques, plutôt que dans un organe entier (Soifer et al., 2007).
1.2. Rôle des miRNAs dans le développement cardiaque et les
maladies cardiovasculaires
Le cœur est le premier organe fonctionnel formé dans l’embryon et tous les évènements subséquents de la vie sont dépendants de sa contraction (Liu and Olson, 2010). Une étude de l’expression des miRNAs impliqués dans les maladies cardiovasculaires a montré que la majorité des 18 miRNAs les plus exprimés dans le cœur avaient des expressions altérées dans les maladies cardiaques chez des adultes (Small et al., 2010). Ceci démontre l’importance des miRNAs dans la régulation de l’expression des gènes modulateurs des maladies cardiovasculaires (Small et al., 2010).
1.2.1. Cardiogenèse
Des analyses systématiques de l’expression spatiotemporelle des miRNAs ont montré que plusieurs miRNAs sont impliqués dans la régulation de l’expression de gènes impliqués dans la cardiogenèse durant des phases précoces de l’embryon (Catalucci et al., 2009; Latronico et al., 2007). Ainsi, miR-1, miR-133 jouent des rôles importants dans la prolifération et la différentiation musculaire (Catalucci et al., 2009; Chen et al., 2006). L’injection
de miR-1 ou de miR-133 dans des Xenopus Laevis à différents stades de développement embryonnaire, induit des changements drastiques dans le développement cardiaque de l’amphibien (Chen et al., 2006): aucun tissu cardiaque n’a pu être observé suite à l'injection de miR-1. En revanche, une injection de miR-133 induit une formation de tissus cardiaques, mais avec une formation très désorganisée (Chen et al., 2006). L’effet inhibiteur de miR-1 sur la myogenèse s’effectue en régulant l’histone deacétylase 4 (HDAC4). MiR-133 aiderait à la prolifération des myoblastes en inhibant le serum response factor (SRF) (Chen et al., 2006).
1.2.2. Hypertrophie cardiaque
Le cœur répond à des dommages chroniques et aigus par une croissance hypertrophique et une diminution de la contractilité cardiaque. Le miR-208a est requis pour le remodelage cardiaque hypertrophique en affectant la contractilité cardiaque (van Rooij et al., 2006). La contractilité cardiaque dépend des deux gènes myosin heavy chain (MHC) α et β qui sont régulés de manière opposée. Le gène MHC α encode pour le miR-208a. Lorsque miR-208a est surexprimé, il active indirectement l’expression du gène MHC β, ce qui diminue la contractilité (van Rooij and Olson, 2007). D’autre part, miR-133 est un régulateur négatif de l’hypertrophie puisqu’il inhibe le remodelage cardiaque (Care et al., 2007). Ce miR-133 agit en ciblant RhoA, une protéine échangeur
de GDP-GTP qui régule l’hypertrophie cardiaque, mais miR-133 cible aussi Cdc42 qui est une kinase impliquée dans l’hypertrophie (Care et al., 2007).
1.2.3. Apoptose et prolifération des cardiomyocytes.
Le cœur a une capacité limitée de régénération. Une perte excessive de cardiomyocytes après un infarctus du myocarde peut diminuer significativement la performance cardiaque (Small et al., 2010). La suppression du gène de miR-199a induit l’apoptose en conditions d’ischémie alors qu’il contribue aux effets bénéfiques du conditionnement préischémique et protège les cardiomyocytes contre les dommages ischémiques (Rane et al., 2009). La sous-expression de miR-199a est requise pour l’augmentation d’expression de sa cible directe le hypoxia-inducible factor (HIF) 1α (Rane et al., 2009). MiR-199a régule aussi Sirt-1 qui inhibe le propyle hydroxylase 2 (PHD2) inhibant à son tour HIF-1α (Rane et al., 2009). L’expression de miR-320 est faible dans les cœurs ayant subi des dommages de reperfusion postischémique (Ren et al., 2009). La surexpression du miR-320 dans ces conditions conduit à une augmentation de la taille de l’infarctus ainsi qu’une augmentation des niveaux d’apoptose (Ren et al., 2009). MiR-320 agit en inhibant directement Heat-Shock Protein 20 (HSP20) qui est surexprimée dans les coeurs de souris suite à des conditions ischémiques (Ren et al., 2009; Small et al., 2010).
1.2.4. Arythmie cardiaque
Les arythmies cardiaques sont des perturbations électriques qui causent des battements cardiaques irréguliers. Les arythmies cardiaques constituent la cause principale de mort subite (Pan et al., 2010). Malgré l’absence de preuves cliniques de l’implication des miRNAs dans les arythmies cardiaques, des études montrent leurs implications dans des modèles animaux. Ainsi, la surexpression de miR-1 dans les cœurs de rats augmente les incidences d’arythmies (Yang et al., 2007). L’effet inverse est observé lorsque l’on inhibe miR-1 (Yang et al., 2007). Des études montrent que cet effet est dû à un remodelage électrophysiologique des canaux potassiques (Terentyev et al., 2009). Le miR-133 a aussi des effets de remodelage électrophysiologique. Une sous-expression de miR-133 est associée à une surexpression de deux canaux ioniques HCN2/HCN4 ce qui induit un développement d’arythmies qui sont observées dans des cas d’insuffisance cardiaque (Catalucci et al., 2009; Ivey et al., 2008; Yang et al., 2007).
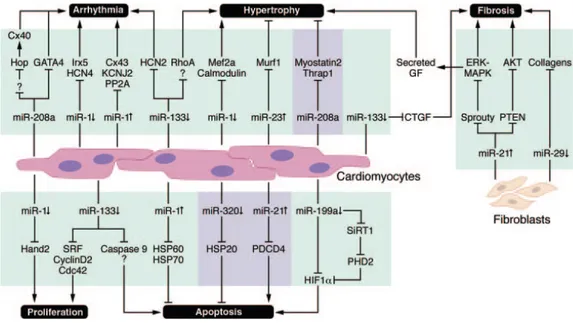
1.2.5. Fibrose
Les miRNAs sont impliqués dans d’autres types cellulaires que les myocytes, dont les fibroblastes (figure 2). Les fibroblastes produisent la matrice extracellulaire de fibres de collagènes à l’entour des myocytes cardiaques. Suite à un stress pathologique ou un infarctus du myocarde, les fibroblastes sécrètent
des protéines de matrice extracellulaire de manière excessive et non proportionnelle (van Rooij et al., 2008a). Plusieurs études montrent que les miR-133, miR-21, miR-29 et miR-30 participent au remodelage de la matrice extracellulaire dans des cas de fibrose (Pan et al., 2010; van Rooij et al., 2008b). Par exemple, des expériences d’inactivation du gène du miR-29 in vivo augmentent les niveaux de collagène dans le cœur (Thum et al., 2008; van Rooij et al., 2008b). L’implication du miR-21 dans la fibrose a également été démontrée: une surexpression du miR-21 dans les fibroblastes augmente la survie de fibroblastes. De plus, l’inhibition du miR-21 chez des souris exposées à une surcharge de pression hémodynamique induit une diminution de l’expression des gènes qui sont très fortement exprimés durant la fibrose et qui codent pour les collagènes (Pan et al., 2010; Roy et al., 2009; Thum et al., 2008).
Figure 2 : Altération de l’expression des miRNAs impliqués dans les maladies reliées aux cardiomyocytes et fibroblastes (Small et al., 2010). AP-1 : activator protein 1; CDC42 : cell division cycle 42; CTGF: connective tissue growth factor; Cx43: connexin 43; Hand2: heart and neural crest derivatives expressed 2; HCN2: hyperpolarization-activated cyclic nucleotide-gated potassium channel 2; HIF1: hypoxia-inducible factor; Hop: homeodomain-only protein; Hsp: heat shock protein; Irx5: Iroquois homeobox protein 5; Kcnd2: potassium voltage-gated channel subfamily D member 2; Kcnj2: potassium inwardly rectifying channel, subfamily J, member 2; MuRF 1: muscle RING-finger protein 1; PDCD4: programmed cell death 4; PHD2: prolyl hydroxylase 2; PP2A: B56a regulatory subunit of protein phosphatase 2a; PTEN: phosphatase and tensin homolog; RhoA: Ras homolog gene family, member A; Sirt1: sirtuin; SRF: serum response factor; and Thrap1: thyroid hormone receptor associated protein 1 (Small et al., 2010).
1.2.6. Angiogenèse
La néoangiogénèse est la formation de nouveaux vaisseaux sanguins. La néoangiogénèse joue un rôle essentiel dans le processus de réparation cardiaque suite à des dommages ischémiques. Dans le cas d’un infarctus du myocarde, la formation de néovaisseaux permet de restaurer le débit sanguin dans la zone ischémique (Kutryk and Stewart, 2003). Ce processus exige une signalisation par des facteurs angiogéniques comme le vascular endothelial growth factor (VEGF) et fibroblast growth factor (FGF) (Scheinowitz et al., 1997; Syed et al., 2004; van Rooij et al., 2008a).
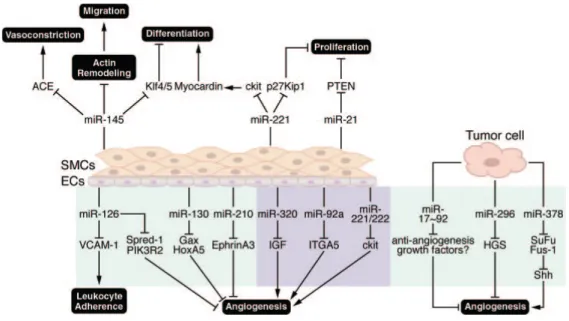
Plusieurs miRNAs jouent un rôle dans l’angiogenèse tumorale comme le miR-296, miR378 et le cluster miR-17~92 (Figure 3). Au niveau endothélial, le rôle de miR-126, qui est spécifique à l’endothélium, est le plus étudié (Wang et al., 2008). L’élimination du miR-126 chez des souris provoque des hémorragies et une mortalité embryonnaire partielle, avec des défauts précoces de la formation des vaisseaux cérébraux et rétiniens. Les souris survivantes sont identiques aux souris contrôles suggérant que miR-126 est un important régulateur du développement, mais pas du maintien à l’âge adulte. Trois semaines post-infarctus, 70 % des souris sauvages (wild-type) ont survécu, alors que seulement 18 % des souris miR126-/- ont survécu; cette mortalité précoce est liée à des défauts de vascularisation cardiaque avec rupture du ventricule (Wang et al., 2008). Parmi les cibles identifiées de miR-126, il y a "vascular cell adhesion protein 1" (VCAM-1) d’où le rôle de miR-126 dans
l’inflammation (Schmidt et al., 2007), sprouty-related, EVH1 domain containing 1 (Spred-1), un régulateur de la voie des MAP kinases (Fish et al., 2008) et PI3K regulatory subunit 2 (PIK3R2) (Wang et al., 2008). Plusieurs études d’angiogenèse se concentrent sur le rôle d’un autre miRNA spécifique de l’endothélium, le cluster miR-17~92. Ce miRNA contrôle l’angiogenèse ainsi que la réparation des tissus endommagés à la suite d’un infarctus du myocarde (Bonauer et al., 2009; Wang and Olson, 2009).
Figure 3. Altération de l’expression des miRNAs impliqués dans les maladies reliées au remodelage vasculaire et à l’angiogenèse. ACE: angiotensin converting enzyme; SuFu: suppressor of fused; Shh: sonic hedgehog; HGS: hepatocyte growth factor–regulated tyrosine kinase substrate; IGF: insulin-like growth factor; ITGA5: integrin-5; PIK3R2: phosphoinositol-3 kinase regulatory subunit 2 (p85); PTEN: phosphatase and tensin homolog; Spred-1: sprouty-related EVH domain-containing protein-1; VCAM-1 (Small et al., 2010).
1.3. Sénescence
La sénescence est un arrêt de croissance cellulaire permanent dans lequel les cellules sont métaboliquement actives, mais incapables de se diviser (Kuilman et al., 2010). La sénescence a tout d'abord été remarquée lorsque des cellules primaires humaines (fibroblastes) extraites de tissus humains mis en culture n’arrivaient plus à proliférer (Hayflick and Moorhead, 1961; Kuilman et al., 2010). Ce phénomène cellulaire est causé par l’attrition progressive des télomères qui accompagne inévitablement la réplication cellulaire, mais la sénescence peut aussi être induite prématurément par un stress (Stress-induced-premature-senescence : SIPS), en absence de raccourcissement ou même de dysfonction télomérique (Kuilman et al., 2010).
1.3.1. Sénescence Réplicative vs SIPS
La sénescence réplicative est observée lorsque les cellules en culture (non cancéreuses ou génétiquement modifiées) in vitro ne se divisent plus et présentent des phénotypes qui accompagnent la sénescence: une morphologie large et plate, très vésiculaire, un arrêt de la synthèse d'ADN et une augmentation de la β-galactosidase acide associée à la sénescence. Ce dernier phénotype n’est pas observé dans toutes les formes de sénescence (Deng et al., 2008). Outre les phénotypes mentionnés, les cellules sénescentes plusieurs changements dans leur sécrétome c’est ce qu’on appelle la
senescence-associated-secretory-phenotype (SAPS) (Coppe et al., 2008; Deng et al., 2008; Kuilman et al., 2010). La sénescence réplicative est largement causée par un raccourcissement des télomères, une structure protectrice de tous les chromosomes eucaryotes, raccourcissement qui accompagne la division cellulaire. Lorsque les télomères ont raccourci jusqu’à un niveau critique, les dommages à l’ADN engendrés poussent les cellules à entrer en sénescence. La télomérase (hTERT) est une enzyme qui allonge le télomère et protège les cellules contre les dommages à l’ADN (Deng et al., 2008; Kuilman et al., 2010). L’induction du SIPS est générée lors de l’utilisation in vitro de faibles doses non toxiques de différentes sources de stress comme les rayonnements UV, le peroxyde d’hydrogène, les agents chemiothérapeutiques et les rayonnements ionisants (Suzuki and Boothman, 2008). Le SIPS est induit à un stade plus précoce que la sénescence réplicative puisque l’attrition des télomères jusqu’au niveau critique n’est pas encore réalisée (Suzuki and Boothman, 2008).
1.3.2. miRNAs impliqués dans la sénescence
Plusieurs études s’intéressent à l’implication des miRNAs dans la sénescence cellulaire. Le miR-146 est surexprimé dans des cellules sénescentes de fibroblastes humains comparés à des cellules quiescentes. Ce miRNA inhibe les modulateurs inflammatoires associés à la sénescence : interleukine 6 et 8 (IL-6 et IL-8) (Bhaumik et al., 2009). D’autre part, le miR-34a est surexprimé
dans les cellules endothéliales de la veine ombilicale humaine (HUVEC) sénescente. Une surexpression de miR-34a induit la sénescence en inhibant la sirtuine 1 (SIRT-1) une déacétylase qui a comme cible le gène suppresseur de tumeur p53 (Ito et al., 2010; Yamakuchi and Lowenstein, 2009). Un autre miRNA agit de façon identique. La surexpression de miR-217 dans des HUVEC, des cellules endothéliales aortiques ainsi que des cellules endothéliales d’artères coronaires humaines induit la sénescence prématurément. Ce miRNA inhibe SIRT-1 modulant ainsi l’expression de Forkhead box protein O1 (FoxO1) et la synthase du monoxyde d’azote endothélial (eNOS ou NOS3). Une inhibition de miR-217 sur ces types cellulaires réduit les niveaux de sénescence et augmente les propriétés angiogéniques des cellules (Menghini et al., 2009). Une étude a évalué les changements d’expression des miRNAs lors de l’induction de SIPS dans des fibroblastes diploïdes et de trabéculum cornéoscléral humain. Cette étude montre que l’expression de 24 miRNAs est significativement changée. Parmi ces miRNAs, miR-16 est sous-exprimé dans les cellules sénescentes (Li et al., 2009).
1.4. Transport actif du glucose
Le glucose est l’une des molécules biologiques les plus abondantes dans la nature. Au cours de l’évolution, les mécanismes cataboliques et anaboliques ont constamment évolué pour utiliser le glucose comme substrat (Thorens and
Mueckler, 2010). Les cellules ont besoin de transporteurs de glucose pour permettre l’entrée de glucose dans les cellules.
1.4.1. Transporteurs de Glucose
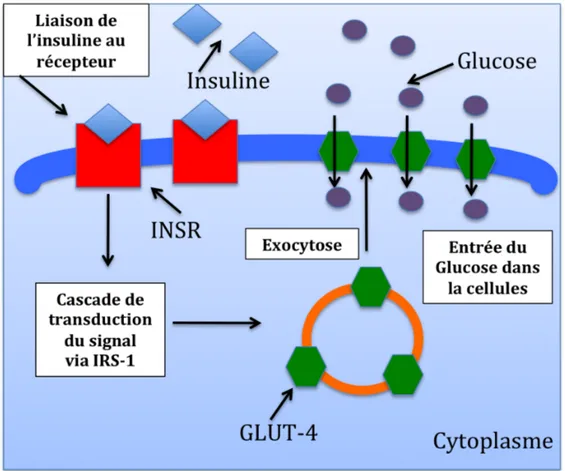
La famille des transporteurs de glucose comprend 14 membres. Ils sont surnommés SLC2A, mais le nom le plus couramment utilisé est GLUT (GLUT-1 à (GLUT-14). La plupart des transporteurs de glucose catalysent un transfert facilité (dépendant de l’énergie) bidirectionnel de leurs substrats (Thorens and Mueckler, 2010). Parmi les transporteurs de glucose, GLUT-1 et GLUT-4 sont les plus étudiés. GLUT-1 est indépendant de l’insuline. Les mécanismes par lesquels il transporte le glucose ne sont encore pas bien élucidés. Le GLUT-4 est un transporteur de glucose dépendant de l’insuline. Une stimulation avec l’insuline active le récepteur à l’insuline (INSR), un homodimère de type tyrosine kinase. Cette activation initie une cascade de signalisation à travers la voie des phosphoinositol 3 kinase (PI3K) et du substrat du récepteur à l’insuline 1 (IRS-1) (Chiu and Cline, 2010). Cette cascade de signalisation engendre le transport de glucose par le trafic des GLUT-4 intracellulaires en trois étapes (Figure 4) : la ségrégation du GLUT-4 dans des vésicules, suivie par la migration de ces vésicules vers la membrane plasmatique, et finalement la fusion de ces vésicules avec la membrane plasmatique (Brady et al., 1999). Une fois le GLUT-4 dans la membrane plasmatique, il pourra transporter le glucose à l’intérieur de la cellule. Le GLUT-4 est prédominant, mais non
exclusivement, exprimé dans les tissus adipeux ainsi que dans les tissus musculaires squelettiques et cardiaques (Brady et al., 1999; Klip, 2009).
Figure 4. Transport actif du glucose par le GLUT-4. Suite à une stimulation par l’insuline, une cascade de signalisation traduit un signal qui permet aux vésicules sécrétrices de GLUT-4 de se localiser vers la membrane cytoplasmique où GLUT-4 permettra l’entrée de glucose dans les cellules.
1.4.2. miRNA et INSR
A ma connaissance, aucune étude à jour n’implique une corrélation directe ou indirecte entre les miRNAs et le récepteur à l’insuline.
1.4.3. miRNA et IRS-1
Contrairement au INSR, l’implication des miRNAs dans la régulation de IRS-1 est plus étudiée. IRS-1 est souvent surexprimé dans la plupart des cancers humains (Baserga, 2009)En effet, IRS-1 est une cible directe de miR-145, ce qui inhibe la croissance des cellules cancéreuses du colon (Shi et al., 2007). D’autre part, deux études montrent que le miR-126 inhibe IRS-1 également au niveau traductionnel (La Rocca et al., 2009; Zhang et al., 2008). Une troisième étude montre que miR-126 est sous-exprimé dans les cellules du cancer du sein et une surexpression de miR-126 inhibe la progression du cycle cellulaire de la phase G1/G0 à la phase S (Zhang et al., 2008). L’inactivation génique de IRS-1 mime les effets de miR-126 (Zhang et al., 2008). Cela suggère que l’effet de la surexpression de miR-126 sur l’arrêt du cycle cellulaire se fait par l’inhibition de IRS-1 (Zhang et al., 2008).
1.4.4. miRNA et GLUT-4
Deux études ont évalué l’implication des miRNAs dans la régulation du GLUT-4. Le miR-133 est impliqué dans le contrôle du métabolisme des myocytes cardiaques par la régulation l’expression de GLUT-4 en ciblant le Krueppel-like factor 15 (KLF15) (Horie et al., 2009). Le facteur de transcription KLF15 active le promoteur de GLUT-4 (Gray et al., 2002). Ainsi, une inhibition de KLF15 par miR-133 diminue les niveaux de GLUT-4. Outre
KLF15, plusieurs protéines ciblent GLUT-4 au niveau transcriptionnel, mais aucune régulation post-transcriptionnelle n’est encore connue. Le miR-223 régule l’expression de GLUT-4, mais en augmentant les niveaux de GLUT-4 et ultimement le transport de glucose des cardiomyocytes (Lu et al., 2010). Cette augmentation peut s’expliquer par l’inhibition d’une protéine inhibitrice de GLUT-4 (Lu et al., 2010). Ces miRNAs peuvent donc avoir des effets indirects sur le diabète.
1.4.5. Rôle des miRNAs dans le diabète
Le diabète mellitus est une maladie chronique caractérisée par une hyperglycémie causée par une déficience en expression d’insuline, par un problème du mécanisme d’action de l’insuline, ou les deux à la fois (2011). Il existe deux types de diabète: le diabète de type I causé par l’absence de sécrétion d’insuline, et le diabète de type 2 causé en partie par une résistance à l’insuline ainsi qu’une sécrétion inadéquate d’insuline compensatrice (Gutteridge, 1999). Ce dernier constitue la majorité des cas de diabète. Le diabète de type 2 est associé à un polymorphisme de type insertion/délétion ACAA dans le 3’UTR du « insulin growth factor 2 receptor » (IGF2R). Une étude montre que le miR-657 est un régulateur direct de IGFR2 dans des cellules hépatiques HEP G2. La séquence complémentaire de ce miRNA se trouve dans la séquence contenant le polymorphisme ACAA, ce qui suggère que miR-657 est un régulateur potentiel du diabète de type 2 (Lv et al., 2008).
D’autre part, une étude a évalué l’expression de miRNA dans le plasma de patients diabétiques. Parmi les miRNAs dont l’expression change, le miRNA endothélial miR-126 diminue fortement chez les patients comparés aux sujets sains (Zampetaki et al., 2010). Ces résultats ont été reproduits dans un modèle de souris hyperglycémiques (Zampetaki et al., 2010). Contrairement au miR-126, le miR-503 est surexprimé dans le plasma de patients diabétiques ainsi que dans des cellules endothéliales en conditions hyperglycémiques (Caporali et al., 2011). Ce miRNA régule directement les gènes « G1/S-specific cyclin E1 » (CCNE1) et « cell division cycle homolog 25 A » (CDC25A) qui sont sous-exprimés dans les cellules endothéliales en conditions hyperglycémiques (Caporali et al., 2011).
1.5. microRNA-16
Le miRNA-16 est l’un des miRNAs les plus étudiés. Il fait partie de la famille des miR-15/107. Cette famille est constituée de 10 membres qui sont exprimés chez l’humain : miR-15a, miR-15b, miR-16, miR-103, et miR-107 sont exprimés chez tous les vertébrés tandis que miR-195, miR-424, miR-497, miR-503 et miR-646 ne sont exprimés que chez les mammifères. Ils ont tous une séquence conservée qui commence, du côté 5’, du premier ou du deuxième nucléotide par AGCAGC (Finnerty et al., 2010) (Figure 5). Cette similarité entre les séquences des différents membres de la famille fait en sorte que plusieurs miRNAs peuvent avoir des cibles communes. Ils ont donc
probablement des rôles cellulaires semblables et complémentaires. En effet, leurs rôles dans différentes pathologies révèlent des similitudes avec différents membres de la famille (Finnerty et al., 2010).
Figure 5. Séquence de la famille des miR-16. Tous ces miRNAs commencent (au premier ou deuxième nucléotide) avec une séquence conservée au 5’ : AGCAGC (Finnerty et al., 2010).
1.5.1. Rôles connus de la famille de miR-15/107
La famille de miR-15/107 est impliquée dans plusieurs processus biologiques : la division cellulaire, le stress et le métabolisme cellulaire ainsi que l’angiogenèse (Finnerty et al., 2010). Ces miRNAs sont aussi impliqués dans plusieurs maladies : les cancers, les maladies neurodégénératives et les maladies cardiaques (Finnerty et al., 2010). MiR-16 est impliqué dans plusieurs
de ces processus. C’est un miRNA qui est reconnu pour son rôle antiprolifératif, mais qui a plusieurs autres fonctions cellulaires.
1.5.1.1. miR-16 et division cellulaire
Un groupe a démontré que miR-16 inhibe la croissance cellulaire ainsi que la progression du cycle cellulaire (Linsley et al., 2007). Cette étude montre que miR-16 module l’expression de plusieurs gènes impliqués dans le contrôle de la progression du cycle cellulaire. MiR-16 agit en inhibant simultanément un groupe de gènes et c’est ce contrôle collectif qui induit un arrêt du cycle cellulaire (Linsley et al., 2007). L’inhibition individuelle de ces gènes n’a pas d’effet sur le cycle cellulaire (Linsley et al., 2007). L’effet du miR-16 observé pourrait ainsi être expliqué par un contrôle collectif et non individuel des gènes régulateurs du cycle cellulaire.
1.5.1.2. miR-16 dans le métabolisme et le stress cellulaire
L’implication du miR-16 dans le métabolisme cellulaire n’est pas encore bien connue. Une étude a évalué l’expression des microRNAs dans des cellules pancréatiques β suite à des changements de concentration de glucose. Parmi les miRNAs dont l’expression augmente suite à une stimulation avec le glucose, on retrouve ceux de la famille de miR-15/107 incluant miR-16 (Tang et al., 2009). Des perturbations du métabolisme peuvent causer un stress cellulaire, mais l’inverse est vrai aussi (Finnerty et al., 2010). Les niveaux de miR-16
augmentent suite à une irradiation UV. Cette induction des niveaux de miR-16 inhibe le gène CDC25a, un médiateur essentiel du point de contrôle de la phase G1-S et régule ainsi la prolifération cellulaire (Pothof et al., 2009).
1.5.1.3. miR-16 et angiogenèse
Le VEGF est l’un des facteurs de survie les plus importants des cellules endothéliales. Son rôle dans l’angiogenèse est essentiel. MiR-16 inhibe directement la traduction de VEGF en ciblant son ARNm (Karaa et al., 2009). Ceci devrait faire de miR-16 un régulateur direct de l’angiogenèse dans les cellules endothéliales, mais aucune étude ne montre cet effet physiologique. D’autres membres de la famille sont impliqués dans l’angiogenèse comme miR-107. Ce dernier est exprimé dans le cancer du côlon humain et il est transcriptionnellement régulé par le gène p53. Ce miRNA inhibe l’expression de « l’hypoxia-inducible factor-1β » (HIF-1β) et affecte la signalisation hypoxique, régulant ainsi la croissance tumorale du cancer du côlon dépendante de l’angiogenèse pathologique nécessaire à la demande métabolique de la tumeur (Finnerty et al., 2010; Yamakuchi et al., 2010)
1.5.1.4. miR-16 et cancer
MiR-16 est considéré comme un suppresseur de tumeur. Les effets cellulaires décrits précédemment sont en lien avec cette considération, mais
plusieurs autres études montrent que miR-16 est impliqué dans l’inhibition de la prolifération cellulaire, l’activation de l’apoptose, la suppression de tumeur in vitro et in vivo (Aqeilan et al., 2010). Plus de 100 études ont exploré ces aspects du miR-16. Brièvement, voici les effets du miR-16 les plus pertinents.
La première évidence de l’implication du miR-16 dans le cancer est dans la leucémie lymphocytaire chronique (CLL). Plus de la moitié des cas ont une altération génétique homozygote ou hétérozygote de la région 13q14.3 de l’ADN (Chiorazzi et al., 2005). Cette observation suggère la présence de gènes suppresseurs de tumeurs dans cette région. Cette région de l’ADN code pour le miR-15/16. Des délétions des gènes de miR-15 et miR-16 sont associées à la CLL. Le miR-15/16 est aussi impliqué dans d’autres types de cancer comme le cancer de la prostate où le miR-15/16 est sous exprimés dans 80 % des tissus cancéreux de patients comparés à des tissus sains (Bonci et al., 2008). D’autre part, les miR-15/16 sont sous-exprimés dans les cellules d’adénomes pituitaires comparées à des cellules pituitaires normales (Bottoni et al., 2005). Des études ont par la suite cherché à trouver par quels mécanismes ce miRNA agit comme suppresseur de tumeur. Un oncogène B-cell lymphome 2 (BCL2) est surexprimé dans les cellules B de la CLL. Cette surexpression est corrélée avec la sous-expression des miR-15/16 dans ces mêmes cellules. MiR-16 agit en induisant l’apoptose en inhibant directement l’expression de BCL2 (Cimmino et al., 2005; Xia et al., 2008). MiR-15/16 cible aussi d’autres oncogènes comme la cycline D1 (CCND1) et le gène WNT3A (Bonci et al., 2008).
2 Raisonnement et objectifs de l’étude
MiR-16 est l’un des miRNAs les plus étudiés. Les divers effets de miR-16 décrits précédemment démontrent la complexité des miRNAs dans le fonctionnement cellulaire par la régulation d’une multitude de cibles directes et indirectes. Un des mécanismes régulés par les miRNAs est la sénescence, un arrêt irréversible de croissance cellulaire. Les cellules sénescentes restent cependant métaboliquement actives et il a été démontré que la sénescence réplicative peut être induite par le glucose (Blazer et al., 2002; Stolzing et al., 2006). Nous avons observé que l’expression de miR-16 est diminuée dans les cellules endothéliales sénescentes. Ainsi, une diminution de miR-16 dans les cellules sénescentes pourrait représenter une tentative de compensation pour contrer la sénescence, qui mène ultimement à une diminution du métabolisme, en augmentant l’entrée de glucose dans les cellules. À ce jour, aucune étude ne montre l’implication de miR-16 dans la modulation du métabolisme cellulaire. Nous avons émis l'hypothèse que miR-16 régule le transport du glucose par la régulation de GLUT-4, un transporteur de glucose dépendant de l’insuline. Mon projet vise à étudier l’implication du miR-16 dans la régulation de l’expression de GLUT-4. D’autre part, j’ai étudié la corrélation existant entre l’expression de miR-16 dans des artères mammaires de patients coronariens et leur glycémie. Finalement, une relation entre la régulation de GLUT-4 par miR-16 et la
glycémie a été établie dans un modèle de souris soumises à une diète riche en calories.
Mon hypothèse de travail est que miR-16 est impliqué dans le métabolisme cellulaire par l’inhibition de l’expression protéique du transporteur de glucose dépendant de l’insuline, GLUT-4. Cette inhibition de l’expression de GLUT-4 par miR-16 diminue l’entrée de glucose dans les cellules et augmente ainsi la glycémie.
3 Matériel et Méthodes
3.1. Analyse in silico
Toutes les constructions et analyse du clonage moléculaire ainsi que le design d’amorces et l’analyse des données de séquençage ont été faits à l’aide du logiciel clone manager suite.
Les prédictions des sites de liaisons des miRNAs aux gènes cibles ont été faites à l’aide de quatre logiciels offerts en ligne :
• www.pictar.mdc-berlin.de • www.targetscan.org • www.mirbase.org • www.microrna.org
3.2. Microarray
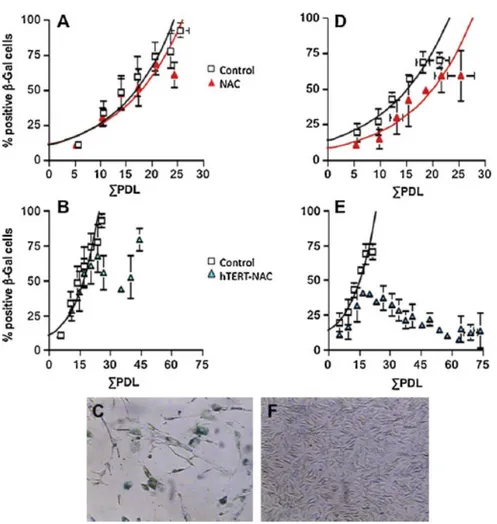
Pour mieux comprendre le principe et la méthodologie utilisés pour le microarray, voici une mise en contexte du sujet. Des résultats préliminaires du laboratoire ont montré que le traitement avec un antioxydant, le N-acétyl-cystéine (NAC) combiné à l’introduction de la télomérase (hTERT) dans des cellules endothéliales en culture d’artères mammaires internes de patients coronariens (hIMAEC, "human internal mammary artery endothelial cells") engendre l’immortalisation dans seulement la moitié (5/9) des cas. Pour l’autre moitié, les cellules ont arrêté de proliférer et sont entrées en sénescence (Figure 6).
Figure 6. Effet du traitement simultané au NAC et hTERT sur les hIMAEC. A, B et C: Cellules qui sont entrées en sénescence suite au traitement NAC. D, E et F, cellules qui ont été immortalisées suite au traitement hTERT-NAC. A et D: Courbe temps dépendant de la moyenne (mean ± SEM, n=5) des niveaux de sénescence dans les cellules traitées au NAC seulement. B et E: Courbe temps dépendant de la moyenne (mean ± SEM, n=5) des niveaux de sénescence dans les cellules traitées au NAC et hTERT. C et F: Photos des deux groupes: cellules sénescentes (B) contre cellules immortalisées (F) du marquage sénescence-associated β-galactosidase (Voghel et al., 2010).
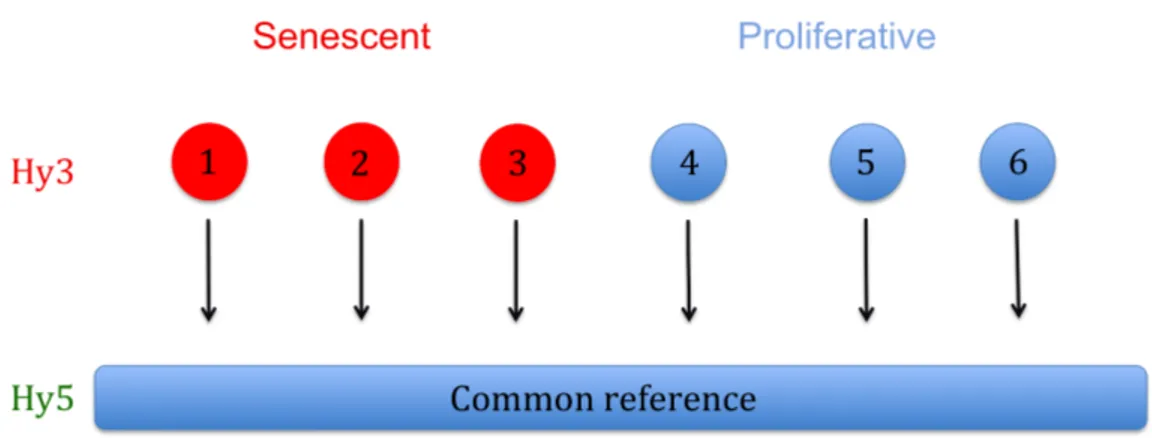
Le but de l’expérience avec le microarray est de trouver des miRNAs exprimés différemment entre les deux groupes (sénescent ou immortalisé). Ainsi, nous avons effectué un microarray de miRNAs sur 3 échantillons de chaque groupe. Chaque échantillon provient d’un patient différent. Le microarray permet d’identifier l’expression de tous les miRNAs connus. La puce de microarray contient dans chaque puits des amorces spécifiques à chaque miRNA qui permettra d’amplifier et ainsi de quantifier le miRNA. Dans chaque puits sont ajoutés deux échantillons marqués différemment (Hy3 et Hy5). Le Hy5 représente la référence commune, c’est le même échantillon qui sera rajouté dans tous les puits de tous les échantillons. Cette référence commune est constituée d’une quantité égale de chacun des échantillons, elle sert donc de normalisateur. Le Hy3 représente les différents échantillons des deux groupes (Figure 7).
Figure 7. Représentation schématique de la méthodologie abordée pour le microarray. Représenté en rouge, les échantillons du groupe sénescent de l’expérience hTERT-NAC sur les cellules endothéliales d’artères mammaires de patients coronariens. Représenté en bleu, les échantillons du groupe sénescent de l’expérience hTERT-NAC sur les cellules endothéliales d’artères mammaires de patients coronariens. Common Reference : quantité égale des 6 échantillons utilisés pour le microarray. Cette méthodologie a été proposée par la compagnie Exiqon qui a effectué le microarray.
3.3. Culture cellulaire
Les cellules embryonnaires de rein (HEK 293, Invitrogen) ont été cultivées dans un incubateur à 37oC dans un mélange gazeux de 5% CO2 / air en présence de milieu de culture DMEM auquel ont été rajoutés 1% pénicilline / streptomycine et 10% FBS (Fetal Bovine Serum) (Gibco). Les cellules ont été passées lorsque confluentes, par incubation à 37oC pendant 8 minutes avec la
trypsin-EDTA (Sigma). Les cellules HEK293 sont originaires d’une souche de cellules tumorales utilisées à des passages élevés.
Les cellules endothéliales de la veine ombilicale (HUVEC, Lonza) ont été cultivées dans un incubateur à 37oC dans un mélange gazeux de 5% CO2 / air en présence dans leur milieu de culture spécifique EBM 2 (Endothelial basal medium, Lonza) additionné des facteurs de croissance endothéliaux (EGM 2, Lonza). Les cellules ont été passées lorsque confluentes par incubation à 37oC pendant 8 minutes avec la trypsin-EDTA pour HUVEC (Sigma). Les expériences sur les HUVEC ont été faites sur des cellules à passage 3 - 6 seulement. Les pétris utilisées pour les cellules endothéliales sont de type Cell+ (Sarstedt) pour une meilleure adhérence.
3.4. Transfection
La transfection des cellules HEK 293 et HUVEC s’est faite selon le même protocole avec optimisation de la transfection par Lipofectamine 2000 (Invitrogen). Une fois que les cellules sont à 90% confluentes et sans sérum pendant 12 hrs dans une plaque à 6 puits, les cellules sont transfectées. Les quantités d’ARN ou ADN transfectées étaient les suivantes :
• miR-16 ou contrôle : 100 nM • AMO-16 ou contrôle : 10 nM • Vecteurs Firefly : 50 ng
• Vecteurs Renilla : 1 ng
L’ADN ou ARN est doucement mélangé avec 250 µL de milieu opti-MEM (Invitrogen). Dans un autre tube, 5 µL de l’agent Lipofectamine 2000 sont mélangés doucement avec 250 µL du milieu opti-MEM, puis sont incubés à température pièce (TP) pendant 5 minutes. Les deux solutions sont ensuite mélangées et incubées à TP pendant 20 minutes. Le mélange est rajouté à 2 mL du milieu spécifique aux cellules. Les résultats présentés des transfections ont été obtenus après une incubation d’une durée de 24 heures.
3.5. Essai viabilité
Les cellules transfectées avec les mêmes conditions décrites précédemment ont été soumises à un essai de viabilité pour confirmer que les produits transfectés n’ont pas d’effets significatifs sur la mortalité. L’essai s’est fait avec le bleu de trypan. Les cellules ont été trypsinisées pendant 5 minutes à 37oC. On rajoute 1 mL de DMEM complet. Les cellules sont centrifugées à 250 g pendant 5 minutes. Le culot est lavé avec 1 mL de PBS et recentrifugé à 250 g pendant 5 minutes. On rajoute 1 mL de PBS et 50 µL de bleu de trypan (0,01%). Les cellules qui seront colorées en bleu représentent les cellules mortes. Le ratio cellules bleu par rapport aux cellules non colorées est compté et représente le % de mortalité.
3.6. Extraction protéique
Les artères mammaires humaines sont directement reçues du bloc opératoire, lors du pontage coronarien du patient, et nettoyées (gras et muscle) dans un délai maximal de 15 minutes après réception de l’échantillon. Les artères sont immédiatement plongées dans l’azote liquide et conservées à -80oC. L’extraction protéique des artères mammaires humaines s’est faite par broyage à l’aide d’un pilon et d’un mortier (Coors), sur glace sèche et dans l’azote liquide. La poudre est ensuite homogénéisée avec un broyeur (Wheaton), puis incubée à 4oC pendant 15 minutes dans un tampon de lyse auquel on rajoute un mélange d’inhibiteurs de protéases fraichement préparé (Protease Inhibitor Cocktail Kit, Thermo Scientific). La composition du tampon de lyse protéique est la suivante: Tris-HCl pH=7,5 50 mM, β-glycérophosphate 20 mM, fluorure de sodium 20 mM, EDTA 5 mM, EGTA 10 mM, Na3VO4 1 mM, Triton 1% (v/v)). Les 15 minutes d’incubation permettent d’extraire les protéines totales: membranes plasmiques, cytoplasmiques et nucléaires. Le lysat est ensuite centrifugé à 4oC pendant 15 minutes à 10000 g, le surnageant est ensuite recueilli.
L’extraction protéique des cellules HUVEC et HEK 293 a été soumise aux mêmes tampons et conditions de lyse que l’extraction protéique des artères. Le milieu a d’abord été aspiré des puits de culture de 35 cm2, puis le tampon de lyse a été ajouté, le tout maintenu sur la glace pendant 15 minutes.
3.7. Immunobuvardage de type Western
La concentration de protéines totales a été quantifiée par la méthode de dosage Bradford (BioRad). Les échantillons protéiques (50 à 100 µg) ont été solubilisés dans un tampon Laemmli : 50 mM de Tris-HCl (pH 6,8), 0,5 M de β-Mercaptoethanol, 2,5 % de SDS, 0,4 M de glucose, 1 mM d’EDTA et 0,05 % de bleu de bromophénol. Les échantillons ont été chauffés à 95oC pendant 4 minutes. Les protéines ont migré par électrophorèse dans un gel de polyacrylamide dénaturant de 10% SDS-PAGE ou un gel dénaturant à gradient de 4-20% SDS-PAGE. Suite à cette migration, les protéines ont été transférées sur une membrane de nitrocellulose (Trans-Blot, BioRad) à 100V pendant 90 minutes à 4oC. Les membranes ont été fixées deux fois pendant 10 minutes dans une solution de fixation: PBS (13,6 mM NaCl, 8 mM Na2HPO4, 2,7 mM KCl, 1,5 mM KH2PO4 pH=7,4), 0,05 glutaraldéhyde, 0,1% Tween, pH=7,4. Les membranes ont ensuite été lavées pendant 10 minutes avec du TBST: Trizma Base 25 mM, NaCl 150 mM, Tween 20 0,1%, pH=7,5. Les membranes ont ensuite été bloquées non spécifiquement par une incubation à TP dans une solution de TBST 0,1%, contenant du lait 5% (Carnation) ou bien dans une solution de TBST 0,1%, contenant de la BSA 5%. Le choix du type de blocage dépend de l’anticorps primaire. Suite à ce blocage, une incubation durant 16 heures à 4oC avec un anticorps primaire dilué avec le choix de TBST/Lait ou TBST/BSA est faite. Les anticorps primaires utilisés sont : INSR (dilution 1/1000, Cell Sgnaling), GLUT-4 (dilution 1/200, Chemicon) et GAPDH
(dilution 1/100000, Ambion). Deux lavages de 10 minutes à TP avec la solution de TBST 0,1% suivie d’un blocage de 10 minutes avec la solution de TBST/Lait ou TBST/BSA précèdent l’incubation avec l’anticorps secondaire adéquat couplé au HRP (Horseradish Peroxidase) dilué 1/10000 dans la solution de blocage pendant 2 heures à TP. Après cette incubation, 3 lavages de 5 minutes au TBST 0,1% précèdent la révélation des protéines. Les protéines ont été exposées à un agent chemiluminescent (PerkinElmer), puis révélé sur des films photosensibles (Kodak Film) suite à différents temps d’expositions.
La quantification des protéines a été faite par densité optique des bandes sur les films à l’aide du numériseur VersaDoc Imaging System 4000 ainsi que du logiciel Quantity One (BioRad).
3.8. Extraction des miRNA et ARN total
L’extraction de l’ARN s’est fait avec la trousse d’extraction mirVANA (Ambion) qui permet l’extraction de l’ARN total (miRNA et ARNm) selon le protocole décrit par le manuel d’instruction. Le broyage des artères s’est fait de la même manière que décrite dans l’extraction protéique. Brièvement, les cellules en culture ou poudre d’artères broyées sont lysées avec une quantité adéquate de tampon de lyse (Lysis/Binding Buffer, Ambion). Les cellules sont homogénéisées avec 1/10 de volume du tampon miRNA Homogenate Additive (Ambion). Une extraction organique avec du phénol-chloroforme acide a lieu