Pour l’obtention du Grade de
DOCTEUR DE L’UNIVERSITE DE POITIERS (Faculté des Sciences Fondamentales et Appliquées)
(Diplôme National - Arrêté du 7 août 2006)
Ecole Doctorale : Sciences pour l’Environnement Gay Lussac. Secteur de Recherche : Chimie théorique, physique, analytique.
Présentée par : Irène MAUPIN
************************
ETUDE DES PROPRIETES CATALYTIQUES DE MELANGES OXYDE/FAUJASITE POUR LE TRAITEMENT DE COMPOSES ORGANIQUES VOLATILS (COV) :
CARACTERISATION – MOBILITE DE L’OXYGENE – OXYDATION ************************
Directeur de Thèse : Patrick MAGNOUX
Co-directeurs de thèse : Thomas BELIN et Jérôme MIJOIN ************************
Soutenue le 5 décembre 2011 devant la Commission d’Examen
************************
JURY
Président :
Jacques BARBIER Jr., Professeur, Université de Poitiers Rapporteurs :
Pascale MASSIANI, Directeur de recherche, Université de Paris VI Stéphane SIFFERT, Professeur, Université du Littoral – Côte d’Opale Examinateurs :
Thomas BELIN, Maître de conférences, Université de Poitiers Xavier CHAUCHERIE, Ingénieur VERI, Limay
Pascal GRANGER, Professeur, Université de Lille
Ce travail a été réalisé au sein de Laboratoire de Catalyse en Chimie Organique (LACCO - UMR CNRS 6503) de Poitiers.
Je remercie la Région Poitou-Charentes pour son soutien financier durant cette thèse. Je tiens tout d’abord à remercier Monsieur Jean-Michel Léger de m’avoir accueillie au sein du Laboratoire de Catalyse en Chimie Organique de Poitiers.
Je tiens à exprimer ma profonde gratitude à Messieurs Patrick Magnoux, Jérôme Mijoin et Thomas Belin de m’avoir accueillie dans leur équipe mais également de m’avoir encadrée durant cette thèse. Je veux leur exprimer ici tout ma reconnaissance pour leurs conseils, leurs disponibilités et leurs sympathies.
Je remercie vivement Monsieur Jacques Barbier Jr. qui a accepté de présider mon jury de thèse. Je le remercie également, ainsi que Monsieur Nicolas Bion, de m’avoir fait profiter de leur expérience et de leur savoir-faire.
Je souhaite également remercier Madame Pascale Massiani, de l’Université de Paris VI, et Monsieur Stéphane Siffert, de l’Université du Littoral – Côte d’Opale, de m’avoir fait l’honneur de juger ce mémoire et d’en être rapporteurs.
Je souhaite aussi remercier Messieurs Xavier Chaucherie, de VEOLIA Environnement, Recherche & Innovation, et Pascal Granger, de l’Université de Lille, pour l’intérêt qu’ils ont accordé à mes travaux et d’avoir accepté de faire partie de mon jury de thèse.
J’adresse mes remerciements les plus sincères à tous ceux qui ont participé à ce travail et plus particulièrement à Jean-Dominique Comparot, Sophie Morisset, Morgane Vilette, Sandrine Arii-Clacens, Christelle Roudault, Lilian Eloy et Julie Rousseau, pour leurs compétences scientifiques mais également pour leur sympathie et leur disponibilité. J’associe également à ces remerciements Monsieur Michel Chauveau pour son aide technique et sa disponibilité, ainsi que Messieurs Jean-Jacques Colin, Claude Rouvier et Tristan Beldi. Je désire également mentionner Mesdames Maryvonne Choumil, Corinne Rouil et Jacqueline François pour la partie administrative et leur bonne humeur.
Je tiens aussi à remercier tout particulièrement Monsieur Romain Beauchet, pour ses conseils pratiques et sa patience au cours de ces travaux, mais également pour sa bonne humeur et sa sympathie.
Je remercie sincèrement pour leur amitié, leur bonne humeur de tous les jours et leurs conseils scientifiques : João Martins, Nuno Fonseca, Marisa Abadeço, Inês Graça, Catarina Rocha Hania Ahouari, Soumaya Hamieh, Karima Ben Tayeb, Ludovic Pinard, Kévin Louis, Anthony Levalant, João Dionísio, Stéphane Marie-Rose, Sonia Carré, Mathieu Hinckel, Sébastien Laforge, Christine Canaff, Nuno Fonseca, Suor Gnep, Solène Robin, Guy Joly, Isabelle Gener-Batonneau, Adeline Trouvé, Emmanuel Birot, Samuel Mignard, Magali Bonne, Sylvain Keav, Yannick Pouilloux, Charly Mve Mfoumou, Modibo Moungeungui, Ornellia Ozenga, Francis Ngoye, Nuno Batalha, Joachim Krou Nguessan, Filipa Madeira, Razika Merabti, Mehrad Tarighi, François Lemoine. Si toutefois, j’avais omis de mentionner quelques personnes, j’espère qu’elles voudront bien m’en excuser.
Je souhaite dire toute ma gratitude aux complices de promotion depuis la licence, Messieurs Benoît Dupuy, Patrick Urchaga, Sébastien Berland, Benoît Tapin et Christophe Poupin. Je tiens aussi à remercier les pièces « rapportées » qui ont contribuées au maintien de la bonne humeur et du moral, Messieurs Thibaut Legigan, Aurélien Flura et Saïd Laassiri.
Enfin, je souhaite remercier ma famille et mes amis de m’avoir soutenue durant ces années et tout particulièrement mes parents.
INTRODUCTION GENERALE
1
Chapitre 1 : Etude bibliographique
5
I. Les Composés Organiques Volatils
7
1. Définition des COV
7
2. La législation
7
3. Les sources d’émission
8
4. Les effets
10
a. Les effets directs 10
b. Les effets indirects 12
i. Les effets sur le climat 12
ii. Les effets sur la santé et l’environnement 13
5. Les traitements
13
a. Les méthodes de récupération 14
b. Les méthodes de destruction 14
6. Le choix d’un traitement
15
II. L’oxydation catalytique de COV
16
1. Le schéma réactionnel
16
2. L’effet de la structure du Composé Organique Volatil
18
3. L’effet de l’eau
19
4. La formation de sous-produits
19
5. L’oxydation de mélanges
20
6. Les limites de la catalyse
23
III. Le mécanisme d’oxydation de l’isopropanol
24
IV. Les catalyseurs d’oxydation
26
1. Les catalyseurs non zéolithiques
26
a. Les métaux nobles 26
b. Les oxydes de métaux 27
2. Les catalyseurs zéolithiques
35
a. Généralités 36
b. Composition 36
c. Les familles de zéolithes 38
i. Le classement général 38
ii. L’influence de la structure sur les réactions catalytiques 38
d. Les propriétés acido-basiques des zéolithes 41
i. L’acidité de Brønsted 42
ii. L’acidité de Lewis 43
iii. La basicité 44
iv. L’influence de l’acido-basicité 45
e. La désactivation et la présence de contaminants 46
f. La zéolithe faujasite (FAU) 47
3. Les catalyseurs mixtes oxydes de métaux-aluminosilicates
49
Références bibliographiques 51
Chapitre 2 : Partie expérimentale
59
I. Préparation des catalyseurs
61
1. Préparation des catalyseurs simples
61
a. Oxyde de cérium (CeO2), oxyde de zinc (ZnO) et oxyde de cobalt (Co3O4) 61
b. Dioxyde de manganèse (MnO2) 62
2. Préparation des catalyseurs « mixtes »
63
a. Mélange mécanique (M) 63
b. Calcination (C) 63
c. Traitement thermique (HT) 64
d. Imprégnations (I) 64
e. Echange cationique (EC) 65
f. Récapitulatif des méthodes de préparation 67
II. Caractérisation des catalyseurs
67
1. Physisorption à l’azote
67
2. Thermodésorption de pyridine suivie par infrarouge
70
3. Diffraction des rayons X
72
4. Analyse élémentaire
74
5. Analyse de coke
75
III. Détermination de la mobilité de l’oxygène
77
1. Capacités de stockage de l’oxygène
77
a. Détermination de la Capacité Maximale de Stockage de l’Oxygène (CMSO) 77 b. Détermination de la Capacité de Stockage de l’Oxygène en conditions
dynamiques (CSO) 79
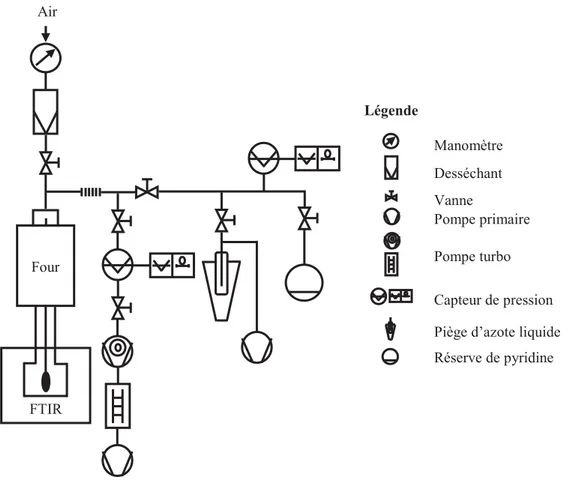
2. Echange isotopique de l’oxygène
81
a. Système de distribution des gaz 81
b. Enceinte réactionnelle 81
c. Système analytique 82
d. Protocole expérimental 82
IV. Tests catalytiques
83
1. Préparation et activation des catalyseurs
83
2. Expérimentation
83
3. Analyse des produits
87
Références bibliographiques 88
Chapitre 3 : Etude préliminaire sur les oxydes et sur la
faujasite
91
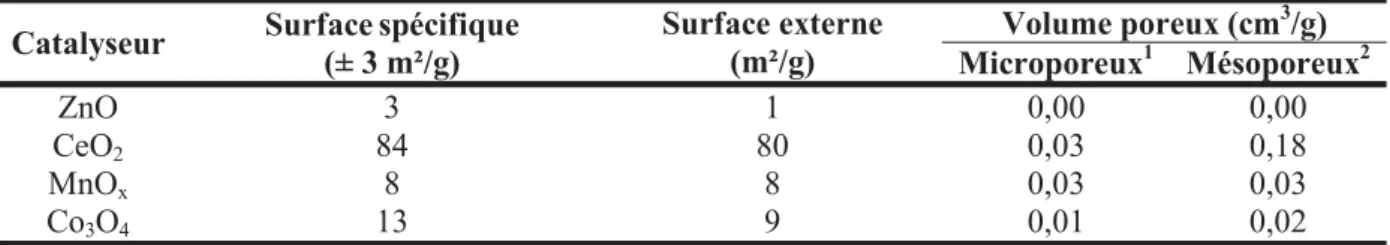
I. Caractérisation des catalyseurs
93
1. Analyse élémentaire
93
2. Physisorption à l’azote
94
a. Les oxydes 94
b. La faujasite 94
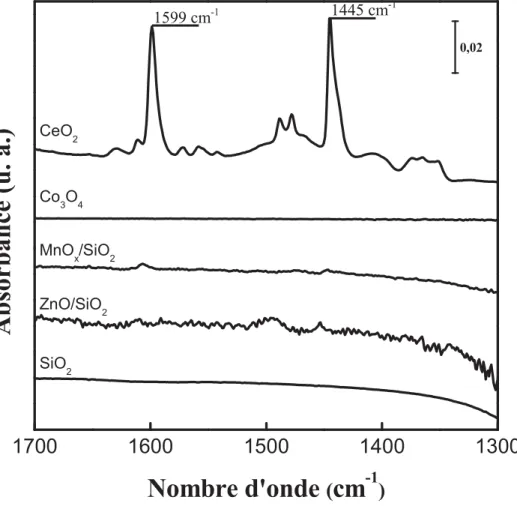
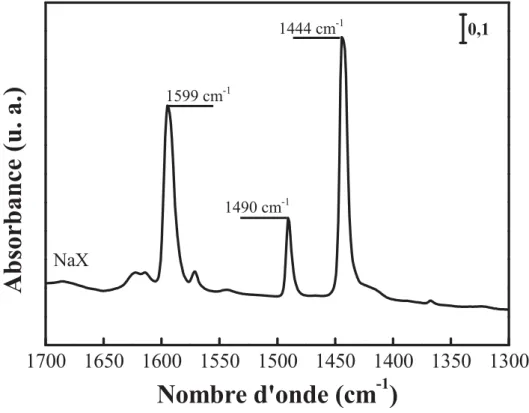
3. Thermodésorption de pyridine suivie par spectroscopie infrarouge
95
b. La faujasite 97
4. Diffraction des rayons X
98
a. Les oxydes 98
b. La faujasite 101
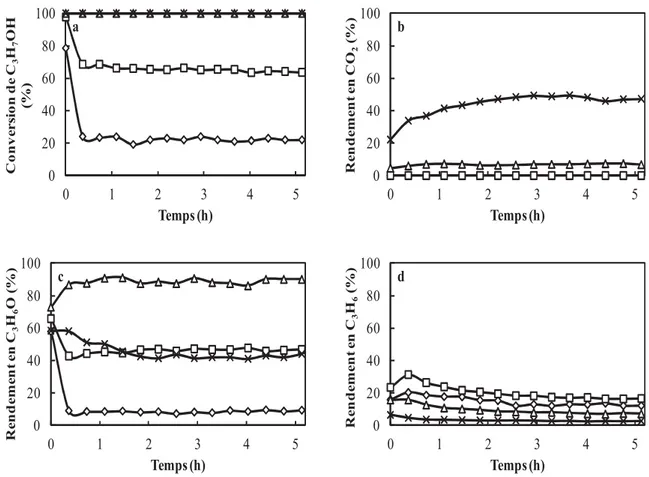
II. Tests catalytiques
102
1. Oxyde de zinc (ZnO)
104
2. Oxyde de cérium (CeO
2) 105
3. Oxydes de manganèse (MnO
2/Mn
2O
3) 106
4. Oxyde de cobalt (Co
3O
4) 107
5. Comparaison des oxydes étudiés
108
6. Conclusions sur les oxydes
110
7. La faujasite
111
III. Conclusion
113
Références bibliographiques 115
Chapitre 4 : Influence des oxydes sur la faujasite
117
I. Caractérisation des catalyseurs
119
1. Physisorption à l’azote
119
2. Thermodésorption de pyridine suivie par spectroscopie infrarouge
121
3. Diffraction des rayons X
122
II. Tests catalytiques
126
1. ZnO/NaX
127
2. CeO
2/NaX 129
3. MnO
x/NaX 130
4. Co
3O
4/NaX 131
5. Comparaison des catalyseurs mixtes oxyde/zéolithe
132
III. Conclusion
134
Références bibliographiques 137
Chapitre 5 : Etude des catalyseurs mixtes cérine/faujasite
139
I. Caractérisation des catalyseurs
141
1. Physisorption à l’azote
141
2. Thermodésorption de pyridine suivie par spectroscopie infrarouge
153
3. Diffraction des rayons X
156
II. Tests catalytiques
160
1. Influence du mode de préparation
161
a. Les catalyseurs contenant 65% de cérine 161
b. Les catalyseurs contenant 50% de cérine 162
c. Les catalyseurs contenant 20% de cérine 164
f. Conclusions sur l’influence du mode de préparation 169
2. Influence de la quantité de cérine
169
a. Les mélanges mécaniques (M) 169
b. Les calcinations (C) 171
c. Les traitementes thermiques (HT) 173
d. Conclusions sur l’influence de la teneur en cérine 175
III. Etude supplémentaire sur un mélange de COV
176
IV. Conlusion
178
Références bibliographiques 180
Chapitre 6 : Mobilité de l’oxygène
181
I. Capacités de stockage de l’oxygène
183
1. Capacités Maximales de Stockage de l’Oxygène (CMSO)
183
a. Etude à 400°C 184
b. Etude à 300°C 185
2. Capacité de Stockage de l’Oxygène en conditions dynamiques
(CSO)
186
a. Etude à 400°C 186
b. Etude à 300°C 188
3. Discussion
189
II. Echange isotopique de l’oxygène
192
1. Echange Isotopique en Température Programmée (TPIE)
193
a. Hypothèse d’un échange des oxygènes de cœur de la cérine 195 b. Hypothèse d’un échange des oxygènes de la zéolithe 196
c. Détermination du mécanisme d’échange 197
2. Echange Isotopique en Isotherme (EII)
199
III. Influence de la mobilité sur la catalyse
203
1. Les catalyseurs mixtes cérine/faujasite
204
2. Les oxydes mixtes à forte capacité de stockage
206
IV. Conclusion
208
Références bibliographiques 211
CONCLUSION GENERALE
213
ANNEXES 221
Fiche JCPDS de ZnO : numéro 00-036-1451
I
Fiche JCPDS de CeO
2: numéro 00-034-0394
II
Fiche JCPDS de MnO
2: numéro 03-065-2821
III
Fiche JCPDS de Mn
2O
3: numéro 00-041-1442
IV
Fiche JCPDS de Co
3O
4: numéro 00-042-1467
V
Introduction
générale
Ce sujet de thèse s’inscrit dans le cadre général de la dépollution atmosphérique et plus particulièrement du traitement d’effluents gazeux de sources stationnaires.
Parmi les différentes émissions, les Composés Organiques Volatils (COV) sont une source majeure de pollution. Les composés organiques volatils regroupent diverses familles de composés : alcanes, alcènes, aromatiques, cétoniques, alcools, acétates, aldéhydes et chlorés.
La volatilité de ces composés leur confère l’aptitude de se propager plus ou moins loin de leur lieu d’émission, entraînant ainsi des impacts directs (toxicité, odeur) et indirects (« smog » et réchauffement) sur l’air. L’inquiétude née de ce constat a poussé de nombreux pays à contrôler leurs émissions de gaz dans l’atmosphère.
Devant l’apparition de textes législatifs de plus en plus restrictifs, l’investissement des industries vers des systèmes préventifs répondant aux critères de chimie verte croît, mais malgré le développement de nouveaux procédés limitant les émissions, l’emploi de méthodes curatives restent indispensable.
Plusieurs procédés ont été proposés pour leur destruction et parmi ceux-ci l’oxydation catalytique de ces polluants en dioxyde de carbone et en eau. L’oxydation catalytique présente de nombreux avantages par rapport à l’oxydation thermique tels qu’un coût moins élevé ou la non-production de NOx. Elle est surtout rentable pour traiter des effluents moins concentrés en polluants, la destruction thermique étant alors discréditée en raison de consommations énergétiques excessives, d’autant plus élevées que la teneur en solvants est faible. Cependant, le choix du catalyseur (activité, sélectivité…) est un paramètre très important afin d’obtenir le succès escompté dans ce type de procédé. Il permettra l’augmentation de la vitesse de réaction et permettra d’opérer à des températures beaucoup plus basses.
Différents types de catalyseurs peuvent être envisagés pour ce type de traitement : les métaux nobles, pour leur fort pouvoir oxydant mais qui frittent à haute température, perdent donc de leur efficacité initiale, ne sont pas aisément régénérables et sont surtout coûteux ; les oxydes de métaux, légèrement moins actifs mais plus résistants à l’empoisonnement (chlore, soufre…) et moins chers. Les zéolithes, également actives en oxydation, présentent de plus une bonne résistivité à l’eau et à l’empoisonnement, la possibilité de retenir les molécules à traiter dans leur porosité, une bonne stabilité et une régénération facile.
L’objectif de cette thèse est d’allier les propriétés des oxydes de métaux et des zéolithes, afin d’obtenir un catalyseur stable, plus actif et plus sélectif lors de l’oxydation de composés organiques volatils, sans avoir recours à un dopage avec des métaux nobles.
L’insertion de clusters d’oxydes au sein de la porosité de la zéolithe et/ou la dispersion de ces oxydes à la surface de la zéolithe, pourraient engendrer un effet de synergie entre les deux solides et conduire à une amélioration des performances catalytiques.
Une idée des interactions existant à l’interface des oxydes et de la zéolithe pourra être obtenue à l’aide de diverses caractérisations (physisorption à l’azote, thermodésorption de pyridine suivie par spectroscopie infrarouge, diffraction des rayons X…).
Ce manuscrit est donc divisé en six chapitres :
Les deux premiers sont consacrés à l’étude bibliographique et aux techniques expérimentales utilisées au cours de ce travail.
Le troisième chapitre est dédié à l’oxydation d’isopropanol sur des oxydes (zinc, cérium, manganèse et cobalt) et sur une zéolithe faujasite NaX. Ce chapitre présente une étude préliminaire des propriétés de ces catalyseurs avant de les utiliser en mélange.
Le quatrième chapitre concerne l’oxydation de cette même molécule sur les catalyseurs mixtes oxyde/zéolithe.
La comparaison des résultats obtenus nous a conduit à traiter, plus en détail, l’oxyde mixte cérine/faujasite au cours de l’oxydation de l’isopropanol. Le cinquième chapitre concerne l’influence du mode de préparation et de la teneur en cérine sur les résultats catalytiques.
Certaines propriétés de la cérine, comme la forte capacité de stockage d’oxygène et la mobilité de ce dernier sur cet oxyde, sont par ailleurs souvent mises en avant pour expliquer les performances catalytiques lors du traitement des composés organiques volatils. Sur les catalyseurs mixtes cérine/faujasite, l’impact de telles propriétés et les phénomènes induits par l’association de ces deux solides sont présentés dans le dernier chapitre.
Etude
I. Les Composés Organiques Volatils
1. Définition des COV
L’arrêté ministériel du 1er mars 1993 considère comme COV tout composé, à l’exception du méthane, contenant du carbone et de l’hydrogène. Ce dernier pouvant être substitué partiellement ou totalement par des atomes de chlore, d’oxygène, de soufre, d’azote ou de phosphore (à l’exception des oxydes de carbone et des carbonates) et se trouvant à l’état de gaz ou de vapeur dans les conditions normales de température ou de pression (20°C et 105 Pa).
Un projet de directive européenne vient compléter cet arrêté en ajoutant que tout produit organique ayant une pression de vapeur supérieure à 10 Pa dans les conditions normales de température et de pression est considéré comme COV. En revanche, la pression de vapeur retenue aux Etats-Unis est plus faible (0,13 Pa) pour les mêmes conditions de température.
Autrefois, le méthane était inclus dans les sources de COV. La tendance actuelle est de le séparer des COV du fait de sa provenance (agriculture et milieu naturel), des flux importants émis dans l’atmosphère et d’une comptabilisation spécifique. En outre, son influence sur l’environnement est différente (effet de serre) de celles des COV qui ont un impact sur la pollution photochimique. Le terme Composés Organiques Volatils Non Méthaniques (COVNM) serait donc à privilégier, mais ne sera pas utilisé dans la suite de ce manuscrit afin de ne pas alourdir le texte.
2. La législation
La réduction de la pollution atmosphérique repose sur des réglementations qui concernent aussi bien les sources fixes, et notamment les installations industrielles, que les sources mobiles comme les transports.
Le phénomène de pluies acides (1970-1980) constitua un souci environnemental majeur en raison des répercussions sur les écosystèmes aquatiques et forestiers, notamment en Europe et en Amérique du Nord. La Convention de Genève de 1979 sur la pollution atmosphérique à longue distance fut le premier traité multilatéral dans le domaine de la prévention atmosphérique prenant en compte cette pollution transfrontalière. Depuis sa signature, plusieurs protocoles ont été adoptés et les engagements pris par la France vont souvent
au-delà des obligations de base qui sont imposées. Un exemple fut le protocole de 1991 à Genève visant à réduire les émissions françaises de COV de 30% entre 1988 et 1999.
La France s’est également engagée, sur le plan international, dans le cadre du protocole de Göteborg (1999), à réduire ses émissions de COV d’environ 40% entre 1999 et 2010 (63% en prenant 1990 pour référence). Ces engagements ont été repris par la directive du 23 octobre 2001 (National Emissions Ceilings, NEC) fixant des plafonds nationaux d’émissions pour certains polluants atmosphériques qui impose une limite aux émissions de COV à respecter en 2010 (1050 kt pour la France). La nouvelle directive (NEC 2010) montre que pour l’ensemble des pays membres (27), les émissions de COV non méthaniques (COVNM) ont diminuées en 2009 par rapport à 1990. Les plus importantes réductions absolues d’émission ont été réalisées par la France et le Royaume-Uni. Cependant, pour 22 des 27 états membres, les émissions de COVNM en 2009 étaient déjà inférieures aux plafonds imposés pour 2010.
La réduction des émissions de COV fait l’objet depuis 2001 d’une action nationale de l’inspection des installations classées (dépendant du Ministère de l’Ecologie, du Développement Durable, des Transports et du Logement, MEDDTL). Cette action vise à identifier les principaux émetteurs industriels (émissions supérieures à 30 tonnes par an) et a permis des réductions importantes, de l’ordre de 42% entre 2000 et 2008, permettant de diminuer les concentrations en ozone dans l’air ambiant et l’impact sur la santé de ces polluants.
Par ailleurs, une directive relative à la réduction des émissions de COV dues à l’utilisation de solvants organiques dans les peintures et vernis décoratifs et les produits de retouche automobiles est à l’étude. Cette dernière permettrait de réduire les émissions françaises de COV de 40 kt environ.
En ce qui concerne les émissions de COV dues au transport routier, les directives « Auto-Oil » de 1998 ont permis de diviser par 5, en 2000, le taux maximum de benzène dans l’essence.
3. Les sources d’émission
Les émissions de COV proviennent de sources naturelles et anthropiques. A l’échelle planétaire, l’émission de COV est principalement d’origine naturelle. La végétation, les feux de forêt ou les animaux sont les principales sources naturelles. En revanche, dans les régions industrialisées et fortement peuplées, ce sont les sources anthropiques qui prédominent.
Ces sources anthropiques sont essentiellement dues à la combustion et à l’utilisation de solvants, dégraissants, conservateurs, … et proviennent donc de sources multiples. Dans le
passé, ce sont les activités industrielles et les foyers de combustion domestiques qui étaient les principales causes de la pollution de l’air dans nos régions. La situation a fortement évolué depuis 20 ans (Figure 1) de par la diminution de ces activités et l’augmentation du trafic et du parc automobile.
e : estimation préliminaire
Source : Centre Interprofessionnel Technique d’Études de la Pollution Atmosphérique, CITEPA, Secten, avril 2011.
Figure 1 : Émissions de COV atmosphériques par secteur en France métropolitaine.
Les émissions de COVNM de la France métropolitaine sont désormais largement sous la barre des 1000 kt atteignant en 2009 le niveau de 878 kt. Ces émissions ont fortement baissé depuis 1988 (- 1753 kt soit une diminution de 67% sur cette période). L’objectif prévu (1050 kt) pour 2010 par la directive NEC est donc pleinement rempli [1].
En 1990, le secteur du transport routier prédominait largement avec 41% des émissions totales alors qu’en 2009, celui-ci ne représente plus que 14% des émissions totales (3ème position).
En 2009, le résidentiel/tertiaire est le premier secteur avec 37% des émissions totales. L’utilisation de solvants à usage domestique ou dans le bâtiment (peintures, colles,…) est la principale source de ces émissions, la combustion du bois dans les petits équipements domestiques y contribue également. Il est à noter que la combustion des énergies fossiles dans les installations de combustion fixes est une faible source d’émissions alors que la biomasse consommée dans les petites installations de combustion domestiques est une source plus importante. 0 500 1000 1500 2000 2500 3000 1988 1989 1990 1995 2000 2001 2002 2003 2004 2005 2006 2007 2008 2009 2010e
É
m
is
si
ons
de
C
O
V
at
m
os
phé
ri
q
u
es
(k
t)
Autres transports Transport routier Agriculture/sylviculture Résidentiel/tertiaire Industrie manufacturière Transformation énergieL’industrie manufacturière occupe la seconde position avec 36% des émissions en 2009, essentiellement du fait de l’utilisation de peinture. Les autres secteurs sont la transformation de l’énergie (4,9%) puis l’agriculture/sylviculture (4,2%) et enfin les autres transports (3,9%).
Toutes les émissions des sources biotiques de l’agriculture et des forêts sont présentées hors total national conformément aux règles de comptabilisation de la CEE-NU/NEC (de 1273 à 1734 kt/an sur la période). Ces émissions « hors-bilan » contribuent cependant, comme les précédentes, aux réactions photochimiques dans l’atmosphère qui conduisent en particulier, à la formation d’ozone troposphérique. Les émissions maritimes internationales, les émissions de la phase croisière ( 1000 m) des trafics aériens domestique et international, ainsi que les émissions des sources non-anthropiques sont également répertoriées « hors-bilan »
4. Les effets
La volatilité des COV leur confère l’aptitude de se propager plus ou moins loin de leur lieu d’émission, entraînant ainsi des impacts directs et indirects sur les animaux et la nature.
a. Les effets directs
En général, les émissions de COV ont un impact direct et important sur l’Homme du fait de leur toxicité (Tableau 1). A chaque COV correspond une toxicité différente même si des similitudes, telles que les propriétés irritantes, sont observées. En réponse au risque potentiel que représentent les COV sur la santé, des normes de qualité d’air et des valeurs guides ont été définies pour un certain nombre de produits volatils. Cependant, le lien entre ces molécules et les maladies contractées n’est pas toujours facile à démontrer. Par conséquent, les valeurs limites, les seuils maximaux et les teneurs cumulées d’exposition proposés sont réajustés régulièrement. Il a néanmoins été prouvé que l’exposition aux COV provoquerait une augmentation des symptômes des maladies respiratoires, des maux de tête, des irritations (nasales et oculaires) et des éruptions cutanées. Ils peuvent également entraîner des troubles digestifs, hépatiques et parfois rénaux, être neurotoxiques, mutagènes, cancérogènes ou toxiques pour la reproduction. Parmi les plus dangereux, citons le benzène, responsable de leucémies ou encore le 1,3-butadiène, reconnu cancérigène.
Tableau 1 : Caractéristiques toxicologiques de quelques solvants [2].
1 VME : Valeur moyenne d’exposition. 2 VLE : Valeur limite d’exposition.
Les VME et VLE sont des valeurs données pour l’hygiène du travail (exposition 8 h/j et pour 40 h/semaine). Par ailleurs, des valeurs d’exposition sont aussi publiées par l’Organisation Mondiale de la Santé (OMS) pour l’air ambiant.
Composé Volatilité Pénétration Pouvoir irritant d’ébriété – Pouvoir
narcotique Toxicité spécifique VME1 VLE2 (ppm) Acétate d’éthyle +++ + ++ + - 400 - Acétone +++ + + ++ 750 -
Benzène +++ ++ + ++ Moelle osseuse Cancérogène 25 5
Dichlorométhane +++ + +++ ++ Intoxication par le CO 100 50 Éthers de glycol à courte chaîne + ++ + + Moelle osseuse Testicule Tératogène 5 5
Éthylène glycol + 0 ++ + Rein (en aigu) 50 -
n-hexane ++ ++ + ++ périphériques Nerfs 50 -
Isopropanol ++ 0 + ++ - 400 -
Méthanol ++ ++ + + Nerfs optiques (en aigu) 1000 200
Méthylbutylcétone (MBK) + + + ++ périphériques Nerfs 5 8 Méthyléthylcétone (MEK) +++ + + ++ 200 - Méthylisobutylcétone (MIBK) + + + ++ 50 - Perchloroéthylène ++ + ++ + Cancérogène ? 50 -
Styrène +++ ++ ++ ++ Moelle osseuse Foie
Cancérogène ?
Tétrahydrofurane +++ ++ +++ + Nerfs Foie 200 -
Toluène ++ + + ++ Tératogène ? 100 150
Trichloroéthylène ++ ++ ++ ++ Cancérogène Cœur 200 75
b. Les effets indirects
i. Les effets sur le climat
Les COV participent à des réactions complexes se déroulant dans l’atmosphère. Schématiquement, il résulte de la présence des COV dans l’atmosphère une augmentation de la formation d’ozone dans les basses couches (troposphère).
Naturellement, l’ozone troposphérique provient de l’oxygène présent dans l’air et d’un atome d’oxygène issu de la dissociation du dioxyde d’azote sous l’effet du rayonnement solaire. Cet ozone réagit ensuite avec le monoxyde d’azote formé pour reformer le dioxyde d’azote complétant ainsi le cycle de Chapman en éliminant l’ozone (Figure 2).
L’introduction de COV dans l’atmosphère entraîne, de par leur dégradation, la formation de réactifs radicalaires. Ces radicaux vont alors réagir avec le monoxyde d’azote pour former du dioxyde d’azote entraînant ainsi une baisse de la consommation de monoxyde d’azote par l’ozone, qui s’accumule alors dans la troposphère. Ceci s’explique par le fait que les réactions photochimiques ont des cinétiques lentes alors que les réactions radicalaires sont rapides. Le cycle de Chapman est alors modifié (Figure 3).
L’ozone troposphérique ainsi formé est un gaz à effet de serre puissant, adsorbant dans l’infrarouge 2000 fois plus que le CO2. Il est ainsi responsable à hauteur de 18% de l’effet de
serre mondial [2].
Figure 2 : Cycle de Chapman en équilibre [2]. NO2 O3 O2 hȞ NO OƔ +
ii. Les effets sur la santé et l’environnement
Une altération de l’eau potable due au transfert des COV de l’air dans les milieux aquatiques (eaux souterraines ou de surface) a également été remarquée. Les COV peuvent être adsorbés sur différents solides (argiles, limons…) et réapparaître, de façon chronique, par désorption lente dans le milieu naturel [2].
Par conséquent, l’ozone troposphérique endommage les arbres et les cultures, irrite les yeux et engendre des maladies respiratoires.
5. Les traitements
Le passage à des procédés totalement propres (chimie verte) s’appuie sur la prévention sur les rejets polluants et les procédés rejetant des COV. Néanmoins, une approche curative s’impose du fait d’installations industrielles déjà en activité et/ou de verrous technologiques.
Les méthodes curatives peuvent être classées en deux catégories distinctes : les méthodes de récupération et les méthodes de destruction.
Figure 3 : Cycle de Chapman perturbé par la présence de radicaux libres [2]. RCOƔ RCO2Ɣ RCO3Ɣ COV + NO2 O3 O2 hȞ NO OƔ +
a. Les méthodes de récupération
Ces méthodes englobent la condensation, l’absorption, l’adsorption et la séparation par membrane.
La condensation des COV présents dans l’air est possible en jouant sur la variation de la pression de vapeur saturante avec la température. Cette technique est utilisable pour des faibles débits, hautement concentrés (> 5000 ppm) en COV ayant une température d’ébullition supérieure à 40°C [2].
L’absorption est un procédé de transfert des COV de l’air vers une phase liquide. Les solutions de lavage utilisées sont généralement de l’eau, des solutions oxydantes ou des huiles lourdes. Des liquides réactifs permettant d’accélérer les transferts par réaction d’oxydation de l’absorbat sont également employés [2].
L’adsorption est un procédé de transfert de la phase gaz vers un solide poreux. Classiquement, la récupération s’effectue alternativement sur deux lits fixes, permettant la régénération thermique de l’adsorbant. Le charbon actif est le matériau le plus communément employé, mais des supports à base de fibres de carbone activé, tissus ou feutres, des polymères ou des zéolithes peuvent également être utilisés [2].
La séparation par techniques membranaires semi-perméables dépend de la pression appliquée et de la sélectivité de la membrane. Cette méthode est généralement couplée avec un système d’adsorption ou de condensation.
b. Les méthodes de destruction
Il existe deux types de méthodes destructrices, les traitements thermiques et les systèmes biologiques. Elles sont surtout utilisées pour des mélanges de COV.
Les méthodes de destruction par voie thermique sont les plus couramment utilisées. L’incinération thermique permet de traiter les faibles comme les fortes concentrations en COV, néanmoins, les températures de travail étant généralement comprises entre 600 et 850°C, l’autothermie du système ne peut être atteinte si la concentration est trop faible.
L’oxydation catalytique, quant à elle, fait intervenir des températures de destruction beaucoup plus faibles (entre 200 et 450°C) entraînant ainsi des économies d’énergie. L’absence de NOx en tant que sous-produits (en effet à faible température il n’y a pas de formation de NOx contrairement à l’incinération thermique qui se réalise à haute température) est un des avantages de cette technique. Enfin, elle permet le traitement de faibles concentrations en COV (entre 100 et 2000 ppm).
A cela, nous pouvons ajouter les techniques à plasma qui présentent l’avantage d’oxyder les COV à température ambiante. Cependant, ce traitement donne souvent lieu à la formation de NOx et d’ozone, qui devront être à leur tour traités en aval.
Récemment, le marché des procédés de traitement a vu apparaître les méthodes de biofiltration utilisant la capacité des bio-organismes (bactéries, levures…) à dégrader les composés organiques. Cependant, les cinétiques de dégradation sont généralement lentes et demandent donc de grandes surfaces filtrantes. Cette technique est plus adaptée au traitement de débits importants à faible concentration et à température ordinaire.
6. Le choix d’un traitement
Aujourd’hui, il n’existe pas de technique universelle pouvant s’adapter à tous les types de rejet gazeux à traiter. Chaque procédé possède son domaine de faisabilité et doit être choisi principalement selon les critères suivants : la qualité de l’effluent à traiter (molécules volatiles, matières en suspension, humidité…), la concentration, le débit, les paramètres physiques (pression, température…) et les paramètres de sécurité (explosivité, réactivité, corrosivité…).
L’humidité peut effectivement être un facteur déterminant, notamment dans le cas de l’adsorption, puisque de fortes compétitions peuvent être observées empêchant le piégeage des molécules. Ce type de compétition existe aussi dans le cas des mélanges de COV, pour lesquels une méthode destructrice sera préférée à une méthode récupératrice.
La présence de matières en suspension (poussières ou graisses dans le cas de gaz chaud) peut considérablement gêner les traitements par un colmatage des filtres.
La destruction par voie thermique est de nos jours la méthode la plus utilisée. En effet, malgré des températures de destruction totale beaucoup plus faible par voie oxydo-catalytique, les industriels restent prudents sur l’emploi de cette méthode destructive probablement du fait de la crainte liée à l’empoisonnement du catalyseur. Toutefois, les systèmes peuvent être équipés d’échangeurs permettant, notamment grâce à la chaleur dégagée par la combustion des COV, l’autothermie du système et par conséquent une diminution du coût de fonctionnement.
Tableau 2 : Techniques de traitement des COV [2].
Procédés Principe Domaines d’application
Incinération
Thermique combustion (T # 750°C) Oxydation par Solvants Fortes et faibles
concentrations Catalytique Oxydation sans flamme (200 < T < 450°C)
Traitements biologiques
Oxydation par voie biologique
Molécules biodégradables et
hydrosolubles Biofiltre sur le garnissage traversé Micro-organismes fixés
par les gaz à épurer Lit bactérien Phase aqueuse circulante, biomasse fixée
Biolaveur Phase aqueuse circulante, biomasse en suspension
Adsorption Transfert de molécules gazeuses dans un matériau poreux Molécules adsorbables Flux importants Absorption Transfert des composés de la phase gazeuse vers une phase liquide Molécules solubles Flux importants
Condensation Refroidissement des vapeurs de solvants
Concentration : 0,5 à 10%vol Faible débit Molécules condensables Séparation par membranes
Transfert sélectif des molécules à travers une couche épaisse à base de silicone
Prétraitement d’effluents gazeux fortement
concentrés
II. L’oxydation catalytique de COV
L’oxydation complète correspond à toute technique de conversion des COV en dioxyde de carbone, eau et différents oxydes ou produits d’oxydation. Elle peut se dérouler à basse température (biofiltration) ou plus élevée (oxydation catalytique ou thermique). Elle est largement utilisée pour réduire les COV présents dans les effluents industriels. Pour des composés simples, elle peut se traduire par l’Equation 1 suivante :
4 CmHnOo + ሺ4 m + n - 2 oሻ O2 ĺ 2 nH2O + 4 mCO2
+ dégagement de chaleur Equation 1
L’oxydation complète des COV par voie catalytique est une application particulière de la catalyse hétérogène. Les réactions d’oxydation se déroulent de manière similaire à l’oxydation thermique si ce n’est que l’énergie d’activation des molécules nécessaire à la réaction est diminuée par la présence du catalyseur.
1. Le schéma réactionnel
Le processus global d’oxydation catalytique peut être subdivisé en plusieurs étapes. Un schéma réactionnel général peut se présenter comme suit :
Transfert de masse (diffusion) des réactifs à partir du fluide vers la surface externe du catalyseur,
Diffusion du réactif à partir de l’entrée du pore à travers les macropores, jusqu’aux micropores sur la surface interne du catalyseur,
Adsorption du ou des réactifs sur les sites actifs libres, Réaction chimique en phase adsorbée,
Désorption des produits,
Diffusion des produits de la surface interne du catalyseur jusqu’à l’entrée (ou la sortie) du pore, à la surface externe du catalyseur,
Transfert de masse des produits de la surface externe au fluide.
Des études effectuées sur des mélanges ont mis en évidence des phénomènes de compétition sur les sites actifs du catalyseur. Il peut s’agir d’une compétition à l’adsorption (physique, ou moléculaire, qui s’apparente à la condensation que subissent les molécules sous l’effet des forces de Van der Waals ; chimique, ou activée, qui, de par sa spécificité, son énergie d’activation et la chaleur dégagée, s’apparente à une réaction chimique) ou bien d’une compétition lors de la réaction avec l’oxygène chimisorbé. En effet, même s’il est admis que la plupart des réactions d’oxydation prennent place en phase adsorbée [3], il se peut, suivant la nature et la polarité des COV, que certains d’entre eux réagissent directement en phase gaz avec l’oxygène chimisorbé sur la surface du catalyseur.
La compétition à l’adsorption a été choisie comme modèle par Barresi et Baldi [4] et Mazzarino et Barresi [5] pour des mélanges benzène et éthenylbenzène. L’adsorption compétitive est par ailleurs l’hypothèse la plus communément adoptée.
Le partage de la surface catalytique par les constituants du milieu peut également s’effectuer sans compétition à l’adsorption, ce qui implique des sites ou des aires présentant une affinité particulière pour chacun des constituants. La réaction d’oxydation suit alors un modèle similaire au mécanisme originel de Mars-van Krevelen [6]. Typiquement, les COV en phase gaz réagissent directement sur les sites oxydés du catalyseur, générant ainsi la réduction des espèces catalytiques à différents degrés d’oxydation. Ce modèle a été utilisé entre autre par Gandwall [7].
Le mécanisme de Mars-van Krevelen est schématiquement le suivant : COV + catalyseur oxydé → catalyseur réduit + produits
Burgos et al. [8] ont mené une étude tendant à prouver la coexistence des deux phénomènes. En étudiant la combustion de méthyléthylcétone (MEK), d’isopropanol et de toluène sur un catalyseur au platine supporté par un monolithe de Al2O3/Al, ils ont montré
qu’en mélange binaire, la conversion de l’isopropanol est affectée par la présence du toluène alors que la destruction du toluène n’est pas influencée par la présence d’isopropanol. Du fait de sa haute polarité, l’isopropanol déplace le toluène de la surface du catalyseur. Cependant, le toluène n’est pas affecté puisqu’il ne semble pas réagir lorsqu’il est adsorbé mais est oxydé directement en phase gaz par l’atome d’oxygène chimisorbé sur le platine (le même que celui utilisé par l’isopropanol adsorbé). En conséquence, le toluène réagit plus vite entraînant une diminution de la conversion de l’isopropanol pour une température donnée. En mélange ternaire, un effet similaire est observé puisque la conversion totale de l’isopropanol se fait à plus haute température tandis qu’aucun changement n’intervient pour la conversion du toluène et de MEK. La plus grande mobilité des molécules réagissant directement en phase gaz conduit à une baisse de conversion des molécules nécessitant l’adsorption à la surface du catalyseur avant l’oxydation.
2. L’effet de la structure du Composé Organique Volatil
Plusieurs travaux ont montré l’importance de la force de la liaison C-H la plus faible d’un composé organique volatil pour son oxydation totale, bien que ce ne soit pas le seul facteur à prendre en compte [9,10]. En effet, plus la force de la liaison ou l’énergie de dissociation est faible, plus l’oxydation du COV sera simple.
Les travaux antérieurs de Sokolovskii [11] démontraient déjà que la température de réaction est contrôlée par les propriétés des liaisons C-H à s’activer et que pour des liaisons peu polaires stables, une température maximale était nécessaire, n’excédant toutefois pas la température de stabilité du produit. Il précise que, généralement, l’activation s’effectue de manière hétérolytique grâce à la présence de paires acides-bases ou de radicaux à la surface du catalyseur. Pour les oléfines ainsi que les groupements allyles, il est nécessaire d’avoir la participation de sites nucléophiles de surface, apportés par les oxygènes du réseau. Ce type de dissociation peut également s’appliquer aux paraffines. Une activation par dissociation homolytique peut également s’observer sur des oxydes par le remplacement d’un atome d’hydrogène par un oxygène.
Il faut toutefois signaler que cette remarque correspond à la première étape de l’oxydation totale. Les alcools qui s’oxydent en aldéhydes puis en acides carboxyliques, et forment ainsi des espèces plus stables, sont donc plus difficiles à oxyder.
3. L’effet de l’eau
L’eau est un produit de la combustion qui se trouve naturellement dans le mélange en cours de réaction. Cependant, afin de mener les expériences dans les conditions réelles de traitement, il est nécessaire d’ajouter de l’eau au mélange initial de COV, puisque l’air à épurer est généralement humide et avec une teneur en eau bien supérieure à celle qui serait obtenue par le seul fait de l’oxydation.
L’eau agit habituellement comme un inhibiteur [12] de la réaction d’oxydation mais son effet est plus ou moins prononcé suivant la nature des COV à traiter.
Par exemple, dans le cas des travaux entrepris par Papaefthimiou et al. [12] sur PtTiO2(W6+), dopé au tungstène, l’eau n’agit pas de la même façon suivant la nature des
composés en mélange (benzène et acétate d’éthyle). En effet, en mélange, l’eau se conduit en réel inhibiteur pour la conversion de l’acétate d’éthyle en CO2, tandis qu’il n’y a pratiquement
pas d’effet sur la conversion du benzène. L’inhibition de la conversion du benzène en CO2
engendré par l’acétate d’éthyle, est en effet plus prononcée que celle de l’eau. En revanche, il est important de signaler que l’eau agit comme un inhibiteur dans le cas de l’oxydation simple du benzène et de l’acétate d’éthyle (augmentation de 20°C dans le cas du benzène pour une conversion en CO2 égal à 90% et augmentation de 45°C pour l’acétate d’éthyle).
Par contre, dans le cas de l’oxydation du chlorobenzène, l’ajout d’eau améliore sa destruction et diminue la formation de sous-produits [13].
L’effet de l’eau sur l’oxydation de COV dépend aussi de la température de réaction et du catalyseur. Ainsi, Tsou et al. [14] ont montré que l’eau influait sur la conversion en CO2 de la
méthylisobutylcétone (MIBK) suivant la température sur un catalyseur zéolithique au platine (0.16Pt/HFAU) tandis qu’aucun effet n’était observé sur un catalyseur au palladium.
Enfin, il est important d’ajouter que des zéolithes très désaluminées ou acides sont hydrophobes. Par conséquent, l’eau agit très faiblement sur la réaction [15]. En revanche, sur les zéolithes basiques, telles que la NaX, qui sont hydrophiles, l’eau peut avoir un rôle, positif ou négatif, prononcé.
4. La formation de sous-produits
Comme nous l’avons déjà énoncé précédemment, l’oxydation de COV ne s’effectue pas directement en dioxyde de carbone et en eau, mais suit des chemins passant par la formation de sous-produits [16], qui ne seront toutefois pas systématiquement détectables.
Plusieurs travaux sur catalyseur PGM (Platinum Group Metals) ont mis en évidence la formation de sous-produits, l’aldéhyde correspondant à l’alcool de départ pour le méthanol, l’éthanol et le n-butanol, respectivement d’après Windawi [17], Mazzarino et Barresi [5], Hermia et Vigneron [18]. L’oxydation de l’aldéhyde conduit à la formation de l’acide correspondant. Seule l’oxydation des alcanes ne conduirait pas, selon Yao [19], à la formation de sous-produits, qu’il n’a pas observés.
Lintz et Wittstock [20] ont également montré la formation de nombreux sous-produits lors de l’oxydation de l’acétate de butyle et de l’acétone sur CuMn2O4.
Il faut ajouter qu’en pratique, les sous-produits formés ne doivent pas percer le lit catalytique dans des conditions normales de température et de vitesse spatiale, puisque la destruction sélective des COV en dioxyde de carbone et en eau reste l’objectif prioritaire.
5. L’oxydation de mélanges
Les mécanismes d’oxydation de COV pris isolément sont déjà fortement compliqués par l’apparition de sous-produits de réaction, constituant déjà en quelque sorte la formation d’un mélange plus ou moins complexe [21]. Bien que les systèmes industriels de traitement des COV concernent essentiellement des effluents gazeux de mélanges de composés, la littérature décrivant l’oxydation des mélanges est limitée. Les différentes études s’accordent à dire que l’oxydation catalytique d’un COV en mélange diffère de son oxydation simple et généralement un effet inhibiteur est observé [5,8,12,14,22,23].
Tichenor et Palozzolo [24] ont travaillé sur l’incinération de onze mélanges constitués de cinq COV en moyenne. Ils ont remarqué que la conversion du n-hexane est significativement diminuée lorsque l’alcane est oxydé dans un mélange de cinq familles différentes (n-hexane, isopropanol, méthyléthylcétone, acétate d’éthyle, benzène) alors qu’elle ne l’est pratiquement pas dans un mélange constitué d’alcanes et d’aromatiques (n-hexane, n-octane, n-décane, benzène, toluène, m-xylène, isopropylbenzène). Cette observation rejoint les résultats obtenus lors de l’oxydation d’un mélange n-hexane/toluène, où ni la conversion du n-hexane, ni celle du toluène ne sont significativement réduites en mélange par rapport aux conversions observées lors de l’oxydation des composés seuls. Ceci s’expliquerait par le fait que le n-hexane ne commence à réagir que lorsque la conversion du toluène atteint déjà 90%.
Les travaux de Barresi et Baldi [4,25], qui portent plus précisément sur des mélanges d’aromatiques, confirment que plus les courbes de conversion des composés sont proches, plus les inhibitions sur les composés considérés sont importantes.
Dans la même idée, Moro-Oka et al. [26] ont déjà mis en évidence la relation entre réactivité et effet inhibiteur. L’expérimentation consistait à ajouter à une concentration donnée de propylène, un second composé à différentes concentrations et de mesurer l’influence de ces ajouts sur la conversion du propylène. L’ordre apparent de la réaction décroît régulièrement de valeurs positives vers des valeurs négatives, dans l’ordre suivant : n-propane > butène > propylène > isobutylène > acétylène. Le n-propane, qui présente le plus grand ordre de réaction parmi les composés testés, n’inhibe pas l’oxydation du propylène. Il apparaît que l’inhibition due à chacun de ces composés augmente lorsque l’ordre de réaction pour ces composés diminue.
Cullis et al. [27] étudièrent l’influence des halométhanes sur l’oxydation catalytique du méthane sur un catalyseur PGM. Il apparaît que plus la concentration de l’halométhane est élevée, plus l’oxydation du méthane est retardée et plus la concentration de son sous-produit de réaction, le formaldéhyde, augmente.
Une autre étude entreprise par Tsou et al. [14] montre que l’effet inhibiteur de l’orthoxylène sur la méthylisobutylcétone (MIBK) doit être dû à la compétition des deux composés sur les sites actifs, l’adsorption de l’orthoxylène sur les sites métalliques par les électrons ʌ de l’aromatique étant certainement favorisée par rapport à la cétone.
Comme en oxydation simple, l’utilisation de différents catalyseurs mène à différentes conversions en COV. Ainsi, Papaefthimiou et al. [23] ont remarqué que sur des catalyseurs tel que Pt/Ȗ-Al2O3 et Pd/Ȗ-Al2O3, le benzène n’était pas converti tant que le butanol était présent.
En contre partie, si le mélange a un effet inhibiteur sur la conversion de ces deux COV sur le catalyseur Pt/Ȗ-Al2O3, l’utilisation de Pd/Ȗ-Al2O3 a un effet promoteur sur la conversion du
butanol.
Même si les études rapportées ont tendance à étudier l’inhibition de conversion sur les différents COV d’un mélange, il existe des exemples d’effets promoteurs. Tichenor et Palozzolo [24], ainsi que Pêcheux et Vigneron [28] ont en effet remarqué que l’acétate d’éthyle s’incinère mieux en mélange.
De la même manière, l’ajout d’un composé non chloré à un composé chloré augmenterait la conversion d’un composé chloré seul. En effet, la présence d’hydrocarbures dans le mélange réactionnel peut engendrer le déplacement du chlore présent à la surface du catalyseur et qui est susceptible d’empêcher l’adsorption d’oxygène impliqué dans le mécanisme d’oxydation [29].
d’heptane éliminait la formation de produits secondaires de l’oxydation de chlorobenzène et provoquait une nette diminution de la température de conversion de ce composé, le T50,
température pour 50% de conversion, diminuant de 305 à 225°C. Cependant, la température de conversion de l’heptane augmente, bien que cette différence entre l’oxydation simple et l’oxydation en mélange soit beaucoup moins importante que pour le chlorobenzène.
En complément de cette étude, les mêmes auteurs se sont ensuite intéressés au chlorobenzène en mélange avec du toluène, du benzène, du cyclohexane, du cyclohexène, du 2-butène et de l’éthylène [29]. Leurs résultats ont montré que l’ajout en excès ou non de ces composés augmentait le taux de conversion du chlorobenzène. En outre, le but-2-ène, le toluène, et l’éthylène sont très réactifs, tandis que les aromatiques tel que le benzène, le cyclohexane, le cyclohexène et le 1,4-cyclohexadiène, qui sont convertis en benzène au cours de la réaction, sont moins réactifs de par la chloration des noyaux aromatiques. L’effet promoteur de ces réactions s’explique principalement par l’élimination du chlore à la surface du catalyseur par les alcanes et les alcènes. L’augmentation de la température produite par la combustion des hydrocarbures et la présence de vapeur d’eau ne paraissent pas dans ce cas suffire à expliquer la baisse de la température de conversion T50.
Gervasini et al. [30] présentent un exemple similaire avec l’oxydation de CCl4 sur du CuCr
et du Mn supporté par de l’alumine. Il apparaît que la réaction d’oxydation est facilitée par la présence de n-hexane et/ou de toluène (entre 100 et 500 ppm), ceci pouvant être expliqué par la capacité de ces espèces à donner de l’hydrogène. La concentration des COV est également un paramètre très important puisqu’en présence d’une grande quantité de toluène ou de n-hexane la conversion en CCl4 diminue. Il y a alors compétition entre les COV sur les sites
actifs entraînant une inhibition.
Un COV peut être oxydé à plus basse température en mélange qu’en oxydation simple du fait de l’exothermicité provoquée à la surface du catalyseur par l’oxydation d’un ou plusieurs autres COV. C’est le cas de l’acétone (produit de l’oxydation de l’isopropanol) qui est oxydé à plus basse température en présence de méthyléthylcétone, de toluène et d’isopropanol que lors de son oxydation seule [8].
L’oxydation des mélanges n’est donc pas facilement prédictible à partir des courbes de conversion individuelles [12]. L’adsorption compétitive régit vraisemblablement les phénomènes d’oxydation mais des phénomènes spécifiques d’inhibition ou de promotion peuvent se produire. Il est d’usage de considérer que l’efficacité d’un catalyseur sur la destruction de COV est déterminée par l’activité en oxydation du composé organique le plus
récalcitrant [12], spécialement quand le composé agit comme un inhibiteur de l’oxydation d’un mélange.
6. Les limites de la catalyse
L’activité du catalyseur diminue au cours du temps suite à des phénomènes physiques et chimiques. Les principales causes de désactivation sont le vieillissement naturel, le vieillissement accidentel ou l’empoisonnement.
Le vieillissement naturel correspond à une diminution de l’activité au cours du temps dans des conditions normales d’utilisation. Ce phénomène résulte d’une passivation thermique, d’une perte d’éléments actifs, de l’altération mécanique du catalyseur et principalement de sa surface spécifique. Aleksiü et al. [31] ont observé que la concentration en métal noble diminue tandis que la masse de matière inerte à la surface augmente, suite à l’abrasion, à la migration de certains éléments du support, et au dépôt de matières inertes.
Le vieillissement accidentel est quant à lui provoqué par une température d’utilisation trop élevée (généralement supérieure à 650°C).
Le vieillissement par empoisonnement est le plus délicat à gérer. Les métaux précieux peuvent être particulièrement sensibles à la présence de certains composés dans l’effluent à traiter. Ils sont les plus actifs mais les plus sensibles aux poisons à cause de leur configuration électronique [32].
Deux types d’empoisonnement peuvent subvenir, par réaction chimique entre l’élément actif et le polluant gazeux ou par masquage.
L’empoisonnement chimique est le plus souvent irréversible. Le masquage réduit la surface spécifique du catalyseur et masque les sites actifs. Ceci peut s’avérer réversible si le masquage s’effectue par adsorption. Par exemple, en faisant passer un courant gazeux à travers le lit catalytique, un catalyseur contaminé au soufre peut progressivement retrouver ses propriétés. Dans le cas d’un masquage semi-réversible, une action de nettoyage s’avérera nécessaire. Si cette action n’est pas efficace alors l’empoisonnement est irréversible.
Les poisons sont malheureusement nombreux et communs aux effluents gazeux industriels courants [33] : phosphore, bismuth, plomb, antimoine, arsenic et mercure sont considérés comme conduisant à des empoisonnements irréversibles rapides. Le fer, l’étain et les composés siliconés sont considérés comme conduisant à des empoisonnements irréversibles et lents, le soufre, les composés halogénés et le zinc comme des poisons réversibles. D’autres types de poison peuvent être cités, tels que les composés organiques lourds qui se déposent ou
catalytique, ou les poussières plus ou moins bien tolérées selon la densité des canaux du catalyseur.
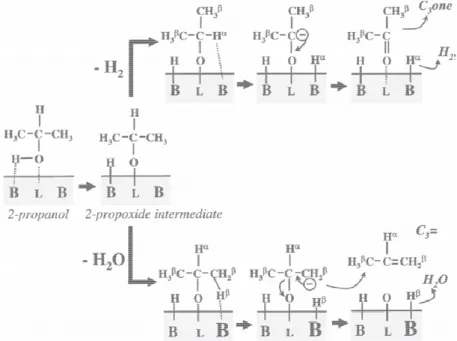
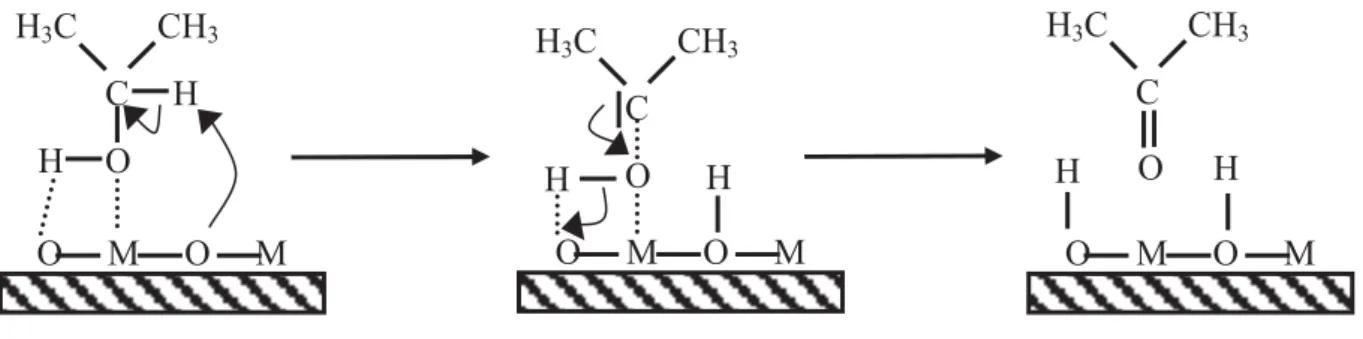
III. Le mécanisme d’oxydation de l’isopropanol
Au même titre que la réaction du 2-méthyl-but-3-yn-2-ol [34], l’oxydation de l’isopropanol [35,36] est utilisée comme réaction test de l’acido-basicité des catalyseurs. Une étude sur la validité de cette réaction-test réalisée par Lahousse et al. [37] précise que la décomposition du méthylbutynol en acétone et acétylène prouve bien la basicité des catalyseurs tandis que la déshydrogénation de l’isopropanol nécessite une activité redox supplémentaire.
De manière générale, la déshydratation de l’isopropanol en propène est associée à la présence de sites acides (Brønsted et/ou Lewis) et la déshydrogénation de l’isopropanol en acétone à la présence de sites basiques.
Cependant, la formation de propène est également observée sur zéolithe basique, ce qui implique de nombreuses controverses [38].
Les travaux de Gervasini et al. [39] sur la décomposition de l’isopropanol sur des séries d’oxydes (alumine : Al2O3, magnésie : MgO et silice : SiO2) proposent divers schémas
réactionnels pour l’oxydation de l’isopropanol sur sites acides et/ou basiques, qui sont résumés dans la Figure 4.
Figure 4 : Schémas réactionnels d'oxydation de l'isopropanol en produits de déshydratation, diisopropyléther et propène, et de déshydrogénation, acétone sur des
sites acides de Bronsted (H+), de Lewis (Aį+) et des sites basiques de Lewis (:B), de force
faible (f), moyenne (m) ou forte (F) [39]. isopropanol propène acétone propène propène diisopropyléther H+ (F) ou Aį+(F) E1 E2 :B(m)(F) + Aį+(m)(F) E1b E1b :B(F) :B(F) + Aį+(f)
La majorité des alcools primaires et secondaires réagit en suivant un mécanisme concerté E2 (bimoléculaire), tandis que les alcools tertiaires réagissent via un mécanisme en deux
étapes E1 (monomoléculaire), ceci à cause de la plus grande stabilité du carbocation
intermédiaire formé. Le mécanisme E1 requiert seulement des sites acides très forts,
responsables de la formation d’oléfines, tandis que le mécanisme E2, impliquant à la fois des
sites acides et basiques, conduit également à la formation d’éther [40]. En effet, la littérature [41] rapporte que des solides purement acides, tels que des silice-alumine ou des zéolithes, donnent plus d’oléfines que des oxydes amphotères qui présentent des sites acides et basiques de force équivalente, tels que l’alumine. La déshydratation de l’alcool peut également s’effectuer via un mécanisme E1b (avec participation) mettant en jeu un carbanion
intermédiaire sur des solides très basiques qui possèdent des sites acides/bases aux forces non semblables [42]. La réaction de déshydrogénation suit également un mécanisme de type E1b,
impliquant le même carbanion intermédiaire, la cétone étant formée par élimination de l’hydrogène Į [42-44].
D’autres études ont montré que le propène pouvait être formé sur des sites basiques forts [45,46]. La Figure 5 propose un schéma réactionnel expliquant la déshydratation et la déshydrogénation de l’isopropanol via un mécanisme E1b.
Figure 5 : Formation de propène et d'acétone via un mécanisme E1b catalysé
basiquement avec L site acide faible de Lewis (cation M+), B site basique de force
moyenne de Brønsted (oxygène combiné dans des paires M-O), B site basique fort de
La formation d’alcène à partir d’alcool sur des catalyseurs possédant des sites uniquement basiques est aussi appuyée par Tanchoux et al. [47] et a été proposé dès 1996 par Aramendía
et al. [48] qui ont étudié la déshydratation et la déshydrogénation de l’isopropanol sur de
l’oxyde de magnésium. Par ailleurs, Conceptiȩn-Heydorn et al. [49] n’ont observé aucune formation d’acétone mais des rendements élevés en propène avec respectivement 62 et 78% sur des zéolithes basiques NaY et CsNaY. Sur NaCsX, des traces d’acétone sont observées. Plus récemment, Beauchet et al. [50] ont proposé que l’oxydation d’isopropanol sur une zéolithe basique NaX pouvait advenir via un mécanisme basique, formant du propène, qui pouvait ensuite être oxydé en aldéhyde et en cétone avant oxydation complète en dioxyde de carbone. Ces mêmes auteurs indiquent que l’oxydation des produits secondaires, propène et acétone, est plus difficile que celle de l’isopropanol.
IV. Les catalyseurs d’oxydation
La pièce maîtresse d’un système d’oxydation est bien évidemment le catalyseur. Il se doit de posséder une activité aussi élevée que possible car elle détermine la température d’oxydation nécessaire pour atteindre une certaine conversion, une spécificité par rapport aux réactifs aussi faible que possible pour ne pas discriminer certaines familles de composés, des pertes de charge faibles, des propriétés mécaniques et thermiques satisfaisantes, une durée de vie aussi longue que possible et un coût acceptable.
1. Les catalyseurs non zéolithiques
Du point de vue de leur activité, les catalyseurs PGM (Platinum Group Metals) sont significativement plus actifs que les catalyseurs BMO (Based Metal Oxide).
a. Les métaux nobles
Du fait de leur fort pouvoir oxydant, les métaux nobles (PGM : Platinum Group Metals) sont très couramment utilisés pour l’oxydation totale. Dans cette catégorie, deux métaux sont principalement utilisés : le platine (généralement plus actif) et le palladium, pour des raisons de stabilité et de volatilité limitée.
De nombreuses publications montrent la supériorité du platine pour l’oxydation catalytique de COV, notamment pour les alcènes [51,52], les alcanes autres que le méthane [19,52], les composés oxygénés [53,54] et les aromatiques [15,51,54].
Moro-oka et al. comparent les activités du platine, du palladium et d’une série d’oxydes en oxydation totale du propène [55], de l’isobutène, de l’acétylène, de l’éthylène et du propane [26]. Le platine est toujours le plus actif alors que le palladium est moins actif que certains oxydes.
L’association des deux métaux pour des catalyseurs bimétalliques peut être aussi envisagée. En effet, Kim et al. [56] montrent que pour une petite quantité de Pt ajoutée au catalyseur Pd/ȖAl2O3, celui-ci est plus actif que les catalyseurs monométalliques Pt/ȖAl2O3 ou
Pd/ȖAl2O3. Ajoutons que l’étude de Skoglunk et al. [51] mentionne un effet promoteur du
platine sur le palladium pour l’oxydation des hydrocarbures alors que l’inverse n’est pas vérifié.
C’est sous leur forme réduite (Pt0 et Pd0) que ces deux métaux nobles sont les plus actifs. La plus grande activité des catalyseurs à base de platine peut être reliée à une quantité plus importante d’espèces réduites Pt0, plus facilement formée que Pd0, par autoréduction pendant la calcination du précurseur de métal Pd ou Pt(NH3)4Cl2 [15].
La température a un effet sur la réduction de la surface d’un catalyseur PGM et donc sur son activité. A partir de 850°C, le catalyseur fond, mais le vieillissement thermique commence dès 650°C, température qui est donc considérée comme une limite à ne pas dépasser. Ce vieillissement se traduit par une diminution de la dispersion du métal à la surface du catalyseur [57].
b. Les oxydes de métaux
L’oxydation catalytique sur les oxydes de métaux (BMO : Based Metal Oxide) a été largement étudiée [11]. Ils sont formés à partir des éléments des groupes VIB et IIB de la classification périodique. Ces catalyseurs sont généralement moins actifs et moins sélectifs en CO2 que les métaux nobles. Cependant, ils sont moins chers et résistent mieux à
l’empoisonnement. Ils ne supportent pas les températures élevées tout comme les PGM car ils deviennent instables à partir de 600°C [58].
Les oxydes métalliques les plus actifs en oxydation totale sont les semi-conducteurs de type P. La conduction se faisant à partir des trous positifs dans ce type de semi-conducteur, les électrons sont très mobiles et permettent d’adsorber facilement l’oxygène en surface sous forme anionique telle que O- [16].
Dans le cas de l’oxydation de COV, l’alumine est très couramment utilisée, en tant que catalyseur ou support. C’est pourquoi nous définirons brièvement la nature des sites présents à la surface de l’alumine.
L’alumine se présente sous une grande variété de formes cristallographiques. Les plus intéressantes pour la catalyse sont les phases Ș et Ȗ du fait notamment de leur surface spécifique élevée (en général entre 100 et 200 m2/g).
La surface d’une alumine non déshydratée se présente comme une monocouche de groupements hydroxyles OH. La déshydratation se produit par condensation de deux hydroxyles voisins, avec formation d’une molécule d’eau qui est expulsée de la surface, et d’un « pont d’oxygène » selon le schéma décrit en Figure 6.
Figure 6 : Formation d'un pont oxygène sur alumine.
Les alumines très déshydratées présentent les propriétés catalytiques les plus intéressantes [59].
Selon Peri [60], il existe à la surface de l’alumine très déshydratée cinq types d’OH isolés (visible en infrarouge), différents par leur environnement en ions oxygène (Figure 7). Peri suppose que tous les Al3+ sont équivalents et qu’ils sont situés dans des sites octaédriques (coordinence 6 : AlVI).
Selon ce modèle, la densité de la charge électronique sur le groupement OH croît avec le nombre de O2- voisins, ce qui signifie que l’acidité diminue avec le nombre de O2- voisins.
H O Al H O Al O Al Al
+
H2OFigure 7 : Modèle de Peri.
3800 cm-1 3733 cm-1 3744 cm-1 3700 cm-1 3780 cm-1 Acidité Ĺ lacune OH O
2-Le modèle de Knözinger et Ratnasamy [61] permet lui de différencier 5 types d’hydroxyles qui, d’après leur charge électrostatique, peuvent être soit acides, soit neutres, soit basiques. Les Al3+ sont, soit dans des sites octaédriques (coordinence 6 : AlVI), soit dans des sites tétraédriques (coordinence 4 : AlIV). Sur le schéma (Figure 8) qui suit, les représentations schématiques des différents hydroxyles sont reliées à leurs vibrations observées en infrarouge.
Le processus de déshydratation laisse un ion oxygène sur la couche de surface, et un ion aluminium incomplètement coordiné exposé sur la couche inférieure. Ce cation se trouve dans un « trou » de la surface qui est déficient en électrons, et par conséquent se comporte en site acide de Lewis, tandis que l’ion oxygène exposé présente des propriétés basiques.
Trois types de sites se trouvent donc à la surface de l’alumine : des ions oxygène, des hydroxyles et des ions aluminium tricoordinés. La force acide ou basique de ces sites, et donc l’activité catalytique de l’alumine, dépendent fortement du degré de déshydratation de la surface. Selon certains auteurs, l’acidité de Lewis suffirait à expliquer toutes les propriétés catalytiques de l’alumine : ainsi l’isomérisation du but-1-ène serait catalysée par ces seuls sites acides de Lewis [62]. D’un autre côté, l’alumine catalyse plusieurs réactions qui nécessitent la participation de sites acides de Brønsted [63-65].
Comparativement, la basicité de l’alumine a été très peu étudiée. Mise en évidence par Scwab et Kral [66], elle a été notamment confirmée par Peri [67] et Medena [68], mais n’est jamais proposée comme responsable de réaction observée sur l’alumine. Les sites basiques de l’alumine sont formés en même temps que les sites acides de Lewis selon le schéma présenté en Figure 9 : Al OH Al OH Al Al OH Al Al OH Al Al Al O H
Ib Ia IIb IIa III
3785-3800 cm-1 3760-3780 cm-1 3740-3745 cm-1 3730-3735 cm-1 3700-3710 cm-1
Acidité Ĺ
![Figure 3 : Cycle de Chapman perturbé par la présence de radicaux libres [2].](https://thumb-eu.123doks.com/thumbv2/123doknet/7943345.266109/23.892.263.637.144.538/figure-cycle-chapman-perturbé-présence-radicaux-libres.webp)